

Abstract
The adhesion forces of cervical carcinoma cells in tissue culture were measured by using the manipulation force microscope, a novel atomic force microscope. The forces were studied as a function of time and temperature for cells cultured on hydrophilic and hydrophobic polystyrene substrates with preadsorbed proteins. The cells attached faster and stronger at 37°C than at 23°C and better on hydrophilic than on hydrophobic substrates, even though proteins adsorb much better to the hydrophobic substrates. Because cell adhesion serves to control several stages in the cell cycle, we anticipate that the manipulation force microscope can help clarify some cell-adhesion related issues.
Many of the activities of mammalian cells in vivo, such as embryogenesis, mitosis, morphogenesis, cell orientation, cell motility, and survival depend on attachment to neighboring cells and the extracellular matrix (1–5). Attachment is often mediated by integrins, transmembrane glycoproteins that bind to ligands such as collagens, laminin, (LAM) or fibronectin (FN) (1, 6). Cell–substrate adhesion involves a cascade of events leading to integrin activation and strong adhesion (6), whereas a force applied to an integrin receptor induces a local strengthening response of the integrin–cytoskeleton linkages (7). Cell attachment is an active process and therefore is temperature-dependent. Umbreit and Roseman (8) concluded from studies of cell-aggregation kinetics that cell–cell attachment involved two steps, an initial recognition event and an active attachment process expending metabolic energy.
In vitro, most mammalian cells are anchorage-dependent and attach firmly to the substrate. Several attempts have been made to quantify cell adhesion. The simplest attachment assay is based on rinsing the surface to remove weakly attached cells from the substratum and counting the remaining cells (9–13). Cells adhering with a force less than the rinsing force are removed, but this force is difficult to control and therefore ill-defined.
Quantitative force measurements have been made using centrifugal assays (14, 15). Here, the discrimination force is better defined, and a probability distribution is obtained by counting the number of remaining cells for a given series of applied forces. This method was recently improved (16, 17), solving the technical problems in applying large forces in the earlier centrifugal assays.
Significant progress has been made in measuring the traction forces of cells. By cultivating cells on an elastic silicon-rubber membrane (18–20) or on protein films formed at fluorocarbon oil–water interfaces (21), it is possible to estimate the forces applied by the cell to the substrate by observing the deformation of the elastic films. Recently, a related technique employing a micromachined array of small pads supported by springs was used to resolve the traction force exerted by the different parts of a locomoting cell (22).
The atomic force microscope (23) has found many uses as a tool in biology. As a microscope scanning biological surfaces, it has characterized adsorbed lysozyme (24), imaged cells (25), and purple membranes (26), has followed the action of enzymes (27) and changes in the local elasticity of cells (28), and has been used in several other applications (29).
The atomic force microscope is a sensitive force transducer because subnanometer deflections of its cantilever can be detected. The cantilever is a micromachined film acting as a spring with typical compliances (from 0.01 N/m to 1 N/m) that result in piconewton force sensitivity. Applied to specific biomolecular recognition events, the forces of biotin–streptavidin (30, 31) and antibody-antigen interactions (32) were directly measured. The technique has also been used for investigating the interaction between complementary strands of DNA oligomers (33).
We recently introduced the manipulation force microscope, an atomic force microscope adapted to measuring the force necessary to dislodge micrometer-sized objects attached to surfaces (Fig. 1). This instrument has been used to measure adhesion forces of protein-covered silica spheres adsorbed to polystyrene surfaces (34). In this paper, we extend this concept to measure the force necessary to displace cells attached to various substrates (Fig. 2).
Figure 1.

The manipulation force microscope employs an inclined atomic force microscope cantilever and laser beam deflection to measure the force when displacing cells adhering to a substrate. The cells are viewed by using an inverted optical microscope.
Figure 2.

