

Abstract
Background
Amplified fragment length polymorphism (AFLP) is a PCR-based technique that involves restriction of genomic DNA followed by ligation of adaptors to the fragments generated and selective PCR amplification of a subset of these fragments. The amplified fragments are separated on a sequencing gel and visualized by autoradiography or fluorescent sequencing equipment. AFLP allows high-resolution genotyping but the lack of a format for databasing and comparison of AFLP fingerprint profiles limits its wider applications in profiling large numbers of biological samples.
Results
A scheme is described to represent a DNA fingerprint profile with a nucleotide sequence-like format in which the information line contains the minimal necessary details to interpret an AFLP DNA fingerprint profile. They include technique used, information on restriction enzymes, primer combination, biological source for DNA materials, fragment sizing and annotation. The bodylines contain information on size and relative intensity of DNA fragments by a string of defined alphabets or symbols. Algorithms for normalizing raw data, binning of fragments and comparing AFLP DNA fingerprint profiles are described. Firstly, the peak heights are normalized against their average and then represented by five symbols according to their relative intensities. Secondly, a binning algorithm based loosely on common springs and rubber bands is applied, which positions sequence fragments into their best possible integer approximations. A BLAST-like reward-penalty concept is used to compare AFLP fingerprint profiles by matching peaks using two metrics: score and percentage of similarity. A software package was developed based on our scheme and proposed algorithms. Example of use this software is given in evaluating novelty of a new tropical orchid cultivar by comparing its AFLP fingerprint profile against those of related commercial cultivars in a database.
Conclusions
AFLP DNA fingerprint profiles can be databased and compared effectively with software developed based on our scheme and algorithms. It will facilitate wider use of this DNA fingerprinting technique in areas such as forensic study, intellectual property protection for biological materials and biodiversity management. Moreover, the same concepts can be applied to databasing and comparing DNA fingerprint profiles obtained with other DNA fingerprint techniques.
Background
DNA markers reflect difference in the DNA sequences of chromosomes derived from different progenitors. They arise as a result of mutations as well as rearrangements in the DNA intervening between two restriction sites, or two priming sites. "DNA fingerprint" refers to applying combined multilocus profile of DNA markers for identification of individual, clonal identical individuals (cultivar) or species. Polymerase chain reaction (PCR) technology has promoted the development of a range of molecular assay systems that detect polymorphism at the DNA level and offer an alternative to the hybridization based method of Restriction Fragment Length Polymorphism (RFLP) [1]. Amplified Fragment Length Polymorphism (AFLP) developed by Vos et al. [2] is based on the amplification of short restriction endonuclease-digested genomic DNA fragments onto which adaptors have been ligated at both ends. Primers complementary to the adaptors and possessing 3' selective nucleotides of one to four bases are used in a selective amplification reaction. The presence or absence of these selective nucleotides in the genomic fragments being amplified provide the basis for revealing polymorphism. Besides being relatively cheap, easy and fast, it is a very robust and reliable technique, especially with the use of fluorescent DNA sequencing equipment. AFLP has been used to uncover cryptic genetic variation of strains, or closely related species in plants, fungi, animals and bacteria [3], genetic mapping [4] as well as study genetic variation within populations [5]. Because of the high level of polymorphism it provides, a few primer combinations will suffice to generate fingerprint with hundreds of highly replicable markers that allow high resolution genotyping any individual or clonally identical individuals (e.g. plant cultivars). However, the lack of solution to database and compare AFLP fingerprint profiles impedes full exploitation of its potentials in areas such as individual profiling, help manage plant breeder rights and management of biodiversity.
In AFLP analysis, amplified DNA fragments are separated by polyacrylamide gel electrophoresis after selective PCR reactions. Radioisotope labeled fragments are detected by either autoradiography followed by densitometer scanning or directly by phosphoimagers. Fluorescence labeled DNA fragment data is acquired by built in fluorescence detectors in genetic analysis instruments or by fluorescent imagers. Software has been developed to optimize raw data collected, mainly through reducing noise while maintaining data integrity; and to size and quantitate DNA fragments with reference to standard samples. Quantity One® from Biorad and Genotyper® by Applied Biosystem are two examples. Least square method that uses regression analysis to build a best-fit size calling curve from size standards is a popular option for sizing DNA fragments. Other choices like point to point, cubi spline interpolation, local southern method and global southern methods often give very similar results. Results from such software are given in tabular form with size and quantitation information but without other minimal information to interpret an AFLP fingerprint profile like biological source of DNA and primer information. Data in such a format is hard to database and compare in an efficient manner. There is the need for a common format or guideline that allows the unambiguous interpretation, and potential reproduction, of an AFLP fingerprinting experiment. Such format should also allow easy databasing and comparing AFLP fingerprint profiles.
