

Abstract
Estrogen receptors (ER) have been localized to the cell plasma membrane (PM), where signal transduction mediates some estradiol (E2) actions. However, the precise structural features of ER that result in membrane localization have not been determined. We obtained a partial tryptic peptide/mass spectrometry analysis of membrane mouse ERα protein. Based on this, we substituted alanine for the determined serine at amino acid 522 within the E domain of wild-type (wt) ERα. Upon transfection in CHO cells, the S522A mutant ERα resulted in a 62% decrease in membrane receptor number and reduced colocalization with caveolin 1 relative to those with expression of wt ERα. E2 was significantly less effective in stimulating multiple rapid signals from the membranes of CHO cells expressing ERα S522A than from those of CHO cells expressing wt ERα. In contrast, nuclear receptor expression and transcriptional function were very similar. The S522A mutant was also 60% less effective than wt ERα in binding caveolin 1, which facilitates ER transport to the PM. All functions of ERα mutants with other S-to-A substitutions were comparable to those of wt ER, and deletion of the A/B or C domain had little consequence for membrane localization or function. Transfection of ERα S522A into breast cancer cells that express native ER downregulated E2 binding at the membrane, signaling to ERK, and G1/S cell cycle events and progression. However, there was no effect on the E2 transactivation of an ERE-luciferase reporter. In summary, serine 522 is necessary for the efficient translocation and function of ERα at the PM. The S522A mutant also serves as a dominant-negative construct, identifying important functions of E2 that originate from activating PM ER.
Steroid action is attributed primarily to the regulation of target genes through nuclear receptor binding and transactivation, subsequently producing cell biological effects (42). However, it has increasingly been appreciated that steroids, such as estradiol (E2), act rapidly through nongenomic mechanisms of signal transduction (4, 16, 41). These signaling mechanisms have important consequences for the effects of steroids on cell biology (14, 40). For E2, these effects can occur after the sex steroid binds to plasma membrane (PM) estrogen receptors (ER) (17, 30), which has been demonstrated by immunohistochemistry (36) and by immunoblotting of isolated PM domains (6). Some signaling effects of E2-ER can result from complex interactions with PM growth factor tyrosine kinase receptors, such as the epidermal growth factor receptor (EGFR) (10).
Although the exact sequence of this receptor has not been reported, the membrane ER appears to be very similar, and perhaps identical, to the nuclear receptor. This is based upon the identification of similarly sized nuclear and membrane ER proteins that result from the expression of a single cDNA (and resulting single mRNA) in CHO cells (32). Also, membrane ER have been localized on vascular smooth muscle, pituitary, and endothelial cells that express endogenous receptors, by using antibodies raised against multiple epitopes of the nuclear ERα (25, 26, 36). However, many questions remain concerning this relatively small population of ER at the cell surface.
The membrane ER has been reported to be G protein linked (32, 50), and E2 binding can activate many signal transduction pathways that emanate from G protein activation. These include kinase and endothelial nitric oxide synthase activation, cyclic AMP (cAMP) and inositol phosphate (IP) generation, and phospholipase C (PLC) stimulation (4, 16, 18, 24, 50). Linkage to G proteins may be direct, as shown in transfected CHO cells expressing ERα or ERβ (32) or in endothelial cells (50), but it has also been reported that E2 activates an orphan G protein-coupled receptor (10). Furthermore, it is not clear whether this receptor spans the cell membrane or is predominately localized within or associated with the membrane bilayer. Membrane ER have recently been shown to exist in discrete caveolar domains of the PM (6, 13). It has recently been found that membrane ERα can physically associate with the caveolar structural coat proteins caveolin 1 and caveolin 2 (31). Caveolin proteins serve as scaffolds, bringing together various signaling molecules within a discrete area of the PM to regulate cytokine-induced signal transduction (3, 27). These include G proteins, nonreceptor and receptor tyrosine kinases (Src, EGFR), and threonine-serine kinases, such as phosphatidylinositol 3-kinase (PI 3-kinase) and Raf. Organization of signaling molecules within a confined space potentially allows E2-ER to modulate a variety of signaling cascades in target cells.
Signal transduction via the membrane ER has increasingly been found to be important for the cell biological effects of this steroid, including the survival and/or growth of breast cancer, bone, and neural cells (5, 7, 14, 24, 34, 49). This receptor has also been implicated in prevention of the inflammatory response to muscle ischemia-reperfusion injury (40), maintenance of the endothelial cell cytoskeleton, and upregulation of vascular cell migration and angiogenesis (33). E2 stimulation of transcription can also be signal dependent, as stimulation of the mitogen-activated protein (MAP) kinase ERK (extracellular regulated kinase) has been shown to be important for transactivation of the c-fos and prolactin genes (9, 45, 46). Transcription in response to E2 generation of cAMP has also been reported (4). The precise structural features of ER that facilitate the translocation of this steroid binding protein to the membrane are not known, but such information is important for understanding of the details of estrogen action at the cell surface.
The studies reported here result from our attempt to understand the localization and function of this protein at the PM. To begin this, we partially determined the amino acid structure of the mouse membrane ERα, isolated from CHO cells transfected to express this protein. We identified a serine residue at 522 that is necessary for the optimal localization and function of the sex steroid receptor at the cell surface. In contrast, mutation of this serine had no effect on nuclear ER number, affinity for E2, or E2-induced transactivation function. We also report that expression of the S522A mutant ERα resulted in a dominant-negative action only at the membrane, in cells expressing wild-type (wt) ERα. This mutant abolished several important effects of E2 in breast cancer and can be used as a reagent to deduce the cellular actions of E2 originating from membrane ERα.
MATERIALS AND METHODS
Isolation and partial sequencing of a membrane ER.