Cell displacement. A cartoon of the cell displacement is shown on the Left, the cells as viewed in the optical microscope are shown in the Center, and the applied force is shown as a function of displacement on the Right. The arrows indicate the approximate position on the force curve when the snapshots were taken. The cells were displaced by aligning the cantilever of the force microscope with the cell. When starting translation of the sample, no force was measured (A). Eventually, the cell made contact with the cantilever, and the force on the cell increased (B). The cell was released from the surface gradually (C), and was finally translated at negligible force when all bonds to the substrate were broken (D), at which point data logging and translation was stopped. In a typical experiment, adhesion force measurements on 200–300 cells were carried out. Each cell was displaced only once. Each force curve was analyzed to find the baseline-to-peak force, ΔF, the force peak width, Δx, and the work, ΔW.
METHODS
Cell Culture.
Cells of the human cervical carcinoma cell line NHIK 3025 (35, 36) were cultured in CO2-independent medium (GIBCO/BRL) containing 15% fetal calf serum. The cells were kept in a humidified incubator at 37°C and released from culture flasks twice per week by using 0.136 g/liter trypsin and seeded on new flasks.
Cell Preparation.
Cells were released with trypsin, centrifuged, and re-suspended in medium with serum. The cells were kept suspended in medium at room temperature for 90 min after trypsinization, at which point the cells were seeded in the experimental wells and the experiment started.
Microsphere Preparation.
Silica microspheres (4 μm; Bangs Laboratories, Carmel, IN) were exposed to a 5% solution of (γ-aminopropyl)triethoxysilane (Fluka) for 2 hr, a 2.5% solution of glutaraldehyde (Fluka) for 2 hr, and 0.5 mg/ml FN (Sigma) in 33 mM Tris-buffered NaCl (100 mM, pH 7.4). The spheres were continuously stirred and were washed between each step.
Surface Preparation.
Experimental wells were made by gluing (RTV 118, General Electric Silicones, Waterford, NY) an 11 × 3 mm Viton O-ring (Busak and Shamban, Ft. Wayne, IN) to a surface cut from a tissue culture dish (Nunc). Two types of surfaces were used: a hydrophilic polystyrene surface (for tissue culture) and a hydrophobic polystyrene surface. Before use, the wells were washed in ethanol and dried under nitrogen gas.
To prepare protein-covered surfaces, BSA (Calbiochem), FN (Sigma), or LAM (Sigma) at 0.5 mg/ml in Tris-buffered saline, pH 7.4, were adsorbed for 20 min at room temperature.
Instrumentation.
Briefly, the experimental well was closed by the glass slide holding the cantilever (Fig. 1). A small gap between the O-ring and the glass slide was held in place by surface tension and allowed for the relative movement of cantilever and sample. The cantilever (Digital Instruments, Santa Barbara, CA) was glued to a standard glass microscope slide (Menzel, Braunschweig, Germany) at 30° to vertical by using a laser for precise angular alignment. A water-filled chamber was glued on top the glass slide to improve the focus at the cantilever. A fiber optic laser (Schäfter & Kirchhoff, Hamburg, Germany), a beam-splitter cube (Melles Griot, Irvine, CA), and a split diode (Advanced Photonix, Camarillo, CA) in an autocollimator arrangement were used to detect the cantilever deflection. The diode signal was amplified, measured with a multimeter (Keithley) at a rate of 500 Hz, and logged by a personal computer. The sample was translated continuously at 2.5 μm/s by a geared motor (Halstrup, Kirchzarten, Germany) that drove a hydraulic micromanipulator (Narishige, Tokyo). The optical detection system was characterized by displacing the cantilever using a 100-μm gold wire glued to the sample holder, and the translation was calibrated by using a high-resolution charge-coupled device camera (Photometrics, Tucson, AZ) attached to the microscope. The cantilever stiffness was measured to 0.34 N/m by using a 13-μm gold wire as standard.
Analysis.
The force peaks were identified by the increased separation of consecutive data points when interacting with the cell. By using a computer program, regions of probable contact were identified when the standard deviation of the force within a sliding window (corresponding to 50-nm translation) increased above a discriminatory level. This level was set to 3× the typical noise in the sample.
For each region of probable contact, the baseline-to-peak force, ΔF, the peak width, Δx, and the area between the force curve and the baseline, ΔW, were calculated (Fig. 2). The region with the largest area was selected as the peak.
The results were then pooled to obtain the median force as a function of time (Fig. 3) or sorted to give cumulative force distributions (Fig. 4).