In this paper, we suggest a simple format containing the minimal-necessary information about a DNA fingerprint (MIADF) which is compatible with all techniques of DNA fingerprinting. A DNA fingerprint profile is represented by a few informational fields and a pure-text signature, with DNA bands represented by a sequence of predefined alphanumeric symbols indicating different relative intensities. The specification of this format and accompanying algorithms for the normalization of raw data, binning DNA fragments and compare and score AFLP fingerprint profiles are described in detail.
Results
Ampsig format
We propose an Amplified-type signature (Ampsig) as an informative, highly specific, yet extremely compact format for a DNA fingerprint profile (Table 1).
Table 1.
Illustration of the Ampsig format with example values
| # | Field | Example Value | Information |
| 1 | Technique | AFLP | Full abbreviation of type of DNA markers, such as AFLP, SSR, RAPD |
| 2 | Experiment | EcoRI_ACA-MseI_CAC | For VNTR, RAPD and SSR markers, primer sequences will be given in full. In this example, a six-nucleotide cutting enzyme EcoRI and a four-nucleotide cutting enzyme MseI were used to digest genomic DNA. PCR reaction was conducted with an EcoRI primer with selective nucleotides ACA, and a MseI primer with selective nucleotides CAC |
| 3 | Source | D Sonia id0011 | Biological source of DNA. The binomial system with abbreviation of genus followed by species name is used to designate biological species. It is followed by a four-digit ID number to identify individual samples. In this example, DNA was isolated from a plant belonging to genus Dendrobium, species Sonia, with a lab-assigned ID 0011 |
| 4 | UnitSize | 1 | Unit size of DNA fragment to define the unit spacing. It can also be interpreted as the sequence resolution. In this example, the 1 indicates that the sequence spacing is 1 base pair. |
| 5 | StartSize | 50 | Size of the first DNA fragment (in base pairs) |
| 6 | EndSize | 90 | Size of the last DNA fragment. This field is redundant, and is kept mainly for proof-reading |
| 7 | Annotation | AFLP profile for D. Sonia | Annotation of the fingerprint (only if required) |
| 8 | Signature | A..B..DC..B.D.....CC...A.....DDA.A....C..D..BB | Alternatively, a system employing the symbols [4,3,2,1] for peaks and [0] for peak-absence may also be used. |
The following is a sample AFLP fingerprint, written out as bar- and line-delimited entries in a FASTA-style text file (alternatively, this data can be stored as ordinary text fields in a relational database):
{AFLP|EcoRI_ACA-MseI_CAC|D Sonia id0011|1|50|101|AFLP profile for D. sonia
B...A.B.D.DD..BB..A..DD.B.A..DC..ABB..BBA..DCB..D..B
The application of Ampsig in AFLP data analysis and algorithms
An AFLP fingerprint comprises of many PCR fragments. Either radioisotope or fluorescent DNA fragment data is obtained depending on whether radioisotope-labeled primers or fluorescent-labeled primers are used for the final selective amplification. The popular ABI PRISM genetic analysis instruments detect DNA fragments by automated fluorescent scanning detection. The GeneScan® analysis software analyzes data to size and quantify DNA fragments and gives result for each sample in a table listing fragment sizes and intensities (see table 2 for an example). Such results are difficult for multiple sample comparison, can't be interpreted without other minimum necessary information, can't be stored in a database readily and contains less reproducible numeric quantitative data (fluorescence counting). They are transformed into Ampsig entries in the following manner:
Table 2.
Example of GeneScan analysis result in a simple text file
| Peak | time | size | height | area | point |
| G,1 | 35.29 | 51.49 | 151 | 544 | 683 |
| G,2 | 35.65 | 53.22 | 296 | 2046 | 690 |
| G,3 | 36.12 | 67.34 | 299 | 1834 | 699 |
| G,4 | 36.32 | 68.52 | 378 | 1529 | 703 |
| G,5 | 36.73 | 69.41 | 765 | 6140 | 711 |
| G,6 | 37.35 | 70.23 | 156 | 963 | 723 |
| G,7 | 37.72 | 71.42 | 171 | 979 | 730 |
| G,8 | 38.03 | 72.64 | 514 | 2982 | 736 |
Normalization
Peak data was obtained in text form using the Export Lane to Raw command on the ABI GeneScan software, and a space-delimited plain text file is obtained, an example is shown in Table 2.