CHO-K1 cells were transiently transfected with a cDNA for the mouse ERα, as previously described (32). This resulted in the expression of both nuclear and membrane receptors. Twenty plates of ERα-transfected CHO cells were scraped and pelleted at 1,000 × g, and pellets were resuspended in 20 mM Tris with 1 mM EDTA, 1 mM dithiothreitol, and protease inhibitors. Cells were then centrifuged at 4°C and 8,000 × g for collection of nuclear receptors, and the supernatant was then ultracentrifuged at 4°C and 100,000 × g for 1 h. The pellet (membranes) was washed and ultracentrifuged again, and membranes were then further separated by sucrose gradient overlay; fractions 3 to 5 contained the buoyant membranes (with caveolae and rafts) that were pooled for all experiments (31). Briefly, membrane samples were first placed in a tube with an equal volume of a solution containing 85% (wt/vol) sucrose, 25 mM A-morpholine-ethanesulfonic acid, and 0.15 M NaCl and were then overlaid with 8.5 ml of 35% sucrose, topped up with 16% sucrose, and centrifuged at 35,000 rpm (105,000 × g) for 18 h at 4°C. Ten fractions (1 ml each) were obtained and either further processed or separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by membrane transfer for immunoblotting. The membrane receptors were solubilized in binding buffer (Pierce) containing 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS; Sigma). The purity of the membranes was confirmed by positive immunoblotting for 5′-nucleotidase and caveolin 1 (membrane proteins) and by the lack of detection of transportin and NTF-2 (nuclear proteins) or β-coatomer protein (endosomal/Golgi protein) (38). This was followed by affinity column purification. Briefly, protein G bound to an antibody against ERα (H222) (11) was cross-linked with disuccinimidyl suberate to make the column. The ERα-containing membrane or nuclear protein was eluted by using proprietary buffers and a proprietary protocol (Pierce). The eluted receptor proteins were dialyzed or concentrated and then analyzed by SDS-PAGE after being run on a 7.5% gel followed by staining. The gel protein bands corresponding to 67 kDa were cut out, trypsin was extracted from the gel, and the bands were then subjected to peptide degradation-mass spectrometry, as previously described (2, 12). This generated peptide sequences from the membrane and nuclear proteins, and these were compared to the known sequences of the classical mouse nuclear ERα.
Site directed mutagenesis and targeting of mouse ERα.
We carried out tryptic peptide matrix-assisted laser desorption ionization (MALDI)-mass spectrometry “sequencing,” as described above. At present this has yielded membrane peptide sequences that identically overlap with 20% of the known nuclear receptor and with 20% of our expressed nuclear receptor, supporting the idea that the two receptors are the same (G. Alton, M. Razandi, A. Pedram, and E. Levin, unpublished data). We identified an overlap sequence from amino acid 508 to amino acid 524 that includes a serine at 522. With surrounding residues, this was identified by computer analysis as a potential (although not a classic) palmitoylation site (HMSN). This sequence is present as amino acids 517 to 520 of the human receptor as well. We then mutated the serine at 522 to alanine in mouse pcDNA3-ERα by PCR using the forward primer 5′-CGGCACATGGCTAACAAAGG-3′. As specificity controls, we also mutated the identified serine residues 10 and 582 to alanine by using the forward primers 5′-CCCTTCACACCAAAGCCGCGGGAATGGCCTTGCTGC-3′ and 5′-GCTCCACTTCAGCACATGCCTTACAAACCTACTAC-3′, respectively. All mutations were confirmed by sequencing at the University of California—Irvine sequencing facility. We additionally subcloned the receptors into a green fluorescent protein (GFP) vector, pEGFPc2 (Clontech, Palo Alto, Calif.), and a multicopy histidine-expressing vector to monitor transfection efficiencies. wt and mutant receptor expression plasmids were then used in studies. To generate nuclear and membrane wt ERα constructs, pcDNA3-mouse ERα was used as a template. Twenty-five cycles of PCR (annealing temperature, 55°C) were performed by utilizing the forward primer 5′GCCGCTAGCACCATGACCATGACCCTTCAC3′ and the lower primer 5′GCCACCGGTCTGATCGTGTTGGGGAAGCCC3′. The PCR product was ligated into pCR2.1 by using the TA cloning kit (Invitrogen, Carlsbad, Calif.) and digested with AgeI and NheI. This fragment was subcloned into AgeI and NheI sites on the pECFP-Nuc and pECFP-mem vectors (Clontech), yielding ER constructs that were predominantly targeted to either the membrane or the nucleus (confirmed by binding and functional studies).
Receptor binding and cell localization studies.
wt and mutant ERα were expressed in CHO cells, and nuclear and membrane fractions were isolated as detailed above and were used for competitive binding assays or signal transduction studies, as previously described (31, 32). Binding studies were repeated at least three times, and the data were used for Scatchard analysis with the LIGAND computer program. Results were combined for statistical comparison by analysis of variance plus Schefe's test. Additional ERα mutants (HE11G, with the A/B domain deleted; HE19G, with the C domain deleted; and HEG0-537, with helix 12 and the F domain deleted) were provided by Paul Webb and expressed in CHO cells.
For cell localization of wt or S522A mutant ERα, we transiently expressed GFP-tagged fusion proteins for each receptor in CHO cells. CHO cells were grown and transfected on coverslips, and localization of the receptors was examined by laser-scanning confocal microscopy. We also colocalized the receptors at the membrane with endogenous caveolin 1 by using an antibody to this protein (Zymed Laboratories, South San Francisco, Calif.). Each section was processed for GFP-ERα (green), caveolin 1 (second antibody conjugated to Texas red), and colocalized caveolin 1 and ERα (yellow).
Signaling studies.
Adenylate cyclase activity in the membrane was determined by measuring cAMP generation, by methods described previously (32), in CHO-K1 cells expressing wt or S522A mutant ERα after the cells had been incubated for 5 min with 10 nM E2. IP generation and ERK (MAP kinase) activation in the CHO cells were also determined as described in detail elsewhere (32). Activation by E2 of an ERE-luciferase reporter in ER-transfected CHO or MCF-7 cells was assessed at 8 h of exposure to 10 nM E2, as previously published (32). Membranes were isolated by sucrose gradient centrifugation (31).
Myristylation, palmitoylation, and PI-PLC studies in CHO-K1 cells.