Figure 3.

Cell-substrate adhesion forces on cell-culture surfaces. Median forces as a function of time for cells seeded on hydrophilic polystyrene (PS-phil) used for cell culture covered with serum protein (A), preadsorbed BSA (B), preadsorbed FN (C), and pre-adsorbed LAM (D) for 23°C (○) and 37°C (•) are shown. The median forces were found by grouping 19 consecutive measurements. The fit to the cell attachment model is shown as a solid line. The fitting parameters are given in Table 1.
Figure 4.

Force distributions for cells in mitosis compared to the cell population. The median force of the cells in mitosis was 23 nN and was 170 nN for the population.
Attachment Model.
To quantify the attachment process, we developed a simple model that used the assumption that the cell had B0 bonds to the surface in equilibrium, that the probability of attaching a bond did not change with time, and that no bonds were formed initially (t = 0). In a small time interval dt, the change in the number of unattached bonds dB(t) must be proportional to the number of unattached bonds B(t) and to the time interval
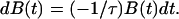 |
1 |
where 1/τ is the probability that a bond will attach in unit time; hence, τ is the typical time for attachment. Thus,
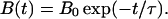 |
2 |
The force is assumed to be directly proportional to the number of attached bonds [B0 − B(t)], hence
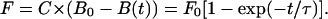 |
3 |
where the saturation force F0 is equal to the proportionality factor C times the equilibrium number of bonds B0. The model was further adjusted to account for a considerable lag time observed at lower temperatures between cell injection and the onset of attachment. The lag time t0 is the time from when the cells are seeded to when the adhesion forces are observed. Thus, we arrive at the function used for fitting the data,
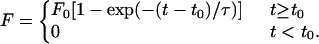 |
4 |
Fitting the Data to the Model.
The data were fit to Eq. 4 by using the Marquant routine (37). This routine minimizes the penalty given by the square of the distance from the data point to the fitting function. Thus, outlying points may have a large influence on the fitting result.
For the data in question, the width of the force distribution was large at every time observed. To reduce the importance of extreme points in the distribution, the data were fit to a set of median forces found by grouping 19 consecutive measurements. The time since the start of the experiment was represented by the mean time of the group. This procedure reduces the number of data points in the fit and may make the fit unstable in some cases. When a poor fit was obtained, parameters found by fitting to the original data set were used.
The median force was used instead of the mean because the median is a more robust measure. In some cases, when the cell adhesion force was high, the cell membrane did not support the applied force and ruptured. Consequently, too small an adhesion force was measured. Thus, the mean force will be underestimated if a single cell ruptures, whereas the median force is underestimated if more than 50% of the cells rupture, providing that the maximum applied force on ruptured cells is larger than the median force. The number of ruptured cells was well within this limit in all experiments.
RESULTS
Cervical carcinoma cells were used to investigate the effect of various adsorbed proteins on hydrophilic polystyrene substrates normally used for cell culture. The cells were seeded either in a medium containing serum on clean surfaces (giving a surface with adsorbed serum proteins) or on surfaces where BSA, FN, or LAM had been preadsorbed.
The median adhesion forces are shown as a function of time in Fig. 3. To characterize the force dynamics, the data were fitted by using a simple model,
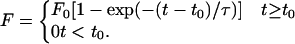 |
described in Methods. This model introduces the parameters F0, t0, and τ, where F0 is the saturation force, t0 is the lag time (the time from when the cells are seeded to when attachment forces are large enough to be measured), and τ is the attachment time (the characteristic time for adhesion of a bond and a measure of how quickly the cell reaches the saturation force). The results obtained by fitting the data to this model are shown in Table 1.
Table 1.