The labels for the 6 columns are peak (peak ID), time (time of detection), size (calculated size in reference to internal size standards), height (height of a peak), area (area of peak) and point (data point at maximum peak height), respectively. In our analysis, we only use data in size and height columns though area data can also be used.
We found that normalizing the height according to the highest peak was inadequate in producing consistent signatures because fluctuations in the amplitude (intensity) of the highest peak have too great an effect in skewing the rest of the data. We finally decided instead on normalizing the waveform by average amplitude of the reported peaks. In this case, one unit would simply represent the average height of the peaks over the waveform's size range.
We visually ranked peaks on gel images based on intensity and concluded that categorizing peaks into five scales is visually achievable without much difficulty (A for very strong intensity, B for moderately strong intensity, C for moderately weak intensity, D for very weak intensity and [.] for absence of peak) and this is minimum necessary to represent quantitative data. Range definitions were adjusted to maximize agreement of computer sorting with visual sorting. Finally we decided the following optimal ranges for scaling peaks (Table 3).
Table 3.
Peak Scales after normalization (1 represents the average peak height)
| Peak symbol | Range (normalized against average height) |
| A | [6.4, +∞) |
| B | [3.2, 6.4) |
| C | [1.6, 3.2) |
| D | (0, 1.6) |
| . | 0 (no peak) |
Binning
Theoretically, an AFLP gel run should only register peaks at integral sizes (since only a round number of base pairs should be present). However, nucleotides composition of gel fragments, uniformity of gel, size markers used, position of tracking lines and method of calibration for a standard curve all contribute to fractional-length fragments. Resolution of one base pair is required for analysis of AFLP results but simple rounding might cancel out bands that contain important information. In the example given in table 2, simple rounding will merge G,4 and G,5 as one band of size 69 bp in spite of the fact that they are clearly two neighboring bands. Thus, the relative distance between peaks as well as their sizes before rounding up should both be considered in order to arrive at a good fit. We developed a binning algorithm based on the familiar physical models of spring and elastic (rubberband) energy. Spring and Rubberband model draws the analogy of a spring maintaining the relative distance between the peaks, and rubberbands pulling the real-valued peaks towards integral points. This is to allow sequence fragments to move as a whole and maintain their relative distance. With this scheme, consecutive fragments less than 2.5 units from each other are clustered into a sequence. If the potential energy function of a binned sequence is defined as the sum of its Spring energy and its Rubberband energy, the 'best fit' simply involves minimizing the fragment's potential energy function.
If simple rounding is used to bin the sequence of fragments (67.3, 68.5, 69.4, 70.2, 71.9 see Figure 1A), it results in the minimum rubberband energy for the system since all peaks are binned to their closest integer values, as shown by the dotted lines). However, its spring energy is quite high (The first spring segment, shown as double lines, is stretched from 1.2 to 2 units in length, while the second segment is compressed from 0.9 to 0 units).
Figure 1.
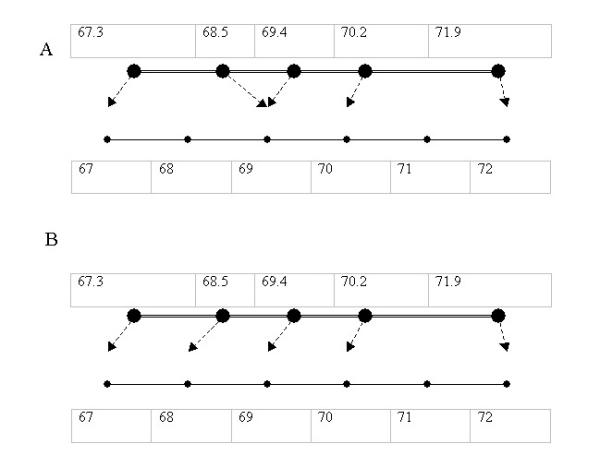
Comparison of two binning models. A sequence of AFLP markers (67,3, 68.5, 69.4, 70.2, 71.9) are binned by: A. Simple rounding B. Spring and rubber band model
Our binning algorithm involves optimizing for the best fit i.e. minimizing on the sum of rubber band energy and spring energy after displacement bands into integers. We then tweaked the formulae to see what adjustments produced the most satisfactory results. By comparing binning results with gel image and electrophoregram of 20 fingerprints, we found that the system worked best when the rubberband energy function was proportional to the cube of the displacement, spring energy function was proportional to the square of the displacement and weightage of the spring energy was tripled (Figure 2). The same fragments will then be binned with this spring and rubber band model as 67,68,69,70,71 (Figure 1B).
