

Abstract
We have combined neutron solution scattering experiments with molecular dynamics simulation to isolate an excess experimental signal that is caused solely by N-acetyl-leucine-amide (NALA) correlations in aqueous solution. This excess signal contains information about how NALA molecule centers are correlated in water, and we show how these solute–solute correlations might be determined at dilute concentrations in the small angle region. We have tested qualitatively different pair distribution functions for NALA molecule centers—gas, cluster, and aqueous forms of gc(r)—and have found that the excess experimental signal is adequate enough to rule out gas and cluster pair distribution functions. The aqueous form of gc(r) that exhibits a solvent-separated minimum, and possibly longer-ranged correlations as well, is not only physically sound but reproduces the experimental data reasonably well. This work demonstrates that important information in the small angle region can be mined to resolve solute–solute correlations, their lengthscales, and thermodynamic consequences even at dilute concentrations. The hydration forces that operate on the microscopic scale of individual amino acid side chains, implied by the small angle scattering data, could have significant effects on the early stages of protein folding, on ligand binding, and on other intermolecular interactions.
Keywords: hydration structure, protein folding
Energy landscape models have defined a “new” view of protein folding for explaining the kinetics and thermodynamics of protein folding (1–3). The free energy surface is postulated to be funnel-like in shape; that is, the energy decreases faster than the diminishment in the number of states, but with a folded structure minimum that is unique and well separated in energy from the nearest non-native state. Both long and short-ranged forces are important because both imply average funnel-like behavior whereas the latter ensure the uniqueness of the native structure minimum. These theoretical conclusions are partly based on highly idealized lattice models of proteins, which have no atomic detail of amino acid side chains and use very nonspecific descriptions of residue-residue interactions, and where individual beads are considered to be several amino acids that have been “renormalized” (1–3). Although the concept of funnel-like energy landscapes is an appealing one, no definitive connection has been made between the landscape model and the genuine physical forces such as hydrogen-bonding or hydration, etc., that may actually give rise to a funneled energy surface.
What is the molecular origin of these free energy biases, and how do we determine them? Our intuition is that amino acid interactions mediated by aqueous solvent are a dominant feature of funneled landscapes in protein folding. We have been especially interested in the idea that solute molecules may influence the structure of water out to a distance of several hydration layers from the surface and that these alterations in water structure may in turn give rise to microscopic long-ranged favorable or unfavorable hydration forces between hydrophobic and hydrophilic solutes, respectively (4–7). We define microscopic long-range hydration forces to mean a significant free energy stabilization of amino acid groups in water beyond the point at which they are in van der Waals contact. This contrasts with simpler hydration models based on minimizing hydrophobic solvent-accessible surface area.
The estimation of the range and magnitude of microscopic hydration forces acting between amino acids will require the development of an approach that is sensitive to both water structure and any thermodynamic forces present because of hydration. Analysis of neutron solution scattering experiments of amino acids in water by using molecular dynamics simulations probed changes in water structure arising from a shift of the main water diffraction peak (5–7). In this paper, we have combined information from experiments and simulation to isolate N-acetyl-leucine-amide (NALA) correlations in water found in the small angle region. After subtracting the simulated terms from the total measured scattering, we isolated scattering caused by NALA correlations and determined a model, gc(r), that best reproduces this excess signal. Once the solute–solute pair correlation function in aqueous solution was determined, it could be related to hydration forces through
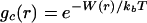 |
1 |
where W(r) is the “potential of mean force” between the two solutes separated by a distance r. The average occurs over all explicit solvent configurations, as well as all orientations and conformations of the two solute molecules. The importance of gc(r), which in turn defines W(r), is that it describes the net correlations between solute pairs, which take into account the complicated solvent environment. The effect of long-range hydration interactions should therefore be immediately evident when the scattering data from aqueous solution are converted into the pair correlation function by modeling or an appropriate Fourier transform.
