

Abstract
Background
Cardiovascular disease accounts for at least 85 percent of deaths for those patients with type 2 diabetes mellitus (T2DM). Additionally, 75 percent of these deaths are due to ischemic heart disease.
Hypothesis
Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress.
Testing of the hypothesis
The vulnerable three arms of the eNOS reaction responsible for the generation of eNO is discussed in relation to the hypothesis: (1). The L-arginine substrate. (2). The eNOS enzyme. (3). The BH4 cofactor.
Implications of the hypothesis
If we view T2DM as a vascular disease initially with a later manifestation of hyperglycemia, we may be able to better understand and modify the multiple toxicities associated with insulin resistance, metabolic syndrome, prediabetes, overt T2DM, and accelerated atherosclerosis (atheroscleropathy). The importance of endothelial nitric oxide synthase, endothelial nitric oxide, tetrahydrobiopterin (BH4), L-arginine, and redox stress are discussed in relation to endothelial cell dysfunction and the development and progression of atheroscleropathy and T2DM. In addition to the standard therapies to restore endothelial cell dysfunction and stabilization of vulnerable atherosclerotic plaques, this article will discuss the importance of folic acid (5MTHF) supplementation in this complex devastating disease process.
Atheroscleropathy and hyperglycemia could be early and late manifestations, respectively, in the natural progressive history of T2DM.
Keywords: ADMA, Atherosclerosis, Folic Acid, Oxidative stress, Reactive Oxygen Species (ROS), endothelial Nitric Oxide (eNO), endothelial Nitric Oxide Synthase (eNOS) (NOS-3)
Background
The historical discovery of the enzyme family nitric oxide synthases (NOS) in 1989 is relatively recent and we now know there are three distinct isoforms of NOS: neural NOS-1 (nNOS); inducible NOS-2 (iNOS); and endothelial NOS-3 (eNOS). These three isoforms were discovered in the above order from 1991–1994 [1]. eNOS, the rate limiting enzyme responsible for the production of endothelial derived nitric oxide (eNO) was sequenced and cloned in 1992 [2,3]. Each NOS enzyme has its own unique amino acid structure sharing some 50 percent homology indicating a common ancestral background. Additionally, each NOS isoform is transcribed from a separate gene (table 1).
Table 1.
Comparisons of the human NOS enzymes
| nNOS NOS-1 | Chromosome 12 | Amino acids 1434 aa 161 kDa | high Ca++ requirement |
| iNOS NOS-2 | Chromosome 17 | Amino acids 1153 aa 131 kDa | low Ca++ requirement |
| eNOS NOS-3 | Chromosome 7 | Amino acids 1203 aa 133 kDa | high Ca++ requirement |
Nitric oxide, voted the molecule of the year by Science Magazine in 1992 [4] has been shown to be an extremely important signaling molecule in the cardiovascular system and in 1998 Furchgott RF, Ignarro LJ, and Murad F. were awarded the Nobel Prize in Medicine and Physiology "for their discoveries concerning nitric oxide as a signaling molecule in the cardiovascular system" (tables 2,3).
Table 2.
The positve protective effects of eNOS
| 1. | Promotes vasodilatation of vascular smooth muscle. |
| 2. | Counteracts smooth muscle cell proliferation. |
| 3. | Decreases platelet adhesiveness. |
| 4. | Decreases adhesiveness of the endothelial layer to WBCs "Teflon effect". |
| 5. | Anti-inflammatory. |
| 6. | Anti-oxidant. It scavenges reactive oxygen species, locally. Acts as a chain – breaking antioxidant to scavenge ROS. |
| 7. | Anti-fibrotic. When NO is normal or elevated MMPs are low and conversely if NO is low MMPs are elevated and active. |
| 8. | NO has diverse anti-atherosclerotic actions on the arterial vessel wall: including antioxidant effects by direct scavenging of ROS – RNS acting as chain breaking antioxidants. |
Table 3.
Janus – faced properties of NO
| GOOD | BAD |
| eNO, nNO | iNO * |
| Chromosomes 7,12 | Chromosome 17 |
| Duration: Seconds to minutes | Duration: Hours to days |
| Signaling molecule. | Killer molecule. |
| Regulated: small bursts | Unregulated: larger bursts and longer duration |
| Cytoprotective | Cytotoxic |
*In host defense mechanisms iNO does just what we want it to, i.e. it kills invading organisms. Inducible NO can also be thought of as inflammatory NO. Paradoxically, in both acute and chronic inflammatory states iNO also kills the native hosts cells in the immediate surrounding area.
This relatively brief and recent history of NOS and NO is in contrast to the ancient history of atherosclerosis which dates to the time of the Egyptians. The theories of atherosclerosis are legion and the atherosclerologists of today have a proud history upon which to build for the future [5].
The endothelial cell, such a central player, responsible for remodeling of the arterial vessel wall in atherosclerosis (intimopathy) and responsible for the synthesis of eNOS and subsequent eNO production will be discussed and related to the accelerated atherosclerosis (atheroscleropathy) associated with type 2 diabetes mellitus (T2DM) [5,8-11].
This review will discuss dysfunction of the eNOS enzyme and its potential to uncouple, resulting in the endothelial cell becoming a net producer of damaging superoxide (O2') instead of the protective eNO (tables 4,5) [6]. The lack of eNO from the endothelial cell is related to the development of atheroscleropathy and is related to the underproduction and / or the excessive consumption of eNO by redox stress. The uncoupling of this complex enzyme will aid in the understanding of the development of atheroscleropathy and the role of oxidative and reductive stress (redox stress) in patients with T2DM [7,8].
Table 4.