Figure 2.
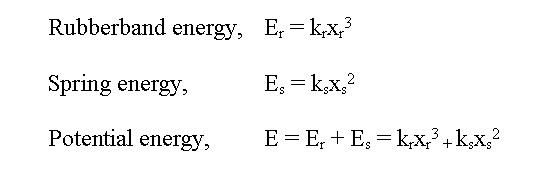
Formula for Rubberband and Spring energy, Er, Es and E are rubberband energy, spring energy and potential energy, respectively. x is the displacement and kr and ks are proportionality constants.
Where x is the displacement and kr and ks are proportionality constants.
Since the system works best when ks/kr = 3, we can set kr = 1 and ks = 3 to give the optimized formula:
E = xr3 + 3xs2
The peaks in the above example will then be binned as shown in Figure 3 and it is easy to appreciate it as the optimal binning.
Figure 3.

Evaluation novelty of an orchid cultivar by using PAPA software. A. After uploading data into the software, the query AFLP fingerprint profile for the cultivar "unknown" is shown, together with other profiles in the database "orchidb1", as a string of letters representing fragments of different relative intensities. Different colors were used for different relative intensities to enhance visual effects. Size bar on top enables easy sizing of DNA fragments. Five columns on the left contain information on score, percentage, sample ID, start size and ending size for a profile. Click to select the "unknown" profile (as indicated by the arrow) to compare with other profiles in the database. B: After database search, profiles are rearranged in the order of their score and percentage against the query profile.
Profile comparison
Consisting of pure text, Amptype signature files can be easily aligned and compared. A method to quantitate similarity is then required. We reason that more intense bands should be given higher weightage due to the fact that they are the most reproducible bands and higher intensity may be attributed to high copy numbers. That higher intensity may also be attributed to comigration of non-allelic fragments also justifies higher weightage. The commonly used methods for calculating genetic distances like DICE, Jaccard and Pearson [6] are based on the assumption that each band carries equal amount of information and are therefore not applicable. We adapted the reward-penalty concept used in BLAST sequence comparisons, i.e. a positive score will be awarded for every matching character, and a penalty will be incurred for every mismatch. Matching peaks of different intensities will be scored less (see table 4).
Table 4.
Reward and Penalty score chart
| Peaks | A | B | C | D | . |
| A | +10 | +3 | 0 | -2 | -4 |
| B | +3 | +6 | +2 | -1 | -3 |
| C | 0 | +2 | +4 | 0 | -2 |
| D | -2 | -1 | 0 | +2 | -1 |
| . | -4 | -3 | -2 | -1 | 0 |
e.g. AB..C.D scored against AB..C.D gives a score of 22 (=10+6+4+2)
Alternatively, for every peak that exists on one sequence but is absent from the other, a penalty is incurred to the score, depending on the peak's intensity. e.g. If CB..A.D is scored against CB....D, a -4 penalty is incurred for the missing A peak.
The scoring will proceed as following:
1) Firstly search for exact matching peaks. All such matching characters are replaced in the string by a dummy character (in this case, a dot '.') to avoid repeated calculation.
2) A second search is then performed for matching peaks of differing intensity.
3) The peaks remaining are all mismatches which contribute to the total score penalty.
4) The final score is simply the score awarded for matching peaks, minus the penalties incurred by unmatched ones.
Score by this method depends on both intensity of matching and width of the scoring window.
We introduce a second scoring method, percentage, to make it easier to comprehend similarity of two fingerprints. It is expressed as a percentage value with a maximum of 100%. Within a designated scoring window, the percentage score of sequence B against sequence A is defined as the ratio of the score of B against A to the highest possible score within the scoring window.