Results of cell adhesion and microsphere adsorption experiments
| Sample | T, °C | F0, nN | t0, ×103 s | τ, ×103 s |
|---|---|---|---|---|
| Cell adhesion | ||||
| Phil/serum | 23 | 121 | 2.4 | 3.7 |
| Phil/serum | 37 | 189 | 0.0 | 3.2 |
| Phil/BSA | 23 | 121 | 1.7 | 3.8 |
| Phil/BSA | 37 | 184 | 0.64 | 3.2 |
| Phil/FN | 23 | 183 | 2.2 | 4.9 |
| Phil/FN | 37 | 204 | 0.86 | 1.1 |
| Phil/LAM | 23 | 108 | 1.7 | 1.9 |
| Phil/LAM | 37 | 105 | 0.72 | 0.66 |
| Phob/serum | 23 | 0.0 | — | — |
| Phob/serum | 32 | 101 | 1.6 | 11.9 |
| Phob/serum | 37 | 87 | 1.9 | 4.7 |
| Phil/no serum | 23 | 45 | 1.7 | 1.7 |
| Phil/no serum | 37 | 45 | 0.44 | 0.63 |
| Phob/no serum | 37 | 37 | — | — |
| Phil/BSA, no serum | 37 | 19 | — | — |
| Microsphere adsorption | ||||
| Phil/clean/FN-sphere | 23 | 186 | — | — |
| Phob/clean/FN-sphere | 23 | >220 | — | — |
| Phil/BSA/FN-sphere | 23 | 2.3 | — | — |
| Phob/BSA/FN-sphere | 23 | 0.27 | — | — |
Median forces were fit to the adhesion model to obtain the saturation force F0, the lag time t0, and the attachment time τ. Samples with no t0 or τ reported were allowed to reach the saturation force before measurement, so only the median force is reported. Each sample is described by the substrate used, where Phil is hydrophilic polystyrene for cell culture and Phob is hydrophobic polystyrene.
On all substrates except LAM, the adhesion force was larger at 37°C than at 23°C. The cells adhered with the strongest force on the FN-covered surface, indicating integrin-mediated adhesion to adsorbed fibronectin. On serum protein and BSA, intermediate forces were measured, whereas on LAM, the saturation force was much lower and not temperature dependent.
The cells also adhered more rapidly for all samples when the temperature was increased; both the lag time, t0, and the typical time of attachment, τ, were reduced. The adhesion force was related to the cell shape. Initially, the cells were rounded and did not adhere. After a lag time, the cells started adhering and gradually spread. This transition in shape corresponded well with the increased force observed (data not shown).
Cells in mitosis were identified by observing the shape of the nucleus. The attachment forces of these cells were reduced compared with the normal cell population, and had a wider distribution (Fig. 4). The measurement did not seem to affect the cells, because mitosis proceeded normally after the cells had been displaced.
Anchorage-dependent cells proliferate on hydrophilic surfaces (such as glass and polystyrene) specially treated for cell culture. On hydrophobic polystyrene surfaces, cells initially attach but later become rounded and leave the surface.
Cells seeded onto hydrophobic polystyrene surfaces in medium containing 15% fetal calf serum did not attach at room temperature (Fig. 5; Table 1). At higher temperatures, the attachment force increased, the lag time decreased, and the forces were smaller than on the corresponding hydrophilic surface.
Figure 5.

Cell-substrate adhesion on hydrophobic polystyrene. Median forces on a hydrophobic polystyrene substrate at 23°C (○), 32°C (□), and 37°C (•) are shown.
Proteins adsorb to most surfaces. It is expected, therefore, that the membrane proteins may adsorb directly to the substrate. To evaluate the strength of such nonspecific adhesion, carefully washed cells were seeded in serum-free medium on both hydrophobic and hydrophilic polystyrene surfaces. The cells adhered quickly, but the saturation force was much lower than when serum was present in the medium (Fig. 6; Table 1). The saturation force did not depend on the temperature and depended little on the choice of polystyrene substrate.
Figure 6.

Direct adsorption to polystyrene. Median forces on a hydrophilic polystyrene substrate for cell culture in a medium with 15% fetal calf serum (dashed lines) and without serum (solid lines) are shown. Samples at 23°C are indicated with open symbols, and samples at 37°C with filled symbols.
Cells also were seeded in serum-free medium on hydrophilic polystyrene with preadsorbed BSA. The saturation force was even lower than in the previous cases, but BSA did not block cell adhesion completely (Table 1).
To assess the strength of the FN interaction with the substrate, FN-covered glass microspheres (4 μm in diameter) were allowed to adsorb for 1 hr on hydrophilic and hydrophobic polystyrene substrates before the adhesion forces were measured.