Lab – garbage acronym
| Factors related to eNOS dysfunction with decreased NO. factors known to uncouple the eNOS enzyme. |
| L – Lipids LDL – Cholesterol elevated both native and oxidized. Elevated Triglycerides of metabolic syndrome. Decreases or impairs dimethylarginine dimethylaminohydrolase (DDAH). Decline in DDAH activity. Increase in ADMA activity. |
| A – Arginine (L-arginine) decreased or impaired: By competitive interaction with ADMA (asymmetric dimethylarginine) an endogenous nitric oxide synthase inhibitor. Elevated in proatherogenic conditions. ACE, Ang II, endothelin, hypertension, insulin resistance, prediabetes, T2DM, and hyperhomocysteinemia. |
| B – BH4 Decreased or impaired Folic acid regeneration of the cofactor BH4 i.e. BH2 → BH4 with 5 methyl tetrahydrofolate (5-MTHF) active form of folic acid. |
| G – Glucose elevation. T2DM. DDAH effect increasing ADMA similar to lipids and elevated homocysteine. |
| A – Arginine (L-arginine) decreased or impaired: By nitrosylation (nitroarginine). By ONOO' peroxynitrite similar to nitrosylation of tyrosine (nitrotyrosine). |
| R – ROS reactive oxygen species. Specifically O2' and ONOO' Decline in DDAH activity results in increased ADMA. |
| B – BH4 decreased or impaired. BH4 oxidized to BH2. Loss of a naturally occurring antioxidant with diminished ROS scavenging capability. |
| A – ADMA Homocysteine impairs DDAH enzyme and increases ADMA. |
| G – Glucose elevated – glycated eNOS. |
| E – Endothelial NOS enzyme decreased and or dysfunctional. Gene polymorphism Glu298 → Asp, additional unidentified gene polymorphisms, CRP causing marked down regulation of eNOS mRNA and protein expression, eNOS enzyme glycation. Last but most importantly . . . ...... |
| eNOS enzyme UNCOUPLING |
Table 5.
eNOS reaction the three critical arms
 |
| IF THE eNOS REACTION IS UNCOUPLED: THEN THE REACTION IS: |
| (L-arginine is not converted to NO and L-citrulline). |
| TREATMENT PARADIGM: |
| (1) Prevent competitive inhibition by ADMA (Reduce substrates: LDL-C, Triglycerides, Homocysteine, and Glucotoxicity formation. Prevent L-arginine from being converted to nitroarginine by nitrosylation. Reducing Redox Stress – A-FLIGHT toxicities reduction of ROS. ARGININE SUPPLEMENTATION. |
| (2) Prevent hs-CRP from decreasing eNOS by lowering hs-CRP and preventing glycation of Enos enzyme. Aggressive treatment of elevated substrates. |
| (3) Add FOLIC ACID not only to lower Hcy and ADMA levels but also to restore BH2, BH3 to the active catalytic cofactor BH4. Use the RAAS acronym and prevent the A-FLIGHT Toxicities (ROS) from developing i.e. decrease the manifold toxicities of MS, IR, PD and overt T2DM. |
| BE AGGRESSIVE AND TREAT TO KNOWN GOALS: |
Definitions
It is important to set forth the authors' working definitions before proceeding with this review.
ATHEROSCLEROSIS is a systemic dysfunctional endothelial, focal occurring, chronic inflammatory, fibroprolifertive, angiogenic, prothrombotic, multifactorial disease of the arterial intima caused by the retention of modified low density lipoproteins, hemodynamic stress, and accelerated by redox stress [5,8-11].
ATHEROSCLEROPATHY: The term used to describe the unique accelerated atherosclerosis observed in and associated with metabolic syndrome, insulin resistance, prediabetes, and overt T2DM which is driven by reactive oxygen species associated with multiple toxicities (table 6) [8].
Table 6.
The manifold toxicities of insulin resistance, metabolic syndrome and T2DM.
| A-FLIGHT toxicities | ||
| A | Amylin (hyperamylinemia)/amyloid toxicity | ROS |
| Ang II (also induces PKC) | ROS | |
| AGEs/AFEs (advanced Glycosylation / fructosylation products) | ROS | |
| Antioxidant reserve compromised | ROS | |
| Absence of antioxidant network | ROS | |
| Ageing | ROS | |
| Angiogenesis (induced redox stress) Arteriogenesis (impaired PAI-1) | ROS | |
| Atherosclerosis – Atheroscleropathy. [ROS beget ROS ] | ROS | |
| F | Free fatty acid toxicity | ROS |
| L | Lipotoxicity | ROS |
| I | Insulin toxicity (hyperinsulinemia-hyperproinsulinemia) (endogenous) | ROS |
| Inflammation toxicity | ROS | |
| G | Glucotoxicity (compounds peripheral insulin resistance) reductive stress | ROS plus |
| Sorbitol / 'olyol pathway | PKC | |
| Pseudohypoxia (NADH/NAD increased) | ||
| H | Hypertension toxicity | ROS |
| t homocysteine toxicity | ROS | |
| T | Triglyceride toxicity | ROS |
T2DM is a heterogeneous, multifactorial, polygenic disease characterized by a defect in insulin's secretion (the beta cell secretory defect) and action (insulin resistance) which results in elevated glucose [8,12].
T2DM may be viewed as a dynamic continuum of five stages (table 9) [12].
Table 9.