Cells were grown on 100-mm-diameter petri dishes in Dulbecco's modified Eagle medium (DMEM)-F12 medium without phenol red. Twenty-four hours after transfection with ERα, the cells were synchronized overnight and then labeled with [3H]palmitic acid (0.5 μCi/ml) or [3H]myristic acid (0.2 μCi/ml) for 2 h. The cells were incubated for 8 h in the presence or absence of 10 nM 17β-E2, washed with cold phosphate-buffered saline, and then lysed in buffer A (50 mM Tris-HCl [pH 7.5], 5 mM EDTA, 100 mM NaCl, 50 mM NaF, 100 μM phenylmethylsulfonyl fluoride, protease inhibitor cocktail, and 0.2% Triton X-100). Nuclear pellets were collected by low-speed centrifugation. Supernatants were centrifuged at 100,000 × g for 30 min to pellet cell membranes. Both pellets were washed twice, once with buffer A and once without detergent. Membranes were further purified by sucrose gradient centrifugation. Membrane and nuclear fractions were denatured in SDS loading buffer followed by gel electrophoresis, fluorography, and autoradiography. For phosphoinositol (PI)-PLC studies, the cells were incubated with 1 U of PI-PLC (Sigma)/ml for 1 h. Cells were washed and lysed, and the membrane and nuclear fractions were collected. Specific, total binding studies were then carried out on 50 μl of nuclear or membrane protein, incubated in DMEM-F12 medium (with no phenol red), bacitracin (1 mg/ml), and 0.5% bovine serum albumin, and with 3H-labeled E2 and unlabeled E2 (10−11 to 10−7 M).
Cyclin D1 protein expression, thymidine incorporation, and kinase activity.
MCF-7 cells were transfected with pcDNA3 (control) or ERα S522A, recovered, then synchronized by serum deprivation for 24 h, and then incubated in the presence or absence of 10 nM E2 for 8 h. In some cells, the soluble MEK inhibitor PD98059 (10 μM) was added to the incubation mixture 30 min prior to the steroid. The cells were then lysed, precleared, boiled, denatured in SDS reducing buffer, and electrophoretically resolved by PAGE. Western immunoblotting was then carried out using a polyclonal antibody (Santa Cruz). Nuclear thymidine incorporation was carried out in nontransfected or transfected MCF-7 cells after synchronization overnight in serum-free medium. All cells were then incubated for 20 h in the absence or presence of 10 nM 17β-E2. In some conditions, the MEK inhibitor PD 98059 (10 μM) was added prior to the steroid. After 20 h, 0.5 μCi of [3H]thymidine was added for 4 more h, as previously described (32). Cells were then washed and incubated for 10 min with 10% trichloroacetic acid at 4°C, followed by additional washes. Cells were lysed with 0.2 N NaOH overnight, and lysates were counted in a liquid scintillation β-counter. For cdk4 activity, MCF-7 cells transfected with pcDNA3 (control) or ERα S522A were incubated with E2 for 6 h and then lysed. The cell lysate was added to a protein A-Sepharose-conjugated cdk4 antibody (Santa Cruz) and then added to in vitro kinase activity tubes containing GST-pRB as a substrate, as previously described (28). This was followed by SDS-PAGE separation and autoradiography. Samples from each condition were assessed for protein loading equivalence, where cdk4 protein was assayed by immunoblotting. For ERK activity assays, transfected or nontransfected CHO, MCF-7, or ZR-75-1 cells were synchronized for 24 h in serum, phenol red, and growth factor-free medium. The cells were then exposed to E2 (10 nM) for 8 min with or without additional substances, and kinase activity was determined by using myelin basic protein (MBP) for the substrate, as previously described (32, 34). For p38β activity, the cells were incubated with E2 (10 nM) for 20 min and then lysed, and the lysate was immunoprecipitated with protein A-Sepharose conjugated to an antiserum for p38β. Immunoprecipitated kinases were then added to the protein ATF-1 for in vitro kinase assays as previously described (33). All experiments were repeated two to three times.
Protein and ER association studies.
Cytosolic fractions of CHO-wt ERα or CHO-ERα S522A were incubated with protein A-Sepharose for 1 h, and supernatants were transferred to fresh tubes containing protein A-Sepharose conjugated to caveolin 1 or ERα antibodies and were incubated for 4 h at 4°C. Immune complexes were washed, boiled, and then separated by SDS-PAGE. After transfer to nitrocellulose filters, the nonspecific proteins were blocked with blocking solution (Bio-Rad) and incubated first with a primary antibody to ERα or caveolin 1 for 2 h and then with a second antibody (Santa Cruz Biotechnology). Bound immunoglobulin G's (IgGs) were visualized using ECL reagents (Amersham) and autoradiography. Portions of the immunoprecipitated ERα or caveolin 1 were immunoblotted for evidence of equal protein loading and equal expression of total ER with the two constructs. In additional studies, MCF-7 cells were transfected to express a GFP-ERα S522A protein or GFP alone. After overnight recovery, the cells were lysed and immunoprecipitated with an antibody to GFP, followed by immunoblotting with antibodies to Src, Ras, and Raf proteins (Santa Cruz). In CHO cells, His-wt ERα or GFP-ERα S522A was singly or doubly expressed. To detect homo- or heterodimerization in these cells, the lysate was immunoprecipitated with an antibody to His, followed by blotting with an antibody to GFP, or in reverse order. All studies were repeated at least three times.
RESULTS
Comparison of wt and S522A mutant ERα binding after expression in CHO cells.
We first isolated the mouse ERα in the PM after expression in CHO cells and partially sequenced the protein by peptide degradation-MALDI mass spectroscopy (Razandi et al., unpublished). We identified a peptide (LAQLLLILSHIRHMSNK) that corresponds to a portion of the C terminus in the known mouse ERα sequence (2), beginning with amino acid 508. Furthermore, HMS (boldfaced in peptide sequence) was noted by computer modeling as a possible, but not classic, palmitoylation site (35). We therefore asked whether the ER was palmitoylated at this site (see below), and we also mutated the critical serine at amino acid position 522 to alanine within the mouse ERα cDNA. Additional S-to-A mutations at residues 10 and 582 were created by site-directed mutagenesis, for comparison to mouse ERα S522A (48) and to support the specificity of any findings.
We then expressed the wt and S522A mutant ERα constructs in CHO cells and carried out competitive binding studies in both nuclear and membrane compartments. By Scatchard analysis of the binding data (Fig. 1A), we found that the receptor affinity for E2 (Kd) and the receptor number (Bmax) were very similar for the two ERα receptors in the nucleus (Table 1). Similar transfection efficiencies were demonstrated using GFP fusion constructs (data not shown). We also determined whether the function of the mutant nuclear ERα differed from that of the wt. We therefore cotransfected CHO cells with either wt or S522A mutant ERα and an ERE-luciferase reporter (32) and determined the response to E2. We found that the two receptors were comparably capable of responding to E2 with an upregulation of reporter activity (Fig. 1B). These data indicate that the replacement of S with A at residue 522 does not affect the quantity of nuclear receptor localization, its binding affinity for E2, or the transcriptional response to the steroid.