In the solution scattering experiments reported (5–7), the mole fraction of solute is quite small: 1 solute per 100 water molecules. We chose to work at these dilute concentrations because we expected the hydration forces we were trying to characterize would be operative in early protein folding when the local concentration of amino acids is relatively dilute and residues are well hydrated. However, the relative weight of observed water–water contributions to the scattering intensity compared with solute–solute contributions is of the order 5000:1, which means the direct observation of solute–solute correlations is not possible because of the weak signal-to-noise ratio of solution scattering experiments.
Nonetheless, the scattering contrast between water and NALA should allow the characterization of solute correlations in solution because the solutes introduce new lengthscales into the water correlations that are due to the size, shape, and type of correlation arising from the solutes. The solute–solute correlations are, in fact, directly related to the excluded volume effect seen in the water correlations, and the primary purpose of this paper is to show how these solute–solute correlations might be determined at dilute concentrations in the small angle region. Our combined experiments and simulation demonstrate the feasibility of detecting the influence of hydration forces on the microscopic scale of individual amino acid side chains at dilute concentrations and encourage more careful study of the information content of small angle solution scattering.
Determining the Solute–Solute Correlations
One of us recently has shown that changes in the measured radial distribution functions can be due to the excluded volume of solutes and not exclusively to water structure that actually has changed because of the presence of “structure-making” solutes (8). It was suggested that the water partial radial distribution functions that are measured in a solution be normalized by a correlation function describing a uniform fluid excluded from a collection of spherical holes. The uniform fluid pair correlation function for the hydrogen–hydrogen (HH) correlations of water molecules, excluded from a collection of spherical holes, is (ref. 8, Eq. 18)
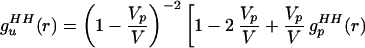 |
2.1 |
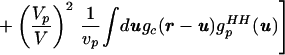 |
where V is the total system volume, Vp is the total volume occupied by the solutes, vp is the volume occupied by an individual solute, gc(r) is the radial distribution function for the solute centers, and gpHH(r) is the solute internal radial distribution function. When gsolutionHH(r) (the HH correlations between water molecules in solution) is normalized by guHH(r), the unwanted correlations caused by the excluded volume effect are removed. The renormalized gsolutionHH(r) then can be compared with gbulkHH(r) to determine whether water has indeed been restructured because of the presence of solutes. As pointed out in ref. 8, a significant approximation is introduced when the solute particles are assumed to be spherical, and we return to this point later.
However, we want to show that Eq. 2.1 can be manipulated further to isolate the solute centers pair correlation function, gc(r), which is especially important in the small angle region. Assume initially that there is an ideal gas of solute molecules in solution, so that gc(r) = 1 for all r. Eq. 2.1 then reduces to
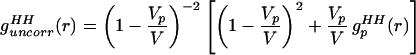 |
2.2 |
and, when Eq. 2.2 is transformed to Q-space, the result is
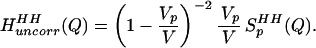 |
2.3 |
Q is the momentum transfer on scattering, given by Q = 4πsin(θ/2)/λ, where λ is the neutron wavelength, and θ is the scattering angle. The simulations discussed in Models and Methods describe the changes in the intermolecular pair correlations of water caused by the presence of one solute, and they therefore include information about lengthscales in the water–water correlations caused by independent or uncorrelated holes in water. This is exactly the information contained in Eqs. 2.2 and 2.3. Furthermore, the simulated estimates of Eq. 2.3 should provide for a more realistic description of the hole shape that approximations of spherical symmetry are unable to capture.