The five stages of T2DM: the natural progressive history of T2DM.
| I. LATENT STAGE:[EARLY] |
| Insulin Resistance: |
| *Genetic Component. |
| *Environmental Component. [Modifiable] Obesity / Sedentary life style. [Nonmodifiable] Ageing. |
| Beta Cell Defect: (Dysfunction) |
| *Genetic....... Abnormal processing, storage, or secretion. |
| *Intracellular extracellular amylin fibril toxicity. Abnormal processing, storage, or secretion. |
| Intra-Islet Endothelial Absorptive Defect: |
| *Heparan sulfate proteoglycan (HSPG) PERLECAN of the capillary endothelial cells avidly attracts amylin (IAPP) and islet amyloid forms an envelope around the capillary. This is in addition to the increase in basement membrane associated with the pseudohypoxia (associated with glucotoxicity) and the redox stress within the capillary. |
| II. TRANSITION STAGE: [MIDDLE] |
| PERSISTENT HYPERINSULINEMIA |
| PERSISTENT HYPERAMYLINEMIA – Continued remodeling of endocrine pancreas. (AMYLOID) Beta cell displacement, dysfunction, mass reduction, and diffusion barrier. |
| III. IGT STAGE: *"Pre-diabetes" HHS ADA term (Impaired Glucose Tolerance) |
| [LATE] *[Start treatment at this time ] [Diagnose earlier: Rejuvenation of the 2 hour glucose tolerance blood sugar 140–199 mg.] |
| *Increased insulin resistance [Feeds forward] > Glucotoxicity [Feeds forward] > Insulin resistance [Feeds forward] > Glucotoxicity : creating a vicious cycle. |
| *Islet amyloid. Increasing beta cell defect. Loss of beta cell mass with displacement. [Remodeling of islet architecture including ECM] Beta cell loss centrally. |
| IV. IFG STAGE: *"Pre-diabetes" HHS ADA term (Impaired Fasting Glucose) |
| [LATER] Blood sugar > 110 mg./dl. < 126 mg./dl. |
| [Impaired Hepatic Glucose Production (HGP)]. Increasing global insulin resistance (HEPATIC) with subsequent gluconeogenesis. Feeding forward in the vicious cycle to accelerate insulin resistance globally |
| V. OVERT STAGE: [TOO LATE] 50% loss of Beta Cell Function. |
| Va. Vb. Vc. Phases I, II, III Mild / Moderate / Severe / Complete. Use medications that do not increase insulin or AMYLIN. Use combination therapy. Start treatment at stage III-IV (IGT) (IFG) |
*Paradigm Shift. Start treatment at the earlier stage of IGT
eNOS and eNO
Chromosome number 7 contains the vascular eNOS gene that encodes the transcription and synthesis of the 1203 amino acid protein enzyme eNOS. eNOS is located uniquely within caveolae of the plasma membrane which allows the monolayer of endothelial cells to compensate if its fellow cellmates become dysfunctional in order to maintain eNO production.
Once conceptualized, that a specific and unique gene (eNOS, iNOS, nNOS) encodes the transcription of NO in a specific organ system, the opposing faces of this highly specialized, small, bioactive, and readily diffusible gas NO can be better understood (table 2, 3) [13].
eNO is a natural occurring, endogenous, chain breaking antioxidant synthesized by the endothelial cell. eNO and superoxide are related to a healthy and dysfunctional endothelium, respectively [8].
THE HEALTHY ENDOTHELIUM IS A NET PRODUCER OF ENDOTHELIAL NITRIC OXIDE [eNO].
THE ACTIVATED, DYSFUNCTIONAL ENDOTHELIUM IS A NET PRODUCER OF SUPEROXIDE [O2'] ASSOCIATED WITH T2DM AND ATHEROSCLEROPATHY
There are at least three arms in the eNOS reaction capable of producing endothelial cell dysfunction with resultant redox stress to the intima and islet resulting in atheroscleropathy and a progressive decline in beta cell function within the vulnerable islet.
Presentation of the hypothesis
Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress.
Testing of the hypothesis
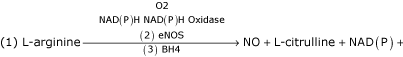
The following eNOS reaction demonstrates the location of the vulnerable three arms of the eNOS reaction responsible for the generation of eNO.
IF THE eNOS REACTION IS UNCOUPLED: THEN THE REACTION IS:
![]()
L-arginine is uncoupled and not converted to NO and L-citrulline (figure 1) (tables 3,4).
Figure 1.

Oxygen reacts with the eNOS enzyme in which the BH4 cofactor has coupled NAD(P)H with L-arginine to be converted to NO and L-citrulline. When uncoupling occurs the NAD(P)H reacts with O2 and the endothelial cell becomes a net producer of superoxide (O2').
THE L-ARGININE ARM
The substrate L-arginine is an essential amino acid and is usually not thought to be rate limiting (with the exception of high demands in a dysfunctional endothelial state such as atherosclerosis or the atheroscleropathy associated with T2DM).
Arginine deficiency is a rare occurrence, however, there can be competitive inhibition by the endogenously produced asymmetrical dimethylarginine (ADMA) and nitroarginine. ADMA is emerging as an important cause of endothelial cell dysfunction.
It is an endogenously produced competive inhibitor of L-arginine which results in a relative deficiency of the natural substrate for the eNOS enzyme to produce eNO. ADMA accumulates by either a process of increased synthesis by the protein arginine N-methyltransferases (PRMTs) or decreased elimination by the enzyme dimethylarginine dimethylaminohydrolose (DDAH) [14-17].
DDAH selectively hydrolyzes ADMA to L-citrulline and dimethylamine, so that the higher the DDAH activity, the lower the ADMA levels. Importantly, inhibition of DDAH activity results in increased ADMA levels. Both native and oxidized LDL-cholesterol have been shown to up-regulate PRMT while causing a decrease in the activity of DDAH [18]. This results in an elevation of ADMA levels contributing to endothelial cell dysfunction and increased oxidative stress to the intima with subsequent accelerated atherosclerosis.