FIG. 1.







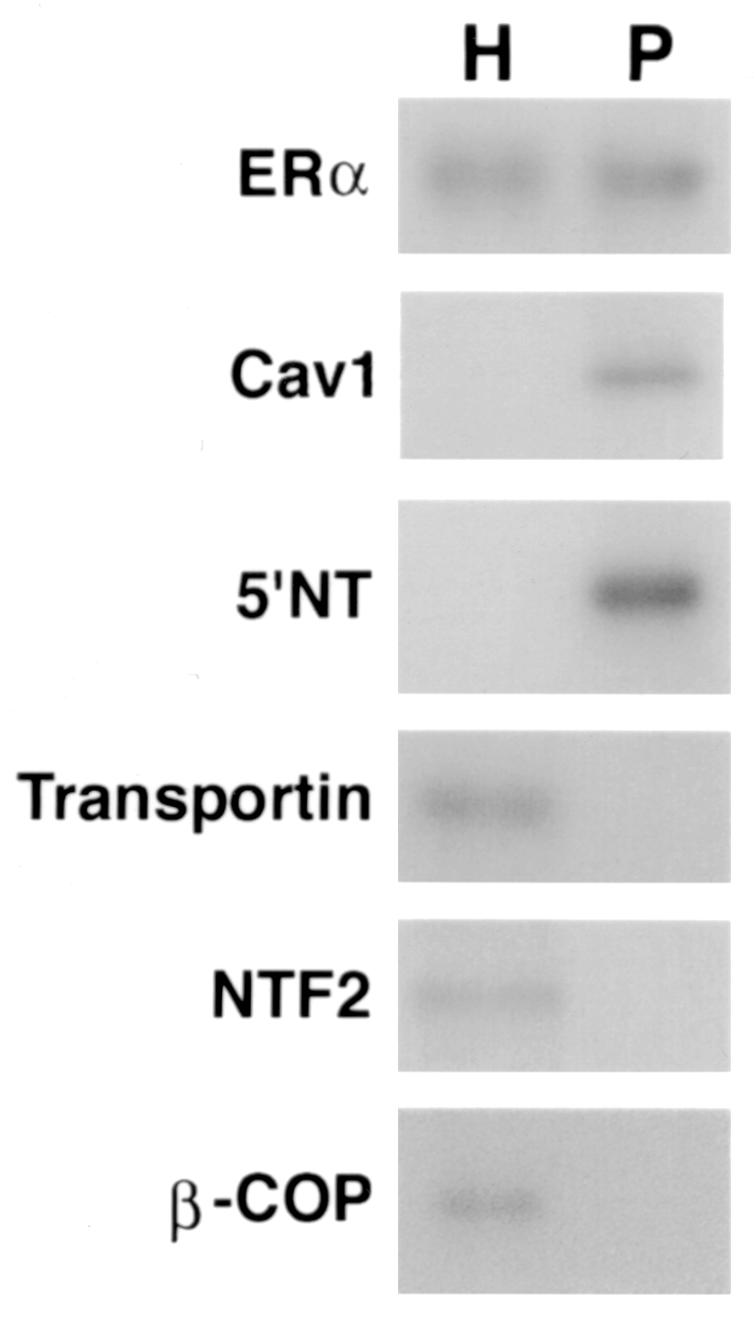

(A) Competition binding of 17β-[3H]E2 to nuclear wt (left) or S522A mutant (right) ERα transfected into CHO-K1 cells. (Inset) Binding to cell membrane ERα. Data are transformed for Scatchard analysis by using the LIGAND program. Data shown here are from a representative study; results from three separate experiments were combined to create Table 1. (B) Transactivation of an ERE-luciferase reporter construct coexpressed in CHO cells with mouse wt ERα or the S522A mutant. Data were determined at 6 h after incubation with either 1 nM E2, 10 nM E2, or no steroid. *, P < 0.05 for the wt or the S522A mutant alone versus the same construct plus E2 (for data combined from three experiments). (C) Serine/threonine residues in the full-length receptor, but not serine 522 in the E domain of ERα, are phosphorylated. Western blotting utilized a specific antibody to serine/threonine residues (Sigma) from lysates of CHO cells transfected to express either the full-length receptor or the E domain targeted to the PM or the nucleus. (D) Membrane localization of GFP-tagged wt ERα or S522A mutant ERα expressed in CHO cells. Arrows indicate a greater membrane localization for wt ERα. Dense nuclear populations for both receptors are seen. (E) Colocalization of wt ERα with caveolin 1 at the membrane, but markedly less colocalization of ERα S522A. Results of a representative study are shown. Arrows indicate differential ER expression (green) at the membrane (panels A and D) and equal caveolin 1 expression (red) (panels B and E). The strong colocalization of wt ERα and caveolin 1 (yellow) (arrow, panel C) is not seen for ERα S522A (arrow, panel F). (F) Total specific binding of labeled E2 to membranes (left) or nuclei (center) in CHO cells expressing either S10A, S582A, or S522A mutant ERα or wt ERα. Data are combined from three experiments. *, P < 0.05 for ERα S522A versus wt ERα or other S-to-A mutant receptors. (Right) Protein blot demonstrating the purity of the membrane preparation. Caveolin 1 (Cav1) and 5′ nucleotidase (5′NT) are integral membrane proteins, while transportin and NTF-2 are nuclear proteins. β-COP is a Golgi protein.
TABLE 1.
Binding characteristics of wt or S522A mutant mouse ERα expressed in the nuclei and membranes of CHO-K1 cells
| Localization | Kd (nM)a | Bmax (pmol/mg of protein)a |
|---|---|---|
| Nuclear | ||
| wt ERα | 0.26 ± 0.02 | 422 ± 38* |
| Mutant ERα | 0.25 ± 0.02 | 395 ± 26* |
| Plasma membrane | ||
| wt ERα | 0.28 ± 0.04 | 17.2 ± 3.8† |
| Mutant ERα | 0.27 ± 0.03 | 6.6 ± 0.26 |
Data are from Scatchard analysis of competitive binding studies and are means ± standard errors of the means for three separate experiments combined.