The adhesion forces on the hydrophobic polystyrene substrate were very strong and outside the dynamic range of the force microscope. On the hydrophilic polystyrene surface, the median force was 186 nN, which is stronger than that observed for BSA, myoglobin, lysozyme, and ferritin in a previous study (34). This shows that FN adheres well to the surfaces used.
To evaluate the blocking efficiency of BSA, BSA was preadsorbed on hydrophilic and hydrophobic polystyrene substrates before the FN-covered microspheres were added. The adhesion forces were greatly reduced in both cases, but the residual force was 10-fold greater on hydrophilic polystyrene (Table 1).
The force acting on the cell was measured as a function of displacement. The force profile contains information on the distribution of attachment bonds to the surface but also depends on the elastic deformation of the cell. The distance over which the force acted was similar to the cell diameter (data not shown), indicating that the cell detached from the surface in a Velcro-like manner, disrupting bonds close to the advancing cantilever (Fig. 2).
DISCUSSION
Several researchers have measured cell adhesion forces. These studies considered, in most cases, the fraction of cells adhering better than a discrimination force. This force may be rather arbitrary (as when using a washing technique) or better defined (as in studies employing a centrifuge). Our study, unlike previous studies, directly measures the time course of cell adhesion forces.
We have chosen to describe the cell adhesion force dynamics by the lag time, t0, which is the time before adhesion forces are observed, and the adhesion time, τ, which is the time over which adhesion saturates. We observed lag times from 0 to 40 min and adhesion times from 10 to 90 min on the hydrophilic substrate. These times always decreased on increasing the temperature and depended on the adsorbed protein layer on the substrate (Table 1). These results are similar to those obtained by different methods for other cell strains. We estimate lag times of 2 min (11) to 20 min (10) and adhesion times of 15 min (15) to 40 min (11) from data found in the literature.
Umbreit and Roseman (8) observed that cell–cell adhesion involved two steps, an initial recognition step (in which a loose association was formed) and a subsequent metabolic step (strengthening adhesion). At low temperatures, the rate of strong cell adhesion was limited by the metabolic step, whereas at 37°C, the recognition event was rate-limiting. In the present study, two-state binding forces were not observed directly for cell–substrate adhesion. However, most cells did not diffuse freely during the initial lag period, indicating that they were loosely bound to the surface even though adhesion forces were not resolved. In any case, the lag and adhesion times depend on the rate at which strong adhesion bonds were formed. These times increased on lowering the temperature, showing that the rate of bond formation was reduced. Hence, the formation of strong adhesion bonds to the substrate is temperature-dependent, in agreement with previous results and expectations.
Different cell types adhere with different forces. Coman (38) used glass needles to measure cell–cell adhesion forces as large as 10,000 nN in normal cervix epithelial cells, whereas his apparatus lacked sensitivity to measure the force between cervical carcinoma cells. However, his data indicate that this force is <2,000 nN. By using a high-speed centrifugation technique, Thoumine et al. (16) exposed epithelial cells to forces tangential to the substrate. These cells started to dissociate from the surface at 100 nN. Recently, Yamamoto et al. (39) measured the adhesion forces of murine fibroblast cells by using a technique employing ideas similar to the manipulation force microscope. Forces between 300 and 400 nN were necessary to remove these cells from a serum protein-covered glass substrate.
Burton and Taylor (20) found that the traction force exerted by fibroblasts on an elastic membrane may be as large as 1,200 nN. Other experiments on fibroblasts have shown comparable forces (22), whereas the traction forces of keratocytes were measured at 20 nN (19).
It has been argued (19) that the traction force is exerted to overcome the adhesion force for locomoting cells, observing that viscous and nonspecific friction forces are negligible. It was proposed that traction forces at the cell front are exerted to mechanically disrupt attachments at the cell tail (19). However, it is unlikely that the force applied by the cell to the substrate is of critical strength with respect to cell-attachment bond disruption at all times. Hence, the traction forces are a lower limit for the force necessary to dissociate the cell bonds from the surface.