There are multiple causes for the elevation in ADMA levels (figure 2). Each of these toxicities relate to the A-FLIGHT acronym responsible for the production of ROS (table 6). Recently, Faldetta CM, Laurenti O, and Desideri G [19] were able to demonstrate in both normal human subjects and T2DM human patients that acute L-arginine infusion decreased plasma total homocysteine concentrations, counteract oxidative stress, and increased the availability of nitric oxide.
Figure 2.

The multiple stressors causing uncoupling of the eNOS enzyme by decreased DDAH and the increased endogenous inhibitor of eNOS: ADMA.
In summary, the relative deficiency of L-arginine due to elevation of ADMA levels contribute to oxidative stress and result in the atheroscleropathy associated with insulin resistance, metabolic syndrome, prediabetes, and overt T2DM.
THE eNOS ARM
Important agonists for the activation of the eNOS enzyme (table 7) play an important role in the production of eNO.
Table 7.
eNOS AGONISTS
| 1. Bradykinin (BK). |
| 2. Phosphorylation by PK-B also called Akt. |
| 3. Mechanical forces – Sheer stress. |
| 4. Acetylcholine (muscarinic receptors). |
| 5. ADP – ATP (purinergic receptors). |
| 6. Vascular Endothelial Cell Growth Factor (VEGF). |
| 7. Histamine. |
| 8. HSP – 90. |
| 9. Acetylation by myristate (N-terminal) by palimitate (cys15 – cys26) Stimulated by BK. |
If there is a complete loss of eNOS enzyme as in the eNOS -/- knockout mouse model of Duplain and Sherrer [20] insulin resistance, hypertension, and hyperlipidemia develop. Their findings indicate that eNOS plays a major role in the regulation of insulin sensitivity. To have an effect on atheroscleropathy and T2DM it would not require a complete knockout of the gene but just one or two changes in the 1203 amino acid protein structure in the eNOS enzyme.
Such a gene polymorphism does exist and may become a major player in the polygenic causes of T2DM and atheroscleropathy:
Glu298 → Asp Gene Polymorphism (Glu298 → Asp GP)
eNOS may be impaired by abnormal structure and or function that occurs in gene polymorphisms.
One of the first significant eNOS gene polymorphisms described to date is Glu 298 → Asp. This amino acid substitution is responsible for a statistically significant increased frequency of hypertension and spastic angina [21]. Also, described in a different set of patients is the occurrence of increased abnormal coronary angiographic findings of atherosclerosis and an association of increased events [22-24].
The Glu298 → Asp GP is responsible for a decrease in basal NO production in healthy subjects according to the publication by Veldman BA et al [25].
This gene polymorphism then could interact with other gene polymorphisms and especially environmental conditions such as smoking, obesity, and the A-FLIGHT toxicities of IR, MS, PD, and overt T2DM. Or as in the hypothesis, could be responsible for the development of IR and MS which then would progress to PD or overt T2DM. We now have a Japanese population, a UK population, and an Italian population that correlates Glu298 → Asp GP to atherosclerosis and CAD events [23-25].
These findings indicate that the eNOS gene is important to the development of accelerated atherosclerosis → atheroscleropathy. Furthermore, these three distinct patient populations indicate that this gene polymorphism is widely distributed throughout diverse patient populations and according to Veldman [25] is present in approximately 10 % of patient populations.
Recently, Noiri E and colleagues have shown that there is a significant association of the Glu298 → Asp GP and end stage renal disease in T2DM which once again points to the importance of the endothelial cell and the eNOS reaction [26].
Since eNOS contains 1203 amino acids there is the possibility of other gene polymorphisms within the eNOS enzyme that have not been identified at this time. Additionally, this gene polymorphism could interact with other gene polymorphisms of other endogenous antioxidant enzymes in the T2DM patient as we know their antioxidant reserve is compromised (tables 6,8) [7,8,12].
Table 8.
Antioxidants: catalytic/enzymatic inactivation of free radicals.
| Enzymatic antioxidants |
| SUPER OXIDE DISMUTASE (SOD)– Location: mitochondrion |
| [O2- + SOD → H2O2 + O2] |
| ecSOD (extracellular) |
| MnSOD (mitochondrial) |
| CuZnSOD (intracellular) |
| CATALASE– Location: peroxisome |
| [2H2O2 + catalase → 2 H2O + O2] |
| GLUTATHIONE PEROXIDASE– Location: mitochondrion/cytosol |
| (Glutamyl-cysteinyl-glycine tripeptide) glutathione reduced -SH to the oxidized disulfide GSSG. |
| (Glutathione peroxidase) [GSH + 2H2O2 → GSSG + H2O + O2] |
| (Glutathione reductase) [GSSG → GSH] at the expense of [NADH → NAD+] and/or [NAD(P)H → NAD(P)+] |
| *NOS (nitric oxide synthase). – Location: membrane |
| Isoforms: |
| (e) NOS (endothelial): good (importance of eNOS uncoupling) LDL native and oxidized. |
| (n) NOS (neuronal): good |
| (i) NOS (inducible-inflammatory): GOOD in host defense. BAD in chronic inflammation, ischemia – ischemia reperfusion injury, acute and chronic as in autoimmunity – T1DM. |
| O2- and nitric oxide (NO) are consumed in this process with the creation of reactive nitrogen species (RNS). |
| O2- + NO → ONOO- (peroxynitrite) + tyrosine → nitrotyrosine. (also causes eNOS uncoupling) |
| Nitrotyrosine reflects redox stress and leaves a measurable footprint. |
| NO: the good; O2-: the bad; ONOO-: the ugly [122] |
| eNO : A chain breaking antioxidant see (table 2) item 6. |
| eNOS uncoupling causes the generation of O2' instead of NO induced by LDL-C, Glucose, O2', and ONOO'. |
| Nonenzymatic antioxidants |
| URIC ACID |
| VITAMIN A, VITAMIN C, VITAMIN E |
| THIOLS |
| APOPROTEINS: Ceruloplasmin and transferrin. Bind copper and iron in forms which cannot participate in the Fenton reaction. [7,8,12] |
As the human genome unfolds there may be other related important gene polymorphisms identified.