P < 0.05 for Bmax of corresponding nuclear versus membrane ERα.
P < 0.05 for Bmax of wt ERα versus S522A mutant ERα.
In contrast, binding experiments revealed that expression of the S522A mutant ERα resulted in 62% fewer receptors (Bmax) in the PM than expression of wt ERα (Fig. 1A insets; Table 1). This was found in three separate binding experiments, where the reduction ranged from 57 to 65%. The binding affinities (Kd) for E2 at the membrane were comparable for wt and mutant receptors. Thus, serine 522 is an important determinant for full membrane localization of ERα.
It is possible that serine 522 is a phosphorylation site, although this would not be a common mechanism for membrane localization. By mass spectroscopy, there was no evidence of phosphorylation on this residue. We also expressed the full-length wt ER, the S522A mutant, or the E domain (ligand binding domain) of wt ERα in CHO cells, targeting the E domain to both nuclear and membrane locations. As seen in Fig. 1C, the entire receptor is phosphorylated at serine/threonine residues, but we find no evidence that the intact E domain is similarly phosphorylated, either when targeted to the membrane or when targeted to the nucleus.
To visualize the receptor at the membrane, we expressed GFP-tagged wt or S522A mutant ERα in CHO cells and detected membrane localization by confocal microscopy. As seen in Fig. 1D, wt receptor expression clearly reveals a population of membrane-localized sex steroid binding proteins, while the mutant receptor does not. Both show a dense nuclear population. We also examined colocalization of the wt or mutant ERα at the membrane with caveolin 1. In Fig. 1E, wt ERα was clearly seen at the membrane (arrow, panel A), in contrast to sparse membrane expression of ERα S522A (panel D). Caveolin 1 was clearly visualized at the membrane (Fig. 1E, panels B and E). Colocalization of membrane wt ERα with caveolin 1 (Fig. 1E, panel C, arrow) was also seen for the S522A receptor (panel F), but the latter showed decreased amounts colocalized, reflecting a decreased number of receptors at the membrane.
We next compared the binding of wt or S522A mouse ERα to that of S10A and S582A ERα constructs expressed in CHO cells (Fig. 1F). Total specific binding of E2 was determined in the nucleus and PM and revealed that both of the two additional mutant receptors were very similar to the wt receptor in both compartments. By comparison, S522A expression again exhibited significantly lower binding at the membrane only. These data indicate the specificity of S522 for ER localization at the cell surface.
Dissection of the contribution of other domains of ERα to membrane localization and function.
It is possible that elements contained within other domains of ERα contribute importantly to membrane localization. In this respect, Schlegel et al. (37) have recently shown that residues 1 to 282 (the A/B and C domains) of human ERα bind to caveolin 1, a largely membrane localized protein that facilitates membrane localization of ER (31) and that, when overexpressed, promotes nuclear ER localization and transcriptional action. We therefore asked whether mutant ERα that lacked either the A/B or the C domain was capable of localizing to the PM and signaling to ERK. We compared the effects of CHO cells expressing these deletion or truncation mutants to those of CHO cells expressing wt ERα. We found that HE11G (with the A/B domain deleted) and H19G (with the C domain deleted) were comparable to wt ERα, both in specific binding of E2 at the cell membrane and in ERK activation by the steroid (Fig. 2). In contrast, a mutant with helix 12 and the F domain deleted, HEG0-537 (truncated at residue 537), specifically bound little E2 in either the nuclear or the membrane compartment and did not support E2 activation of ERK. Thus, the A/B and C domains do not contribute to membrane ER localization and signaling by E2. However, deleting a small but important region within the E domain (in conjunction with loss of the F domain) has a profound effect on E2 binding to any ER pool, as well as on membrane function. Further understanding of the specific residues within the E domain that are required for ER localization at the membrane will require the characterization of a very extensive series of conservative mutations within this region. These results support a focus on the E domain for further understanding of the compartmentalization of ER.
FIG. 2.

(A) Binding of E2 to the nuclei and membranes of CHO cells expressing either HE11G (A/B domain deleted), HE19G (C domain deleted), or HEG0-537 (helix 12 and F domain deleted) ERα or wt ERα. The study was repeated. (B) ERK activation in response to E2 in CHO cells expressing either wt ERα or a deletion mutant. Activity was determined after 8 min of incubation with 10 nM E2. MBP was used as a substrate for ERK activity. Total ERK protein is shown on the immunoblot beneath the activity results.
ERα S522A is less capable than wt ERα of signaling from the membrane.
It was important to ascertain whether the loss of membrane receptors resulting from expression of the S522A mutant also affected signal transduction. We therefore compared ERK (MAP kinase) activation by E2 in CHO cells expressing wt or mutant ERα. E2 significantly stimulated ERK activity after 8 min of exposure to CHO cells expressing wt ERα (Fig. 3A, left). However, ERK activation in response to E2 was reduced by 68% in CHO cells expressing ERα S522A (relative to activation in cells expressing wt ERα) (Fig. 3A, left; compare lanes 2 and 4). Activation of ERK by E2 was further compared in CHO cells that expressed wt or S10A or S582A mutant receptors. Consistent with the binding data, the additional serine mutants were nearly identical to the wt in activation of this MAP kinase (Fig. 3A, right).
FIG. 3.





(A) (Left) ERK activity is stimulated by E2 in CHO cells expressing wt ERα but less so in CHO cells expressing S522A mutant ERα. *, P < 0.05 for E2 versus the control (mouse ERα [mERα] without E2) in three combined experiments; +, P < 0.05 for ERK response to E2 in CHO cells expressing wt ERα (lane 2) versus ERα S522A (lane 4) in three combined experiments. (Right) Comparable stimulatory effects by E2 on ERK activity in CHO cells expressing wt ERα or the S10A or S582A mutant (lanes 3 to 8). Lanes 1 and 2 show that the intrinsic ERK activity of CHO cells expressing the empty vector, pcDNA3, cannot be stimulated by E2, due to a lack of endogenous ER. *, P < 0.05 for control versus E2. (B) Generation of cAMP in response to E2 in CHO cells expressing wt or S522A mutant ER. (C) IP3 generation in response to E2 in the above cells. Bar graph data are means ± standard errors of the means from triplicate determinations per experiment and are from two (cAMP) or three (IP3) combined studies. (D) Targeting ER to the membrane but not the nucleus allows E2 to rapidly activate ERK. CHO cells were transfected to express either nontargeted wt mERα or wt mERα targeted to the nucleus (mERα-ECFP-nucl.) or the membrane (mERα-ECFP-memb.). Cont., control. The study was repeated.