We then manipulated guHH(r) in Eq. 2.1 to separate the uncorrelated and correlated contributions:
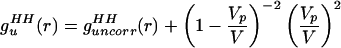 |
2.4 |
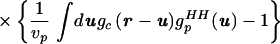 |
and transformed Eq. 2.4 to Q-space:
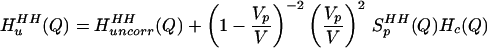 |
2.5 |
We can easily determine Vp and SpHH(Q) from a simulation of a single NALA solute in water and do not have to assume that the solutes are spherical in shape. These more realistic estimates of the uncorrelated quantities, i.e., water correlations arising from a collection of uncorrelated NALA-shaped holes, can be subtracted from the experimental data to isolate an experimental signal that is due to the correlated quantities, the second term in Eq. 2.5.
MODELS AND METHODS
Experimental Materials and Methods.
N-acetyl-l-leucine-amide was obtained from Bachem, and D2O was obtained from Cambridge Isotope Laboratories (Cambridge, MA). Samples were prepared as 1.0 ml of D2O added to 0.5 mmol of dry reagent (5, 6). Companion, or “matched,” solvent samples were prepared by the addition of sufficient H2O to imitate the hydrogen–deuterium exchange that occurs between the solute and the solvent (5, 6). Solution and pure water scattering experiments were carried out with the Sandals detector at the ISIS spallation source at Rutherford Appleton Laboratory (6). The container for the sample was a null-matrix alloy composed of zirconium and titanium in a ratio such that the average coherent scattering length is zero. The path length was 1 mm, with a wall thickness of 1.1 mm. The container was sufficiently wide to allow the use of a circular neutron beam with a diameter of ≈32 mm. All systematic corrections on data collected at the Sandals station were performed at ISIS, including corrections for sample transmission, multiple scattering, and inelasticity effects (9). The resulting data have been reported and described elsewhere (6, 7). The excess scattering, Iexcess(Q), was obtained by taking the difference between the scattering intensity measured for the solution and that measured for the matched solvent (5–7).
The Isolation of the Experimental Signal Due to Solute Correlations.
The measured scattering intensity from an aqueous solution arises from a sum of intensities due to intermolecular and intramolecular correlations:
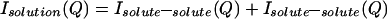 |
3.1 |
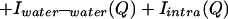 |
where the last term refers to scattering interference between atoms on the same molecule. The scattering contributions of each of the intermolecular terms is a sum of weighted structure factors, H(Q):
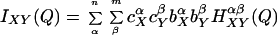 |
3.2 |
where
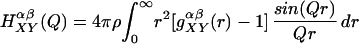 |
3.3 |
and X and Y correspond to solute or water, the indices α and β refer to sums over atoms within a given molecule, c is the atomic fraction, b is the scattering length for an atom in the solute or solvent molecule, and ρ is the atomic density.
We define a function, Isimulated(Q), that is composed of the following terms:
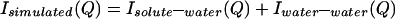 |
3.4 |
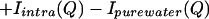 |
All of the quantities in Eq. 3.4 are determined by simulation or calculation and represent uncorrelated NALA molecules in solution. Previously reported molecular dynamics simulations of one NALA solute in water (6, 7) provide the various atomic pair correlation functions, h(r) = g(r) − 1, that then are Fourier transformed to give H(Q). Isolute–water(Q) and Iwater–water(Q) are obtained by using Eq. 3.2, but with concentration factors consistent with the solution scattering experiment, i.e., scaled by a factor of ≈5 that is equivalent to the volume prefactor of S(Q) in Eq. 2.3. To obtain Iintra(Q), we averaged the molecular structure factor over many possible molecular conformations, weighting each one by its probability of occurrence, by using a library of conformations (10). A total of 589 different conformers taken from 10,491 occurrences in the database were used to calculate the molecular structure factor for NALA (5).
The experiments are reported as a difference between scattering from solution and that of pure water:
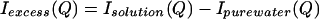 |
3.5 |
We propose to isolate an experimental signal caused by the correlated NALA solutes, Isolute–solute(Q), by subtracting Isimulated(Q) (Eq. 3.4) from Iexcess(Q) (Eq. 3.5) that has been obtained from the neutron scattering experiments. This remaining signal,
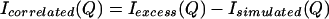 |
3.6 |
arises from scattering of water molecules excluded from the solute regions in which the solutes themselves are correlated in some way that is yet to be discovered (which is the purpose of this paper). Therefore, the intensity defined in Eq. 3.6 arises from the second term in Eq. 2.5, and model gc(r)’s can be used to fit the remaining signal.