The BH4 ARM
Tetrahydrobiopterin (BH4) is the necessary cofactor for coupling L-arginine to the NAD(P)H oxidase enzyme in order for L-arginine to be oxidized to NO and L-citrulline. The importance of this cofactor and its mechanism is just starting to emerge.
If this cofactor is not functioning properly the entire eNOS reaction uncouples and the endothelium becomes a net producer of superoxide [O2'] through the uncoupled NAD(P)H oxidase reaction. In addition to BH4 being the necessary cofactor it also serves as a naturally occurring antioxidant capable of scavenging ROS such as superoxide O2' and perioxynitrite [ONOO']. If BH4 is deficient in supply or function the eNOS reaction not only is capable of uncoupling with subsequent excess [O2'] and [ONOO'] but also is at a disadvantage in that there would be less BH4 to scavenge these local occurring reactive oxygen species being produced in excess amounts [27].
It is important to note that a direct effect of folic acid is to keep this important cofactor BH4 intact so that it can maintain coupling of the eNOS reaction.
The lowering of homocysteine is an indirect effect of folic acid aiding in the decreased production of ADMA due to the effect of Hcy on DDAH. Folic acid given to patients with T2DM have an improvement of endothelial relaxation before Hcy has time to be lowered. It works almost immediately before the Hcy is lowered [28,29].
When you look at the three arms of importance for the eNOS reaction there are two possible changes in the treatment paradigm (in addition to lowering the substrates of the A-FLIGHT toxicities (table 6) and the RAAS acronym [8]) for these patients who suffer from endothelial dysfunction associated with atheroscleropathy, IR, MS, PD, and T2DM.
First, we need to take a global risk reduction approach (by treating each risk factor in the A-FLIGHT toxicities acronym in order to decrease the clinical substrates of lipids, hypertension, and glucotoxicity without elevating endogenous insulin or amylin) in order to decrease the progression of the disease. Second, we should now include in the treatment paradigm the use of folic acid and arginine supplementation. T2DM is now considered to be a coronary risk equivalent and therefore all patients with T2DM should be on folate supplementation, as well as, the non-diabetic atherosclerotic patient.
Those patients with persistent endothelial cell dysfunction after the standard goals have been reached should consider arginine supplementation and possibly should be considered even sooner in the treatment paradigm if there is a history or evidence of symptomatic peripheral arterial disease, congestive heart failure and / or diastolic dysfunction [17,30,31].
Folic acid – 5MTHF
This water soluble B vitamin has been recently gaining considerable attention due to its positive role in cardiovascular disease, neurodegenerative disorders, neural tube defects, and cancer. Folic acid plays an important role in the remethylation of homocysteine and is thus capable of lowering elevated levels of homocysteine (figure 3) [32].
Figure 3.

Hcy (demethylated Methionine) can enter the remethylation cycle with the aid of Folic Acid (5-Methyl-tetrahydrofolate) and the Folate cycle, the enzyme Methionine Synthase, and the necessary Cofactor B12. Folic Acid in addition to being a methyl-donor can serve as an electron or hydrogen donor to restore the required BH4 Cofactor to recouple the eNOS enzyme necessary for the production of eNO from L-arginine. Hcy may also enter the Transsulfuration Pathway with the assistance of Cystathionine Beta Synthase (CBS) enzyme and the necessary Cofactor B6 to form Cystathionine and Cysteine which can then be excreted in the urine or converted to the very important antioxidant Glutathione. The substrate Folic Acid and the Cofactors B6 and B12 are very important not only for improving hyperhomocysteinemia but also important for the recoupling of the BH4 cofactor to the eNOS enzyme for the production of eNO.
Deficiency or impairment of folate metabolism is associated with hyperhomocysteinemia (hHcy), hypomethylation (the decreased one carbon unit transfer to purines and pyrimidines for DNA repair and biosynthesis), DNA damage, and impaired cell proliferation, malignancies, and impaired eNO production [33-37]. 5-MTHF is an electron donor, a hydrogen donor, and a methyl donor.
Each of these attributes confers a unique role to its function. In regards to the eNOS reaction it supplies both hydrogen and electrons to BH2 and BH3 to restore BH4 (the necessary cofactor for the eNOS reaction). This attribute allows the eNOS reaction to re-couple and restores the capability of this reaction to be once again a net producer of eNO and restores endothelial dependent vasodilatation in T2DM and hyperlipidemia [38]. Recently, the combination of folic acid, vitamin B12, and vitamin B6 was able to demonstrate a reduction of in-stent restenosis and recurrent events in up to one year follow up in the Swiss heart study [39-41].
The eNOS hypothesis
Is T2DM a vascular disease with a dysfunctional eNOS enzyme reaction, resulting in endothelial cell dysfunction and associated diminished eNO production? Is atheroscleropathy an early manifestation and hyperglycemia a later manifestation?
The following quote is pertinent to this discussion.
"Diabetes must take its place alongside the other major risk factors as important causes of cardiovascular disease. In fact, from the point of view of cardiovascular medicine, it may be appropriate to say: "diabetes is a cardiovascular disease" [42].