We then examined cAMP generation, reflecting adenylate cyclase activation in the membrane, and found that E2 was 57% less capable of generating this cyclic nucleotide in S522A mutant-expressing than in wt ERα-expressing CHO cells (Fig. 3B). Generation of cAMP often arises from Gαs stimulation, which was previously demonstrated in response to membrane ER activation by E2 (32). Finally, we measured IP generation (Fig. 3C) and found a significant (53%) difference in production between cells expressing the two types of ERα. IP generation commonly results from the activation of Gαq, which was previously shown to be stimulated by E2 activation of membrane ER expressed in CHO cells (32). These data indicate that the reduction in membrane ER levels seen with S522A protein expression has significant functional consequences, and they further support the idea that E2 activates signal transduction through the membrane (and not the nuclear) receptor. To further support the latter concept, we subcloned the full-length wt ERα into vectors that contain membrane or nuclear localization signals and also express a GFP fusion protein (ECFP; Clontech). We then expressed in CHO cells either nontargeted wt ERα or wt ERα targeted either to the membrane or to the nucleus. As seen in Fig. 3D, expression of the nontargeted wt ERα and especially the membrane-targeted receptor supported rapid ERK activation by E2. In contrast, there was no activation of ERK in CHO cells expressing nucleus-targeted ER. Combined with previous experiments targeting the E domain to the membrane or nucleus (31), these data show that it is the membrane ER that supports rapid kinase activation in response to E2.
Palmitoylation, myristylation, and glucosylphosphoinositol (GPI) anchor studies addressing ER localization at the membrane.
A number of posttranslational (or cotranslational) processes have been found to facilitate the movement and anchoring of proteins in the PM. To determine whether any of these alterations helped explain how ER localized to the membrane, we examined lipid modifications of ER. We expressed the wt mouse ERα in CHO cells and then labeled the cells with [3H]palmitate or myristic acid. As expected, there was no uptake or incorporation of either lipid into the nuclear ERα. Furthermore, we found that there was no incorporation of myristate into the membrane ERα, consistent with the lack of a consensus myristylation site determined either from our partial sequence of membrane ERα or from viewing the known full-length sequence (48). Similarly, a possible but nonclassic palmitoylation site was identified from a peptide corresponding to a region in the mouse ERα C terminus, encompassing serine 522. However, in the membrane, there was no specific incorporation of palmitate into ERα in either the presence or the absence of E2 (data not shown). Other modifications, such as farnesylation or geranylgeranylation, usually require concomitant palmitoylation and occur at the very end (usually the N terminus) of the protein. No such sites were identified. Thus, we conclude that ERα is probably not posttranslationally lipidated to effect membrane translocation.
Addition of GPI to a protein in the Golgi complex serves to anchor such modified proteins in the extracellular leaflet of the PM (21). The PI-PLC enzyme cleaves GPI-modified proteins and therefore releases these membrane proteins into the culture medium, so that they cannot be detected by binding ligand at the cell surface. We found that treatment of the ER-expressing CHO cells with this phospholipase did not change the binding of E2 to ER in the PM. Furthermore, the anchoring of ER in the outer leaflet of the PM would generally preclude its localization in the caveolae, a membrane domain where ER has now been detected (6, 13). Thus, it is unlikely that ER undergoes this posttranslational (cotranslational) modification.
Interactions of wt or S522A mutant ERα with caveolin 1.
It was recently reported that endogenous ER physically associates with the caveolin 1 protein in both the PM and cytosol of endothelial, vascular smooth muscle, and MCF-7 cells (31). Furthermore, expression of full-length caveolin 1 in MCF-7 or Caco-2 cells facilitates the movement of ER from the cytosol to the PM. Thus, ER-caveolin binding is important for the ability of ER to localize to the PM. We therefore examined whether the S522A mutant receptor bound to caveolin 1 comparably to wt ERα. This was accomplished in CHO cells, where we expressed the two ER constructs and utilized the endogenous caveolin in these cells. The ER was immunoprecipitated from the CHO cells, followed by immunoblotting for caveolin 1 (and vice versa), and association was examined in the absence of E2. In the cytosol of CHO cells, expression of either wt or S522A mutant ERα resulted in the receptor complexing with endogenous caveolin 1. However, the association of ERα S522A with caveolin 1 was 60% lower than that of wt ERα (Fig. 4). Importantly, as shown, the total ER levels expressed from the two vectors were comparable. These data are compatible with the idea that S522 is important for binding to caveolin 1, a protein that facilitates the membrane localization of the steroid binding protein (31). Thus, we have identified a mechanism to explain why ERα S522A is poorly localized to the PM.
FIG. 4.

ERα and caveolin 1 (CAV-1) association in the cytoplasm. CHO-K1 cell cultures (100-mm-diameter dishes) were transfected with 10 μg of wt or S522A mutant ERα plasmid DNA. The cells were lysed, and immunoprecipitation for caveolin 1 was carried out, followed by immunoblotting for ERα (left panels); or the order was reversed (right panels). Results shown are representative of three experiments. Caveolin 1 and ERα immunoblots are shown (lower panels) to demonstrate equal gel protein loading and equal expression of the two ER. mERα, mouse ERα.
Expression of S522A inhibits endogenous membrane ER function.