RESULTS
Fig. 1A shows the ISIS measured excess scattering Iexcess(Q) (triangles) and the sum of the simulated water–water, water–solute, and intramolecular contributions that comprise Isimulated(Q) (squares). Fig. 1B exhibits the difference between the experimental curve and simulated curve, Eq. 3.6, which is nonzero over the range 0.25 Å−1 < Q < 1.25 Å−1. The importance of the contents of Fig. 1B is that it represents the excess signal caused solely by solute–solute correlations in aqueous solution, which is equivalent to the second term in Eq. 2.5. The form of gc(r) between NALA molecules is what we must determine to reproduce Icorrelated(Q) in Fig. 1B.
Figure 1.

(A) ISIS neutron scattering data for a 0.5 M solution of NALA in D2O (triangles) and the simulated contributions from water–water, water–solute, and intramolecular scattering (squares). The data is in units of barns/steradian per solute molecule. (B) The experimental signal caused by solute–solute correlations, Icorrelated(Q), that is obtained by subtracting the two curves in A. (C) Simulations of excess scattering intensity for a single NALA in water (squares), a single 6.2-Å Lennard-Jones sphere in water (triangles), and a single 8.8-Å Lennard-Jones sphere in water. This comparison allows us to estimate that a single NALA in water corresponds roughly to a 6.0- to 6.5-Å sphere.
Fig. 1C establishes that the simulated excess scattering for one NALA (full atomic representation) in water is roughly equivalent to the simulated excess scattering for a single 6.0- to 7.0-Å sphere in water, and that an 8.0- to 9.0-Å diameter sphere is too large when compared to 0.25 Å−1 < Q < 3.0 Å−1. Fig. 1C also illustrates that the larger sphere, will exhibit more rapid oscillations in inverse Q-space. For example, the steepness in Iexcess(Q) at the smallest angles is greater, and the position of the first minimum deepens (and eventually shifts to increasingly smaller Q) as sphere size increases. The interpretation becomes more complicated than a simulation of a single “hole” if there is a distribution of hole sizes. This understanding provides a rough guide as to how various model gc(r)’s can be judged for their ability to reproduce Icorrelated(Q). The solute–solute peak positions of gc(r) have the most influence on how rapidly the scattering changes at the smallest angles considered and the position of the minimum in Icorrelated(Q). The use of gc(r) to describe the correlations of NALA molecules explicitly assumes that all atomic detail has been erased and that the NALA molecules are instead represented as spheres with an effective radius. This description of NALA is reasonable for analyzing smaller scattering angles that probe lengthscales at which the detailed atomic positions are not resolvable.
In what follows, we consider three qualitatively distinct solute centers pair correlation functions, gc(r): gas, cluster, and aqueous. Although the entire space of solute–solute gc(r)’s has not been explored exhaustively, we argue that all physically motivated gc(r)’s have been considered with these three hypothetical functions; they, in fact, have qualitatively different peak positions. Once the qualitatively distinct solute–solute correlation functions are thus “enumerated,” further constraints on the values of peak positions and peak heights are imposed by the (approximately) known solute and water diameters, the density of solutes in solution, and constraints imposed by Icorrelated(Q) itself.