These traction force experiments showed that cells are bound to the substrate by bonds stressed as a result of cytoskeleton contraction. This should be taken into account when interpreting data from manipulation force microscopy. We expect that it should be easier to dissociate bonds already under load. If this is the case, the manipulation force microscope measures the force required in addition to the instantaneous “traction force” to dislodge the cell.
The forces observed with the manipulation force microscope (Table 1) are similar to those measured by using other methods but depend strongly on the details of the experiment. As shown in the theoretical work of Chang and Hammer (40), the force necessary to remove a bound microsphere depends on the direction with which the force is applied and the nature of the bonding to the substrate. We conclude that larger forces must be employed to lift the microsphere directly from the surface than to push it forward. This also may be the case for cell–substrate adhesion.
The speed at which the cell is displaced also may influence the force measured. Recent studies have shown that the integrin links to the cytoskeleton respond to external force stimuli within seconds (7). It also has been proposed that the rate at which an attachment bond is disrupted influences its rupture force (41). The manipulation force microscope is well suited to displacement-speed studies, but such experiments have not yet been carried out.
Integrin-mediated adhesion to FN receptors is well documented (6), and a larger attachment force following an increased FN concentration on the substrate is expected. However, the close similarity between the BSA- and the serum protein-covered substrate (Table 1) was a surprise. The low interaction forces between the FN-covered microsphere and the BSA-covered substrate show that FN does not bind directly to BSA. However, it is known that proteins may displace other proteins of lower affinity (42); thus, FN and other proteins in solution may displace BSA, giving an attachment ligand density similar to that on the cell culture substrate. Previous work has shown that BSA has a low affinity for hydrophilic polystyrene but a high affinity for hydrophobic polystyrene (34). As shown in Table 1, BSA blocked the adsorption of FN-covered microspheres 10-fold better on the hydrophobic surface. This indicates that BSA was displaced on hydrophilic, but not on hydrophobic, polystyrene. Supporting this conclusion, cells seeded on a BSA-covered substrate in a serum-free medium adhered with low forces (Table 1). The residual force may be the result of membrane proteins displacing BSA.
On the LAM-covered hydrophilic substrate, the adhesion forces were less than on serum protein and the saturation force was independent of temperature. However, the forces were stronger than for cells seeded in medium with serum on hydrophobic or clean polystyrene substrates. The low forces observed indicate that LAM was not displaced from the surface by serum protein. Hence, receptors for LAM were present at low density.
When cells were seeded directly on polystyrene substrates that had no adsorbed protein, they adhered with low forces on both hydrophobic and hydrophilic substrates. Based on earlier observations of protein adhesion to these surfaces (34), we expect that membrane proteins adsorb directly to the substrates but with much higher affinity on hydrophobic polystyrene. However, similar forces were measured on both substrates and at both 23°C and 37°C on hydrophilic polystyrene (Fig. 6), indicating that the membrane proteins were pulled out of the cell membrane and not away from the surface. In cases where the cells adhered specifically to adsorbed proteins, the forces necessary to remove them were much larger. This observation may be explained by a strengthening of the integrin–cytoskeleton links on specific attachment, as observed by others (7, 43).
Cell adhesion forces on a protein-covered hydrophobic polystyrene substrate has not, to our knowledge, been studied previously. Our results (Fig. 5, Table 1) show that this force was strongly temperature-dependent and lower than the force on hydrophilic polystyrene substrates but was higher than for cells adsorbed on clean polystyrene. Because protein binds better to hydrophobic than to hydrophilic polystyrene, a layer of adsorbed serum protein must be present on both surfaces. The reduced adhesion force must therefore be caused by an altered protein composition on the substrate resulting from a change in the relative affinities or from morphological changes in the adsorbed proteins. If the number of integrin ligands on the surface were reduced, a reduction in attachment force would be expected. However, the cells did not adhere on hydrophobic polystyrene at 23°C, whereas adhesion on hydrophilic polystyrene still was considerable. We therefore propose that the difference in force observed is caused by partial denaturation and/or changes in protein orientation on the surface. This may lead to frustration of the binding sites, increasing the activation energy of binding, which explains the relatively slow dynamics of cell attachment to the hydrophobic polystyrene surface. In addition, the binding sites of some of the integrin types may be inaccessible in the protein layer. Work on melanoma cells has shown that lack of αvβ3 integrin binding induce apoptosis within 3 days (4). Similar mechanisms may be at play on the hydrophobic substrate, because the cells initially attach but become rounded and leave the surface after a few days.