Implications of the hypothesis
Could it be that one of the polygenic causes of T2DM involve the eNOS enzyme? Such as one or more gene polymorphisms resulting in a defective but functioning eNOS enzyme. This would certainly help to explain the earlier discussed atheroscleropathy associated with T2DM.
This gene polymorphism could interact with the environment to produce an even more defective eNOS enzyme with increasing endothelial cell dysfunction. Smoking, obesity, hyperlipidemia, hypertension, sedentary life style are all known risk factors and all too often they cluster creating the metabolic syndrome with a markedly elevated tension of redox stress not only in the islet but also within the intima of the arterial vessel wall. A single amino acid substitution in the primary structure could have a considerable effect on the structure and function of this omnipotent enzyme and its reaction producing eNO.
Here is an example where a defective gene could be made much more impotent if there were additional stresses placed on it through a stressor molecule such as ADMA. This endogenously produced competitive inhibitor of L-arginine (as well as an endogenous inhibitor of the eNOS reaction) would cause the eNOS reaction to uncouple and become a net producer of superoxide even before the glucose becomes elevated, thus stressing the islet and the intima before hyperglycemia is detected. This is why we should consider stress testing (by utilizing the 75 gram oral 2 hour post prandial glucose tolerance test) the patient in order to detect impaired glucose tolerance (stage III table 9) in order to reduce the oxidative and postprandial reductive stress to these organs in order to spare them from the devastating natural progressive history of T2DM and atheroscleropathy. Exercise induced skeletal muscle glucose transport (GLUT 4) is eNOS dependent [43-45]. If the production of eNO were defective due to gene polymorphism and environmental interaction there would be increasing peripheral insulin resistance. This is just what Duplain and Scherrer were able to demonstrate in their eNOS -/- model: Insulin resistance, hypertension, and hyperlipidemia.
In humans with gene polymorphism the eNOS enzyme may be capable to withstand the many years of redox stress before the defect in eNOS becomes evident. However, if the stressors were of sufficient magnitude such as seen in the adolescent youth with sedentary life style plus high carbohydrate, high fat western diet (over nutrition) the manifestation of type 2 diabetes may become evident before the youth has reached the more normal middle age in life. This is just what we are experiencing in the USA. Not only are the adolescent youth developing T2DM at an alarming rate, but also, the age of onset of T2DM in adults is becoming younger and younger. It is entirely possible that these abnormal gene polymorphisms could be very old and have never caused a problem until there was sufficient changes in the environment (the thrifty gene hypothesis: gene – environmental interaction).
Peripheral insulin resistance would certainly place an undo burden on the pancreas with elevated levels of endogenous insulin and amylin with a chronically activated RAAS at the local organ-tissue levels.
The additional heightened redox stress and the A-FLIGHT toxicities associated with insulin resistance and metabolic syndrome would only aggravate the underlying eNOS polymorphisms by placing an undo burden on the islet and the intima.
Conclusion
"YOU CAN'T SEE THE FOREST FOR THE TREES"
If we approach T2DM as initially being a vascular disease for many years with glucotoxicity being a later manifestation, then as clinicians and researchers we may be more likely to view this disease in a global perspective.
The Steno-2 study by Gaede P et al. [46,47] just published, revealed a significant reduction in cardiovascular and microvascular events of nearly 50%. Their intensified intervention against multiple risk factors in patients with type 2 diabetes was quite impressive.
Therefore, in T2DM we should consider looking at the multiple toxicities and how they interact with each other to form "the whole disease" and return metabolic and redox homeostasis to the organism.
"We must protect the forests (the whole disease) for our children, grandchildren and children yet to be born. We must protect the forests for those who can't speak for themselves such as the birds, animals, fish and trees (and our patients)." --- Chief Edward Moody, Qwatsinas, Nuxalk Nation.
Competing interests
None declared.
Authors' Contributions
MH and ST collaborated equally on this article. MH wrote and edited this article. ST collaborated on writing and editing this article.
Abbreviations
Accelerated atherosclerosis (atheroscleropathy), type 2 diabetes mellitus (T2DM), nitric oxide (NO), endothelial nitric oxide (eNO), nitric oxide synthase (NOS), neural NOS-1 (nNOS), inducible NOS-2 (iNOS), endothelial NOS-3 (eNOS), superoxide (O2'), tetrahydrobiopterin (BH4), asymmetrical dimethylarginine (ADMA), dimethylarginine dimethylaminohydrolose (DDAH), protein arginine N-methyltransferases (PRMTs), nicotinamide dinucleotide phosphate reduced (NAD(P)H), glutamic 298 → aspartyl gene polymorphism (Glu298 → Asp GP), perioxynitrite [ONOO'], coronary artery disease (CAD), insulin resistance (IR), metabolic syndrome (MS), prediabetes (PD), homocysteine (Hcy), hyperhomocysteinemia (hHcy), renin-angiotensin-aldosterone-system (RAAS), multiple metabolic toxicities of IR, MR, PD, and T2DM producing reactive oxygen species (A-FLIGHT acronym), reactive oxygen species (ROS), 5-methyl tetrahydrofolate (5MTHF), folic acid (5MTHF), glucose transporter 4 (GLUT 4), gene knockout (-/-).
Acknowledgments
Acknowledgements
The authors would like to acknowledge James R. Sowers, M.D. FACP, Professor and his department (Departments of Medicine and Cell Biology Division of Endocrinology, Diabetes and Hypertension, State University of New York Health Science Center, Brooklyn, New York) for their inspiration in writing this manuscript.
Contributor Information
Melvin R Hayden, Email: mrh29@usmo.com.