Percent (A,B) = 100% × score(A,B) / Max {score(A,A), score(B,B)}
This score is symmetric since Score(A,B) = Score (B,A)
Example of use: novelty of plant variety
Based on the scheme and algorithms described above, we have developed a software for databasing and managing AFLP DNA fingerprint profiles. The web-interfaced and user-friendly software can upload raw data files generated by GeneScan software, normalization is conducted during the uploading process. It also provides the flexibility of manual imput, edit or delete operations of fingerprint profiles. A profile can be compared with all fingerprint profiles in the same database, resulting in a list of profiles in the order of score and percentage of similarity. It can be used for profile matching as well as for those purposes requiring evaluating difference of a fingerprint profile against all others in a database. One such use is to prove novelty of new plant varieties to satisfy DUS requirements (novel, distinct, unique and stable) under Plant Breeder Rights. In this example (Figure 3), AFLP fingerprint of one new orchid cultivar is compared with those of related commercial Dendrobium cultivars in a database with >70 profiles. The search result is shown as a list of fingerprint profiles in the order of their similarity to this fingerprint (score and percentage similarity). The new cultivar is found closest to cultivar Dendrobium Tay Swee Keng with a score of 297 out of maximum possible score of 418 and 72.8% similarity. The result makes very good sense since Dendrobium Tay Swee Keng was one parent for this new cultivar. Since the AFLP DNA fingerprint profile of this new cultivar has no stronger similarity to any other profile in this database, we conclude that it is a novel cultivar judging from its unique AFLP fingerprint profile.
Discussion
DNA fingerprinting reveals differences in genomic DNAs, is independent of developmental stage and environmental factors, therefore such information is not necessary to interpret a DNA fingerprint profile. Besides the minimum necessary information we suggest in this paper, other factors will also affect DNA fingerprinting patterns to a lesser extent. These factors include the set-up of a laboratory, skill level of technicians, DNA quality, type of labeling (radioisotope or fluorescent dye), methods of detection and data analysis software, among several others. We reason that these factors affect mostly data quality but are not critical to interpret and for others to repeat an experiment.
The Ampsig format is simple, compact, yet keeps the essence of DNA fingerprint profiles. It allows manual editing using any text editor. It is an open format to accommodate results from other DNA fingerprinting techniques like Random Amplified Polymorphic DNA (RAPD) [7] and RFLP with minimum changes in experiment field to include contents specific to each technique. For RFLP technique, we suggest to include information on restriction enzymes, probe used in the form of accession number for public databases like genebank. For RAPD, Variable Number Tandem Repeats (VNTR) [8] and Simple Sequence Repeats (SSR) markers [9], primer sequences can be given in full in the experiment field. Some of the information fields can serve as a unique accession or 'key' fields in high performance database systems, and can also be split up into separate fields to enable useful indexing. The UnitSize field keeps the format flexible enough to adapt to different type of DNA markers. A UnitSize of 1 base pair is suitable for AFLP, SSR and DNA Amplification Fingerprinting (DAF) [10] while a UnitSize of 100 base pairs may be necessary for RFLP and VNTR markers. The sizes of the fragments can be easily deduced from its StartSize, Signature length and UnitSize definition. The seemingly redundant EndSize for the sequence is also recorded as a validation check, eg. to ensure that the full length of the Signature is indeed present.
Normalization of raw data reflects our understanding that relative intensity of a band is more reproducible than its absolute counts. Absolute values of fluorescence counting or density value after autoradiography for each DNA fragment are much less important to a DNA fingerprint profile than relative intensity. These values tend to fluctuate even in different batches of experiment by the same researcher. Normalization of raw data makes a profile more robust and repetitive.
The five scales we suggested match well with our visual sorting. More scales can be adapted if more detail of relative intensity needs to be recorded. Alternatively, it can also accommodate the practice of using binary symbols (1,0) for the presence or absence of bands if less information is required.
The Ampsig format makes large-scale data analysis and comparison possible because the data can be easily stored and indexed for fast retrieval in any high-performance SQL database server. Software can also be developed to perform in-depth analysis of additional related data along with their Ampsig fingerprints.
We introduce a binning algorithm based on physical springs and rubberbands to position the sequence fragments into their best possible integer approximations. Theoretically, the entire run of peaks may well be treated as a single sequence for binning purposes but this proved to be too computationally intensive without significant improvement in the quality of the results. We have verified that this algorithm reduces variation among different samples and provides consistently reproducible compact biological signature sequences in our lab.