Burton and Taylor (20) showed that the traction force exerted by the cell on the substrate during mitosis varied greatly during this process but was generally lower than for locomoting fibroblast cells. By using the manipulation force microscope, we selected a subset of cells in mitosis from a large population and found that the cell attachment force was reduced, as anticipated (Fig. 4).
Manipulation force microscopy gives detailed information, unobtainable by other methods, on the force applied to the cell as a function of displacement. The force was at a maximum when the cantilever dislodged the region close to the nucleus from the surface, indicating that the adhesion was stronger here. However, interpretation of this data is complicated because of cell deformation. This problem may be overcome in the future by combining manipulation force microscopy with video microscopy techniques to correlate the measured force with cell shape.
In conclusion, we have measured the adhesion force of individual cervix cells to various substrates by using the manipulation force microscope and obtained a reasonable agreement with results obtained by other methods.
Cell adhesion is important in several processes, and we expect that this cell parameter is sensitive to a wide range of external stimuli aside from the temperature and the protein composition on the substrate. Particularly, substances modulating cell affinity are of great interest in the treatment of cancer (4), and their effects may be studied in detail by using the manipulation force microscope. We also anticipate that the force microscope may readily be combined with other techniques, such as immunological methods and staining, for further experiments on cell adhesion.
Acknowledgments
This work was supported by the Norwegian Research Council and Vista, a research collaboration between the Norwegian Academy of Sciences and Letters and Den norske stats oljeselskap (Statoil).
ABBREVIATIONS
- FN
fibronectin
- LAM
laminin
References
- 1.Gumbiner B M. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 2.Klymkowsky M W, Parr B. Cell. 1995;83:5–8. doi: 10.1016/0092-8674(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 3.Lauffenburger D A, Horwitz A F. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 4.Mason M D, Allman R, Quibell M. J R Soc Med. 1996;89:393–395. doi: 10.1177/014107689608900710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchison T, Cramer L. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 6.Hynes R O. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 7.Choquet D, Fesenfeld D P, Sheetz M P. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 8.Umbreit J, Roseman S. J Biol Chem. 1975;250:9360–9368. [PubMed] [Google Scholar]
- 9.Walther B A, Öhman R, Roseman S. Proc Natl Acad Sci USA. 1973;70:1569–1573. doi: 10.1073/pnas.70.5.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin K, Höök M, Öbrink B, Timpl R. Cell. 1981;24:463–470. doi: 10.1016/0092-8674(81)90337-8. [DOI] [PubMed] [Google Scholar]
- 11.Liao N-S, St. John J, Du Z, Cheung H T. Exp Cell Res. 1987;171:306–320. doi: 10.1016/0014-4827(87)90164-9. [DOI] [PubMed] [Google Scholar]
- 12.Dillner L, Dickerson K, Manthorpe M, Puoslathi E, Engvall E. Exp Cell Res. 1988;177:186–198. doi: 10.1016/0014-4827(88)90036-5. [DOI] [PubMed] [Google Scholar]
- 13.Chan B M, Kassner P D, Schiro J A, Byers H R, Kupper T A, Helmer M E. Cell. 1992;68:1051–1060. doi: 10.1016/0092-8674(92)90077-p. [DOI] [PubMed] [Google Scholar]