Suresh C Tyagi, Email: styagi@physiology.umsmed.edu.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein CJ, Glatt CS, Bredt DS, Snyder SH. Cloned and expressed macrophage nitric oxide synthase contrasts with the brain enzyme. Proc Natl Acad Sci U S A. 1992;89:6711–6715. doi: 10.1073/pnas.89.15.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, Nerem RM, Alexander RW, Murphy TJ. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90:2092–2096. doi: 10.1172/JCI116092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE., Jr The molecule of the year. Science. 1992;258:1861. doi: 10.1126/science.1470903. [DOI] [PubMed] [Google Scholar]
- Hayden MR. Atherosclerosis and plaque Angiogenesis: a malignant transformation. Submitted paper Pathology and Clinical classification of Vulnerable Plaque. May 2001, http://www.vp.org http://www.vp.org/ResourceCenter/Pete_Hayden_Angiogenesis.html.
- Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.CIR.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC. Islet redox stress: the manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP J Pancreas (online) 2002;3:86–108. http://www.joplink.net [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC. Intimal Redox Stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovascular Diabetology. 2002;1:3 doi: 10.1186/1475-2840-1-3. http://www.cardiab.com [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC. Arterial vascular remodeling: the endothelial cell's central role. Mo Med. 1998;95:213–217. [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC. Chapter. Atherosclerosis: Implications of angiotensin II and the AT-1 receptor. In: Dhalla NS, Zahradka P, Dixon I, Beamish R, editor. Angiotensin II Receptor Blockade: Physiological and Clinical Implications. Vol. 2. Kluwer Academic publishers. Boston, Ma; 1998. pp. 233–243. [Google Scholar]
- Hayden MR, Tyagi SC. Arteriogenesis: Angiogenesis within Unstable Atherosclerotic Plaques – Interactions with Extracellular Matrix. Curr Interv Cardiol Rep. 2000;2:218–227. [PubMed] [Google Scholar]
- Hayden MR. Islet amyloid, metabolic syndrome, and the natural progressive history of type 2 diabetes mellitus. JOP J Pancreas (online) 2002;3:86–108. http://www.joplink.net [PubMed] [Google Scholar]
- Kroncke KD, Fehsel K, Kolb-Bachofen V. Nitric oxide: cytotoxicity versus cytoprotection – how, why, when, and where? Nitric Oxide. 1997;1:107–120. doi: 10.1006/niox.1997.0118. [DOI] [PubMed] [Google Scholar]
- Stuhlinger MC, Abbasi F, Chu JW, Lamendola C, McLaughlin TL, Cooke JP, Reaven GM, Tsao PS. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA. 2002;287:1420–1426. doi: 10.1001/jama.287.11.1420. [DOI] [PubMed] [Google Scholar]
- Cooke JP, Oka RK. Atherogenesis and the arginine hypothesis. Curr Atheroscler Rep. 2001;3:252–259. doi: 10.1007/s11883-001-0068-x. [DOI] [PubMed] [Google Scholar]
- Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation. 2002;106:987–992. doi: 10.1161/01.CIR.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. doi: 10.1161/01.atv.20.9.2032. [DOI] [PubMed] [Google Scholar]
- Nash DT. Insulin resistance, ADMA levels, and cardiovascular disease. JAMA. 2002;287:1451–1452. doi: 10.1001/jama.287.11.1451. [DOI] [PubMed] [Google Scholar]
- Faldetta CM, Laurenti O, Desideri G, Bravi MC, DeLuca O, Marinucci MC, De Mattia G, Ferri C. L-arginine infusion decreases plasma total homocysteine concentrations through increased nitric oxide production and decreased oxidative status in type II diabetic patients. Diabetologia. 2002;45:1120–1127. doi: 10.1007/s00125-002-0854-1. [DOI] [PubMed] [Google Scholar]
- Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Saito Y, Kajiyama N, Yoshimura M, Shimasaki Y, Nakayama M, et al. Endothelial nitric oxide synthase gene is positively associated with essential hypertension. Hypertension. 1998;32:3–8. doi: 10.1161/01.hyp.32.1.3. [DOI] [PubMed] [Google Scholar]
- Shimasaki Y, Vasue H, Yoshimura M, Nakayama M, Kaugiyama K, Ogawa H, Harada E, Masuda T, Koyama W, Saito Y, Miyamoto Y, Ogawa Y, Nakao K. Association of the missense Glu298Asp variant of the endothelial nitric oxide synthase gene with myocardial infarction. J Am Coll Cardiol. 1998;31:1506–1510. doi: 10.1016/S0735-1097(98)00167-3. [DOI] [PubMed] [Google Scholar]
- Hingorani AD, Liang CF, Fatibene J, Lyon Monteith AS, Parsons A, Haydock S, Hopper RV, Stephens NG, O'Shaughnessy KM, Brown MJ. A common variant of the endothelial nitric oxide synthase (Glu298 → Asp) is a major risk factor for coronary artery disease in the UK. Circulation. 1999;100:1515–1520. doi: 10.1161/01.cir.100.14.1515. [DOI] [PubMed] [Google Scholar]
- Colombo MG, Andreassi MG, Paradossi U, Botto N, Manfredi S, Massetti S, Rossi G, Clerico A, Biagini A. Evidence for association of a common variant of the endothelial nitric oxide synthase gene (Glu298 → Asp polymorphism) to the presence, extent, and severity of coronary artery disease. Heart. 2002;87:525–528. doi: 10.1136/heart.87.6.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman BA, Spiering W, Doevendans PA, Vervoort G, Kroon AA, De Leeuw PW, Smits P. The Glu298Asp polymorphism of the NOS 3 gene as a determinant of the baseline production of nitric oxide. J Hypertens. 2002;20:2023–2027. doi: 10.1097/00004872-200210000-00022. [DOI] [PubMed] [Google Scholar]