Fig. 2A shows two hypothetical examples of gc(r). The first is a gas of Lennard-Jones spheres where gc(r) is determined as
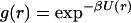 |
4.1 |
and
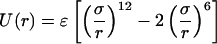 |
4.2 |
with σ = 5.0 Å and ɛ = 1.6 kcal/mol. We note that we varied σ (data not shown) but found that the experimental data always was reproduced poorly for values σ < 4.75 Å and σ > 6.0 Å. This is reassuring because Icorrelated(Q) (Fig. 1B) is robust enough to discriminate against unreasonable NALA molecule sizes. The effective size of a single NALA molecule in solution is ≈6.25 Å (Fig. 1C), and it would be expected that the effective size of NALA becomes more compact when two or more NALA molecules are in contact or even separated by a water layer. In the range 4.75 Å < σ < 6.0 Å, the value of σ becomes less sensitive, and the ɛ value more sensitive, for bringing about productive adjustments in the model to best reproduce the excess signal. When the ɛ parameter was adjusted outside the range of 1.25 kcal/mol < ɛ < 1.8 kcal/mol, the experimental data also was described poorly. The represented gc(r) is typical of a strongly associated, dilute gas.
Figure 2.

(A) Model gc(r)’s describing gas and cluster forms of NALA correlations in solution. The gas phase gc(r) corresponds to a Lennard-Jones interaction between solute molecules represented as spheres of σ = 5.0 Å with ɛ = 1.6 kcal/mol. The cluster gc(r) is the same as the gas phase except for a second peak position at 2σ. (B) A comparison of the excess experimental signal with the simulated solute–solute scattering derived from the various models of gc(r) shown in A. The comparison emphasizes that the gas and cluster forms of gc(r) are probably not viable representations of the solute–solute correlations.
The second model gc(r) in Fig. 2A is meant to exhibit ordering of NALA molecules as a cluster or liquid, i.e., peak positions at σ, 2σ, 3σ, etc. This form of gc(r) was determined by adding to Eq. 4.1 gaussians centered at 2σ, 3σ, etc.,
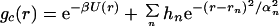 |
4.3 |
where rn = (n + 1)σ, and hn, αn, σ, and ɛ were treated as adjustable parameters to best reproduce the data. We found reasonable optimality for σ = 5.0 Å and ɛ = 1.6 kcal/mol (again noting that a decent fit was unobtainable for σ < 4.75 Å, σ < 6.0 Å, ɛ > 1.25 kcal/mol, and ɛ < 1.8 kcal/mol). We found that a value of αn = 0.5 for all n was needed to place the center of a given Gaussian. The value of h1 was determined to be ≈2.0; anything >2.0 shifted the minimum of the scattering to a value of Q that is too small whereas anything much <2.0 did not differentiate the liquid from the gas. We found that contributions beyond n = 1, i.e., solute–solute correlations >10 Å, caused significant deviations from the experimental signal at the smallest angles (Q < 0.5 Å−1). Fig. 2B shows a comparison of the excess experimental signal due to solute–solute correlations and the simulated scattering for the gas and liquid models of gc(r). Neither the gas phase or cluster forms reproduce the full range of experimental signal considered (0.25 Å−1 < Q< 1.25 Å−1) and essentially shows the limitations of a model gc(r) based on a gas or clustering description of NALA molecules.
The final form of gc(r) that we consider is one that provides for positive correlations of NALA molecules at contact and separated by one or more water layers. This aqueous form of gc(r) is inspired by the observation that smaller hydrophobic groups are stabilized in solution at relative distances corresponding to being in contact and separated by one water layer (11–16). The presence of a solvent-separated minimum or minima imply that hydrophobic solutes in water are correlated over longer distances than that arising from just reducing exposed surface area (i.e., only being stabilized at contact). Fig. 3A shows two aqueous gc(r)s: one exhibiting a solvent separated peak at dsolute + dwater and a second aqueous form showing the same solvent-separated peak and a second peak at even longer distances.
Figure 3.