- 14.McClay D R, Wessel G M, Marchase R B. Proc Natl Acad Sci USA. 1981;78:4975–4979. doi: 10.1073/pnas.78.8.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lotz M E, Burdsal C A, Erickson H P, McClay D R. J Cell Biol. 1989;109:1795–1805. doi: 10.1083/jcb.109.4.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thoumine O, Ott A, Louvard D. Cell Motil Cytoskeleton. 1996;33:276–287. doi: 10.1002/(SICI)1097-0169(1996)33:4<276::AID-CM4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Channavajjala L S, Eidsath A, Saxinger W C. J Cell Sci. 1997;110:249–256. doi: 10.1242/jcs.110.2.249. [DOI] [PubMed] [Google Scholar]
- 18.Harris A K, Wild P, Stopak D. Science. 1980;208:177–179. doi: 10.1126/science.6987736. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Leonard M, Oliver T, Ishihara A, Jacobson K. J Cell Biol. 1994;127:1957–1964. doi: 10.1083/jcb.127.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burton K, Taylor D L. Nature (London) 1997;385:450–454. doi: 10.1038/385450a0. [DOI] [PubMed] [Google Scholar]
- 21.Keese C R, Giaever I. Exp Cell Res. 1991;195:528–532. doi: 10.1016/0014-4827(91)90406-k. [DOI] [PubMed] [Google Scholar]
- 22.Galbraith C G, Sheetz M P. Proc Natl Acad Sci USA. 1997;94:9114–9118. doi: 10.1073/pnas.94.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binnig G, Quate C F, Gerber C. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 24.Radmacher M, Fritz M, Cleveland J P, Walters D A, Hansma P K. Langmuir. 1994;10:3809–3814. [Google Scholar]
- 25.Butt H-J, Wolff E, Gould S, Northern B D, Peterson C, Hansma P. J Struct Biol. 1990;105:54–61. doi: 10.1016/1047-8477(90)90098-w. [DOI] [PubMed] [Google Scholar]
- 26.Müller D J, Schabert F A, Büldt G, Engel A. Biophys J. 1995;68:1681–1686. doi: 10.1016/S0006-3495(95)80345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radmacher M, Fritz M, Hansma H G, Hansma P K. Science. 1994;265:1577–1579. doi: 10.1126/science.8079171. [DOI] [PubMed] [Google Scholar]
- 28.A-Hassan E, Heinz W F, Antonik M D, D’Costa N P, Nageswaran S, Schonenberger C-A, Hoh J H. Biophys J. 1998;74:1564–1578. doi: 10.1016/S0006-3495(98)77868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansma H G, Hoh J H. Annu Rev Biophys Biomol Struct. 1994;23:115–139. doi: 10.1146/annurev.bb.23.060194.000555. [DOI] [PubMed] [Google Scholar]
- 30.Lee G U, Kidwell D A, Colton R J. Langmuir. 1994;10:354–357. [Google Scholar]
- 31.Florin E-L, Moy V T, Gaub H E. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 32.Dammer U, Hegner M, Anselmetti D, Wagner P, Dreier M, Huber W, Güntherodt H-J. Biophys J. 1996;70:2437–2441. doi: 10.1016/S0006-3495(96)79814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee G U, Chrisey L A, Colton R J. Science. 1994;266:771–773. doi: 10.1126/science.7973628. [DOI] [PubMed] [Google Scholar]
- 34.Sagvolden G, Giaever I, Feder J. Langmuir. 1998;14:5984–5987. [Google Scholar]
- 35.Nordbye K, Oftebro R. Exp Cell Res. 1969;58:458. (abstr.). [Google Scholar]
- 36.Oftebro R, Nordbye K. Exp Cell Res. 1969;58:459–460. (abstr.). [Google Scholar]
- 37.Press W H, Flannery B B, Teukolsky S A, Vetterling W T. Numerical Recipes. Cambridge, U.K.: Cambridge Univ. Press; 1986. [Google Scholar]
- 38.Coman D R. Cancer Res. 1944;4:625–629. [Google Scholar]
- 39.Yamamoto A, Mishima S, Maruyama N, Sumita M. Biomaterials. 1998;19:871–879. doi: 10.1016/s0142-9612(97)00248-2. [DOI] [PubMed] [Google Scholar]
- 40.Chang K-C, Hammer D A. Langmuir. 1996;12:2271–2282. [Google Scholar]
- 41.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elgersma A V, Zsom R L J, Lykelma J, Norde W. J Colloid Interface Sci. 1992;152:410–428. [Google Scholar]
- 43.Wang N, Ingber D E. Biophys J. 1994;66:2181–2189. doi: 10.1016/S0006-3495(94)81014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]