- Noiri E, Satoh H, Taguchi J, Brodsky SV, Nakao A, Ogawa Y, Nishijima S, Yokomizo T, Tokunaga K, Fujita T. Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension. 2002;40:535–540. doi: 10.1161/01.HYP.0000033974.57407.82. [DOI] [PubMed] [Google Scholar]
- Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Current Drug Metabolism. 2002;3:159–173. doi: 10.2174/1389200024605073. [DOI] [PubMed] [Google Scholar]
- Doshi SN, McDowell IF, Moat SJ, Payne N, Durrant HJ, Lewis MJ, Goodfellow J. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation. 2002;105:22–26. doi: 10.1161/hc0102.101388. [DOI] [PubMed] [Google Scholar]
- Van Etten RW, De Koning EJ, Verhaar MC, Gaillard CA, Rabelink TJ. Impaired NO-dependent vasodilation in patients with Type II (non-insulin-dependent) diabetes mellitus is restored by acute administration of folate. Diabetologia. 2002;45:1004–1010. doi: 10.1007/s00125-002-0862-1. [DOI] [PubMed] [Google Scholar]
- Boger R, Bode-Boger S, Thiele W. Restoring vascular nitric oxide formation by L-arginine improves the symptoms of intermittent claudication in patients with peripheral arterial occlusive disease. J Am Coll Cardiol. 1998;32:1336–1344. doi: 10.1016/S0735-1097(98)00375-1. [DOI] [PubMed] [Google Scholar]
- Maxwell AJ, Anderson BA, Cooke JP. Nutritional therapy for peripherial arterial disease: A double blind, placebo-controlled, randomized trial of HeartBar. Vasc Med. 2000;5:11–19. doi: 10.1191/135886300676721584. [DOI] [PubMed] [Google Scholar]
- Tyagi SC. Homocyst(e)ine and heart disease: Pathophysiology of extracellular matrix. Clin Exper Hypertens. 1999;21:181–198. doi: 10.3109/10641969909068660. [DOI] [PubMed] [Google Scholar]
- Miller A, Mujumdar V, Palmer L, Bower JD, Tyagi SC. Reversal of endocardial endothelial dysfunction by folic acid in homocysteinemic hypertensive rats. Am J Hypertens. 2002;15:157–163. doi: 10.1016/S0895-7061(01)02286-5. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Tummalapalli CM, Aru GM, Tyagi SC. Mechanism of constrictive vascular remodeling by homocysteine: role of PPAR. Am J Physiol Cell Physiol. 2002;282:C1009–1015. doi: 10.1152/ajpcell.00353.2001. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Aru GM, Tyagi SC. Induction of oxidative stress by homocyst(e)ine impairs endothelial function. J Cell Biochem. 2001;82:491–500. doi: 10.1002/jcb.1175. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Hayden MR, Tyagi SC. Homocyst(e)ine induces calcium second messenger in vascular smooth muscle cells. J Cell Physiol. 2000;183:28–36. doi: 10.1002/(SICI)1097-4652(200004)183:1<28::AID-JCP4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Miller A, Mujumdar V, Shek E, Guillot J, Angelo M, Palmer L, Tyagi SC. Hyperhomocyst(e)inemia induces multi organ damage. Heart Vessels. 2000;15:135–143. doi: 10.1007/s003800070030. [DOI] [PubMed] [Google Scholar]
- Stanger O. Physiology of folic acid in health and disease. Current Drug Metabolsim. 2002;3:211–223. doi: 10.2174/1389200024605163. [DOI] [PubMed] [Google Scholar]
- Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic Acid, vitamin B12, and vitamin B6 on clinical outcome after percutaneous coronary intervention: the swiss heart study: a randomized controlled trial. JAMA. 2002;288:973–979. doi: 10.1001/jama.288.8.973. [DOI] [PubMed] [Google Scholar]
- Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Association of plasma homocysteine with restenosis after percutaneous coronary angioplasty. Eur Heart J. 2002;23:726–33. doi: 10.1053/euhj.2001.2962. [DOI] [PubMed] [Google Scholar]
- Schnyder G, Roffi M, Pin R, Flammer Y, Lange H, Eberli FR, Meier B, Turi ZG, Hess OM. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med. 2001;345:1593–600. doi: 10.1056/NEJMoa011364. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, Mitch W, Smith SC, Jr, Sowers JR. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999;100:1134–1146. doi: 10.1161/01.cir.100.10.1134. [DOI] [PubMed] [Google Scholar]
- Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. AJP-Endocrinology and Metabolism. 1998;274:E692–E699. doi: 10.1152/ajpendo.1998.274.4.E692. [DOI] [PubMed] [Google Scholar]
- Kapur S, Bedard S, Marcotte B, Cote CH, Marette A. Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action. Diabetes. 1997;46:1691–1700. doi: 10.2337/diab.46.11.1691. [DOI] [PubMed] [Google Scholar]
- Shiuchi T, Cui TX, Wu L, Nakagami H, Takeda-Matsubara Y, Iwai M, Horiuchi M. ACE inhibitor improves insulin resistance in diabetic mouse via bradykinin and NO. Hypertension. 2002;40:329–334. doi: 10.1161/01.HYP.0000028979.98877.0C. [DOI] [PubMed] [Google Scholar]
- Gæde P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med. 2003;348:383–393. doi: 10.1056/NEJMoa021778. [DOI] [PubMed] [Google Scholar]
- Solomon CG. Reducing cardiovascular risk in type 2 diabetes. N Engl J Med. 2003;348:457–459. doi: 10.1056/NEJMe020172. [DOI] [PubMed] [Google Scholar]