(A) Model gc(r)’s describing aqueous forms of NALA correlations in solution. The aqueous model of gc(r) is the same as the gas but has peak positions at dsolute + dwater, where d represents the diameter of a water molecule, and a second aqueous form with an additional peak at dsolute+ 2dwater. (B) A comparison of the excess experimental signal with the simulated solute–solute scattering derived from the various models of gc(r) shown in A. The aqueous forms are clearly in good agreement with the excess experimental signal, but the data is inconclusive as to how far apart the solutes are positively correlated in solution.
The gc(r) exhibiting one solvent-separated peak is evaluated with Eq. 4.3 but with r1 = 7.8 Å. Again, σ = 5.0 Å, ɛ = 1.6 kcal/mol, and αn = 0.5 values were used for all n, and h1 = 4.0 was the best value for reproducing the excess experimental signal. We also considered a gc(r) with an additional peak beyond the solvent-separated peak for which the solutes are separated by two water layers, to determine whether the solute–solute correlations are even longer ranged. This was accomplished by adding a second Gaussian at r2 = 10.6 Å with h2 = 3.0. Fig. 3B shows a comparison of the excess experimental signal due to solute–solute correlations and the simulated scattering for the aqueous models of gc(r). Clearly, the aqueous forms of gc(r) are a better description than either the gas or cluster forms (Fig. 2A). The better reproducibility of the experimental data by using the first aqueous form of gc(r) seems to suggest that there are no solute–solute correlations beyond the first solvent-separated minimum. We would estimate from the first aqueous form of gc(r), weighted by the volumes of spherical shells, that the presence of contact and solvent-separated configurations of NALA molecules are equally likely in solution.
The agreement is not perfect, especially at the smallest angles considered. Fig. 1A shows that the simulations can reproduce the experiment in the region 1.5 Å−1 < Q < 3.0 Å−1 with reasonable quantitative agreement (6, 7), so we have some confidence in the solute–water and water–water correlations obtained from simulation. Modeling errors arising from the use of empirical force fields will always be an uncertainty, however. Another possible source of error is the fact that solute–water correlations may change from those calculated for a single solute in water, when the solutes themselves are in contact and/or solvent-separated. We have simulated the intensity contribution from solute–water correlations arising when two NALA molecules are in contact, and we have found no significant changes. Another potential source of error is the fact that the intramolecular scattering is evaluated from a rotamer library based on globular proteins and may have different weights of side chain conformers than those exhibited in solution. However, this is unlikely to be in significant error because protein surface residues, with side chains extending into solvent, would be strongly weighted in the library of protein structures. More careful experiments on a small angle diffractometer would likely help us resolve Iexcess(Q) better for Q < 0.25 Å−1, to better quantify the steepness of the rise at small angle.
We also have simulated a potential of mean force curve for two Lennard-Jones spheres with σ = 6.17 Å and ɛ = 0.35 kcal/mol in SPC (single point charge) model of water (Fig. 4), for a study that is independent of our current goal of analyzing the small angle data. The potential of mean force curve at relative separation of spheres below ≈5.5 Å can be fit with Eq. 4.2, with σ = 5.5 Å and ɛ = 1.0 kcal/mol, which is qualitatively consistent with the aqueous forms of gc(r) in Fig. 3A. This independent calculation provides further support that our aqueous gc(r)s are reasonable and that the excess experimental signal at small angles has meaningful information about solute–solute correlations.
Figure 4.

A simulation of the free energy of association for two 6.2-Å Lennard-Jones spheres (ɛij = 0.35kcal/mol) in water. This comparison shows that the aqueous models of gc(r) (Fig. 3A) that reproduce the excess signal (Fig. 3B) are sensible.
DISCUSSION
In this work, we have combined neutron solution scattering experiments with molecular dynamics simulation to isolate an excess experimental signal that is solely due to solute–solute correlations. In particular, we have subtracted off simulated quantities that describe uncorrelated solutes in water from an experimental signal, to leave an excess signal that contains information about correlated solutes in water. Various model pair distribution functions for NALA molecules, gas, cluster, and aqueous forms of gc(r), were tested for their ability to reproduce this excess experimental signal. We have found that the excess experimental signal is adequate enough to rule out gas and cluster pair correlation functions. The aqueous form of gc(r) that exhibits a solvent-separated minimum, and possibly longer-ranged correlations as well, is not only physically sound but reproduces the experimental data reasonably well. An independent potential of mean force calculation finds a similar aqueous gc(r) to that determined here by a fit of the excess experimental signal.