For the purpose of comparison, we give more weightage to more intense bands and adapt the reward-penalty concept used in BLAST sequence comparisons, with gap penalties set high, because the position of peaks (DNA fragment sizes) are absolute, and deviation should be penalized heavily. The absolute score is the sum of scores awarded for matching peaks minus penalties for unmatched peaks. It evaluates the level of similarity of two fingerprint profiles. Our suggestions of award/penalty points are empirical and may not be the optimal. Another scoring method, percentage scoring, is easier to comprehend and compare similarity within a defined window. Results can be used to build a similarity matrix among multiple samples and be used for phylogenic analysis.
Rigid adaptation of this scoring method may result in poor scoring of DNA fingerprint profiles with a similar pattern but with slight deviations in position. To overcome this problem, modifications can be incorporated to allow a single position deviation while searching for matching peaks, but the score awarded should be reduced.
These concepts have been put into practice and continue to be feature-updated in the form of a software package titled Public Ampsig Peak Analysis (PAPA) that uses SQL for databasing fingerprints. The open and non-binary (text) form can be shared across many current applications and formats including XML.
In the example, we demonstrate utility of our scheme and algorithms by using this software to evaluate novelty of a new plant cultivar by comparing its AFLP fingerprint profile with those of related commercial hybrids in the database. Caution needs to be taken to accept such conclusion of "novelty" since it is directly affected by number of profiles in the database and cut off value for "novelty". Moreover, profiles in this database were obtained by using only one AFLP primer combination, conclusion may vary with data from other primer combinations or with combined data of several primer combinations.
Authors' contributions
Y Hong conceived of and coordinated the study, participated in algorithm design and prepared manuscript. A Chuah designed the algorithms, performed optimization of the formula and helped in preparing the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We would like to thank Dr. Wong Lim Soon of the Kent Ridge Digital Labs and Dr Laszlo Orban of Temasek Life Sciences Laboratory for their invaluable suggestions and critiques.
Contributor Information
Yan Hong, Email: hongy@tll.org.sg.
Aaron Chuah, Email: aaron@tll.org.sg.
References
- Bostein D, White R, Skolnick M, Davis R. American Journal of Human Genetics. 1980;32:314–331. [PMC free article] [PubMed] [Google Scholar]
- Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, et al. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995;23:4407–4414. doi: 10.1093/nar/23.21.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller UG, Wolfenbarger LL. AFLP genotyping and fingerprinting. Trends In Ecology And Evolution. 1999;14:389–394. doi: 10.1016/S0169-5347(99)01659-6. [DOI] [PubMed] [Google Scholar]
- Lionneton E, Ravera S, Sanchez L, Aubert G, Delourme R, Ochatt S. Development of an AFLP-based linkage map and localization of QTLs for seed fatty acid content in condiment mustard (Brassica juncea). Genome. 2002;45:1203–15. doi: 10.1139/g02-095. [DOI] [PubMed] [Google Scholar]
- Steinger T, Haldimann P, Leiss KA, Muller-Scharer H. Does natural selection promote population divergence? A comparative analysis of population structure using amplified fragment length polymorphism markers and quantitative traits. Mol Ecol. 2002;11:2583–90. doi: 10.1046/j.1365-294X.2002.01653.x. [DOI] [PubMed] [Google Scholar]
- Sneath PHA, Sokal RR. Numerical taxonomy: the principles and practice of numerical classification. WH Freeman and Company. 1973.
- Welsh J, McClelland M. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 1990;17:7213–7218. doi: 10.1093/nar/18.24.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Leppert M, O'Connell P, Wolff R, Holm T, Culver M, Martin C, Fujimoto E, Hoff M, Kumlin E, et al. Variable number of tandem repeat (VNTR) markers for human gene mapping. Science. 1987;235:1616–1622. doi: 10.1126/science.3029872. [DOI] [PubMed] [Google Scholar]
- Love JM, Knight AM, McAleer MA, Todd JA. Towards construction of a high resolution map of the mouse genome using PCR-analysed microsatellites. Nucleic Acids Res. 1990;18:4123–4130. doi: 10.1093/nar/18.14.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anolles G, Bassam BJ, Gresshoff PM. DNA amplification fingerprinting using very short arbitrary oligonucleotide primers. Biotechnology. 1991;9:553–557. doi: 10.1038/nbt0691-553. [DOI] [PubMed] [Google Scholar]