We then asked whether the expression of ERα S522A interferes with the function of endogenous E2-ER signaling from the membrane. To test this hypothesis, we transiently expressed ERα S522A in MCF-7 and ZR-75-1 breast cancer cells. It was previously shown that E2 induced rapid signaling from membrane ER in these cells (33). In MCF-7 cells transfected to express pcDNA3 (control), E2 caused a twofold activation of ERK activity via the endogenous membrane ER (Fig. 5A, left; compare lane 1 with lane 2). In contrast, ERα S522A-expressing cells responded to E2 with 61% less activation of ERK (Fig. 5A, left; compare lanes 1 and 2 with lanes 3 and 4). Comparably, ERα S522A expression resulted in a 70% decrease in ERK activation in E2-treated ZR-75-1 cells (Fig. 5A, right; compare lanes 1 and 2 with lanes 3 and 4). We also determined that activation of ERK by epidermal growth factor (EGF) or IGF-1 in MCF-7 cells (Fig. 5B, first three columns) was not significantly affected by expression of the mutant ER (last three columns). This demonstrates the specific action of ERα S522A to impair only E2-ER signaling, and it also indicates that signaling by the two growth factors does not require an intact membrane ER signaling system. To further establish the specificity of these results, we expressed the S10A mutant in MCF-7 cells. There was no difference in ERK activation in response to E2 between cells expressing only endogenous ER (pcDNA3 transfected) and the same cells additionally transfected with ERα S10A (data not shown).
FIG. 5.




(A) Expression of ERα S522A inhibits E2 activation of ERK. MCF-7 (left) or ZR-75-1 (right) breast cancer cells (which express endogenous ER) were transfected transiently to express either pcDNA3 (control) or S522A mutant ERα. The cells were then exposed to 10 nM E2 for 8 min, after which they were lysed, and immunoprecipitated ERK was assayed for activity by using MBP as a substrate. Precipitated ERK protein is shown in the lower gels, and the bar graphs each reflect three experiments combined. *, P < 0.05 for pcDNA3 in the absence versus the presence of E2; +, P < 0.05 for comparison of E2 treatments of pcDNA3-expressing versus ERα S522A-expressing cells. (B) ERα S522A does not impair EGF or IGF-1 activation of ERK. Data from three experiments are combined. *, P < 0.05 for pcDNA3-transfected or ERα S522A-expressing MCF-7 cells in the absence versus the presence of EGF or IGF-1. (C) E2 comparably activates an ERE-luciferase reporter in untransfected MCF-7 cells and MCF-7 cells transfected to express ERα S522A. Bar graph shows results for three experiments combined. *, P < 0.05 for pcDNA3- or ERα S522A-transfected MCF-7 cells without versus with E2.
The inability of ERα S522A to fully localize to the membrane contrasted with the normal amount and function of nuclear ER when this construct was expressed in CHO cells. We therefore determined the specificity of S522A to serve as a dominant-negative protein in MCF-7 cells for membrane but not nuclear ER. MCF-7 cells were transiently transfected with an ERE-luciferase reporter in the presence or absence of coexpression of ERα S522A or pcDNA3. In pcDNA3-expressing cells, E2 caused a dose-related, 2.5-fold maximal stimulation of luciferase function (Fig. 5C). When the S522A mutant was expressed in these cells, transactivation of this reporter by E2 was comparable. This indicates that expression of the mutant ERα did not affect endogenous nuclear ER function.
To understand the cell biological effects of S522A expression and the role of the endogenous membrane ERα, we examined the ability of estrogen to promote cyclin D1 protein expression and cdk4 activation in MCF-7 cells. It has previously been shown that, in response to growth factors, signaling through ERK to Ets protein phosphorylation transactivates the cyclin D1 promoter and stimulates cyclin D1 protein synthesis (1, 15). E2 has been shown to stimulate cyclin D1 transcription and protein synthesis (47). We found that E2 was capable of increasing cyclin D1 protein levels nearly threefold (Fig. 6A). This was significantly related to ERK activation, since the MEK inhibitor PD98059 substantially prevented this effect. It was previously shown that PD98059 completely blocked E2 activation of ERK in MCF-7 cells (34). Importantly, expression of ERα S522A inhibited the E2-induced increase in cyclin D1 protein levels by 68%. Thus, the ability of ERα S522A to inhibit ERK activation arising from the endogenous membrane ER greatly contributed to the inhibition of the increase in cyclin D1 protein levels.
FIG. 6.

(A) Cyclin D1 expression is increased in response to E2 and is dependent on ERK activation in wt MCF-7 cells. MCF-7 cells transfected to express ERα S522A show a lower response to 100 nM E2. Data are representative of three experiments, which were combined for the bar graph. *, P < 0.05 for pcDNA3-expressing cells without versus with E2; +, P < 0.05 for cells incubated with pcDNA3 plus E2 versus the same condition plus 10 μM PD 98059, or versus cells cotransfected with ERα S522A. (B) cdk4 activity is significantly downregulated by ERα S522A in MCF-7 cells. Cells were transfected with pcDNA3 or the mutant ER and exposed to 10 nM E2 for 6 h. cdk4 kinase was immunoprecipitated, and an in vitro assay of activity was accomplished by using Rb protein as a substrate. Bar graph data are from three experiments combined. (C) E2-stimulation of p38β activity in endothelial cells is inhibited by ERα S522A. Transfected endothelial cells were incubated with E2 for 20 min, and p38β activity was determined against the substrate protein ATF-1. Data are from three experiments.
We then determined the effect of E2 signaling on cdk4 activity. We found that E2 stimulated the important phosphorylation of the retinoblastoma (Rb) protein by this kinase 2.5-fold (Fig. 6B). Inactivation of Rb results from its phosphorylation mainly by cyclin D1-cdk4 and possibly by cyclin E-cdk2 and allows G1/S progression in many cell types (39). Expression of S522A resulted in a 70% decrease in the ability of E2 to activate cdk4 activity. The results indicate that signaling from the membrane ER is important for a G1 event that is essential to breast cancer cell cycle progression.
We also assessed G1/S progression, determined by thymidine incorporation into DNA. It has previously been shown in MCF-7 or ER-expressing CHO cells that E2 stimulation of DNA synthesis is partly regulated through the ERK signaling pathway (5, 32). We found here that E2 caused a 70% increase in thymidine incorporation into DNA, a marker of the S phase. Two-thirds of this increase was blocked by PD98059. Thus, E2 utilizes several mechanisms to stimulate G1/S progression, but ERK activation is the most important. Upon expression of ERα S522A, E2-induced thymidine incorporation was significantly reduced, by 50%. Thus, expression of the mutant ERα affirms the participation of the membrane steroid receptor in this event.