The characterization of the range and magnitude of hydration forces between individual amino acid side chains, and the connection to water structure, is a step toward defining the role of hydration in protein folding. The conclusion that the experimental data is consistent with some free energy of stabilization when nonpolar side chains are separated by one or more water layers is perhaps under-appreciated for its potential importance in how a biased landscape might arise because of longer-ranged hydration effects. The collapse to a partially hydrated globule, in which nonpolar groups actually are stabilized at longer relative distances incorporating a water molecule or layer, would result in less steric hindrance for rearrangements to the correct tertiary fold. Once the water of hydration is removed (corresponding to configurations stabilizing hydrophobic groups at contact), the longer ranged forces would no longer play a role, and the roughness of the energy landscape that ensures a single global minimum would be restored.
Acknowledgments
We thank Bob Glaeser for many interesting discussions and his careful readings of the manuscript. T.H.-G. gratefully acknowledges support from the Air Force Office of Sponsored Research (Grant FQ8671-9601129) and U.S. Department of Energy (Contract DE-AC-03-76SF00098) and acknowledges the National Energy Research Supercomputer Center for computer time. J.M.S. is supported by a National Science Foundation Graduate Research fellowship. A.P. acknowledges support from the W. M. Keck Program in Biomedical Research, National Institutes of Health training grants in molecular biophysics and biotechnology, and the support of the Committee on Research of the University of California at Berkeley.
ABBREVIATION
- NALA
N-acetyl-leucine-amide
References
- 1.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 2.Shakhnovich E I. Curr Opin Struct Biol. 1997;7:29–40. doi: 10.1016/s0959-440x(97)80005-x. [DOI] [PubMed] [Google Scholar]
- 3.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 4.Head-Gordon T. Proc Natl Acad Sci USA. 1995;92:8308–8312. doi: 10.1073/pnas.92.18.8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pertsemlidis A. Ph.D. thesis. Berkeley: Univ. of California; 1995. [Google Scholar]
- 6.Pertsemlidis A, Saxena A, Soper A K, Head-Gordon T, Glaeser R M. Proc Natl Acad Sci USA. 1996;93:10769–10774. doi: 10.1073/pnas.93.20.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Head-Gordon T, Sorenson J M, Pertsemlidis A, Glaeser R M. Biophys J. 1997;73:2106–2115. doi: 10.1016/S0006-3495(97)78241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soper A K. J Phys: Condens Matter. 1997;9:2399–2410. [Google Scholar]
- 9.Soper A K, Howells W S, Hannon A C. Rep. No. 89–046. Didcot, U.K.: Rutherford Appleton Laboratory; 1989. [Google Scholar]
- 10.Dunbrack R L, Jr, Karplus M. Nat Struct Biol. 1994;1:334–340. doi: 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- 11.Pratt L R, Chandler D. J Chem Phys. 1977;67:3683–3704. [Google Scholar]
- 12.Pratt L R, Chandler D. J Chem Phys. 1980;73:3430–3433. [Google Scholar]
- 13.Pratt L R, Chandler D. J Chem Phys. 1980;73:3434–3441. [Google Scholar]
- 14.Geiger A, Rahman A, Stillinger F H. J Chem Phys. 1979;70:263–276. [Google Scholar]
- 15.Pangali C, Rao M, Berne B J. J Chem Phys. 1982;81:2982–2990. [Google Scholar]
- 16.Zichi D A, Rossky P J. J Chem Phys. 1985;83:797–808. [Google Scholar]