It would be important to know if the dominant-negative effect of the S522A mutant ERα extended to other cells. It was previously shown that E2 activates p38β MAP kinase in endothelial cells through endogenous membrane ER and that this leads to the angiogenic and cell survival effects of E2 in these cells (33). Here, we report that E2 activation of p38β in endothelial cells is 60% reduced when ERα S522A is expressed (Fig. 6C). Thus, this mutant ER may be a useful tool for determining the contributions of the membrane ERα to various cell-signaling and biological functions.
Mechanisms of ERα S522A inhibition of endogenous ER function.
How might ERα S522A inhibit endogenous ER function? One possibility to explain the dominant-negative effect of ERα S522A is that it might heterodimerize with wt ER. Dimerization is necessary for ER to transactivate genes (43), and heterodimerization between ERα and ERβ has been reported to inhibit ERα function (29). Since the S522A mutant does not translocate effectively to the PM, it could potentially bind and sequester the endogenous receptor, thus interfering with its signaling function. To assess possible heterodimerization, we expressed GFP-tagged ERα S522A and His-tagged wt ERα in CHO cells and performed pulldown studies. After lysis, the cell extracts were immunoprecipitated and blotted with anti-GFP and anti-His antibodies, in both orders. After E2 treatment of cells where either or both tagged forms of the receptor were expressed, we found evidence for homodimerization and heterodimerization of wt and mutant ERα (Fig. 7A). As specificity controls, the fourth lanes show a lack of ER when His-tagged wt ERα is expressed and immunoprecipitated but blotting is done with an antibody to GFP (Fig. 7A, left), and when GFP-tagged ERα S522A is expressed but blotting is done with an antibody to His (Fig. 7A, right). Furthermore, S522A mutant ERα was as capable as wt ERα of dimerizing to wt ERα (Fig. 7A, left). These data indicate that ERα S522A can bind to wt ERα, thereby potentially sequestering or otherwise limiting endogenous receptor signaling from the membrane.
FIG. 7.





(A) Homo- and heterodimerization of wt and ERα S522A after expression in CHO cells. Doubly or singly transfected cells were first exposed to 10 nM E2 for 10 min and then lysed, and the lysate underwent immunoprecipitation (IP) with an antibody to His or GFP, as indicated, eventually followed by blotting with an antibody to GFP (left) or to His (right). Proteins were separated by SDS-PAGE under nonreducing conditions (native gel). Molecular weight markers indicate the different sizes of the GFP-tagged wt ERα and His-tagged ERα S522A homodimers and the intermediate size of the heterodimer. (B) Expression of S522A prevents wt ERα localization at the membrane. CHO cells were transfected with equal amounts of plasmids encoding GFP-tagged wt ERα plus His-tagged wt ERα (10 μg of total DNA/100-mm-diameter dish) or with GFP-tagged wt ERα plus His-tagged ERα S522A. Localization of receptors was determined by confocal microscopy. (C) wt and S522A mutant ERα equally associate with Ras, Raf, or Src at the membrane. (Left) CHO cells were transfected to express either receptor, and after normalization, equal receptor protein aliquots were confirmed by Western blotting. Equal protein aliquots were used for immunoblots to determine wt or mutant ERα association with Ras or Raf. The study was carried out in the presence of 10 nM E2 for 10 min and was repeated. Immunoprecipitation with no antibody (ab) or an irrelevant antibody to the endothelin-1 peptide did not yield a protein band. (Right) MCF-7 cells were transfected with His-tagged ERα S522A (lanes 2 and 3) or with His-tagged pcDNA3 (lane 1) and were incubated or not with 10 nM E2 for 10 min. The cells were lysed and immunoprecipitated for ERα (lane 1) or for His (lanes 2 and 3), then immunoblotted for Src. Expression of His-tagged pcDNA3 did not coprecipitate Src (data not shown).
To further examine this mechanism, we determined the membrane localization of wt ERα in CHO cells transfected to express equal amounts of either (i) GFP-tagged wt ERα plus His-tagged wt ERα or (ii) GFP-tagged wt ERα plus His-tagged ERα S522A. As seen in Fig. 7B, expression of ERα S522A substantially decreased the membrane localization of GFP-tagged wt ERα. By contrast, nuclear receptor expression was not different. Thus, ERα S522A inefficiently translocates to the cell surface and also prevents wt ERα PM localization after heterodimerization. These results provide a mechanism for the dominant-negative action of the mutant ERα, but other effects are tenable.
Membrane wt ERα and S522A mutant ERα bind equally to signaling molecules.
It is also important to consider that mutation of serine 522 to alanine might disturb the inherent ability of membrane ER to associate with important signaling molecules. This could contribute to the differential signaling from the membrane by wt versus S522A mutant ERα. To investigate this, we transfected CHO cells to express either wt or S522A mutant ERα. We immunoprecipitated the ER from membrane preparations and then normalized the proteins for equivalent amounts of receptor(s), as indicated by Western blotting (Fig. 7C). We then took separate (normalized) aliquots of immunoprecipitated wt ERα or ERα S522A and immunoblotted the aliquots for Ras or Raf. We found that the two receptors associated equally with the Ras or Raf signaling molecules in the presence of the steroid (Fig. 7C, left). Control immunoprecipitations without an antibody or with an irrelevant antibody to endothelin-1 (ET-1) did not bring down a Ras or Raf protein band. Especially important is the association of ER with Src, which has been reported to bind to tyrosine 537 of ERα (24). We examined the effect of expression of ERα S522A on subsequent Src association with ER in MCF-7 cells. As seen in Fig. 7C (right), endogenous ERα associated with Src comparably to the S522A mutant receptor, and this was unaffected by E2. This suggests that there is no alteration of association with important signaling molecules by ERα S522A that can account for the differences in signaling from the membrane. Thus, we believe that it is primarily the membrane receptor number that determines the differences in signaling.
DISCUSSION
The presence of a PM ER in human cells that enacts signal transduction and thereby contributes to the cellular effects of E2 is increasingly accepted (14, 33, 34, 40, 49). E2 signaling through PI 3-kinase in vivo rescues muscle from ischemia-reperfusion injury (40), while activation of ERK prevents breast cancer (34) or osteoblast (14) cell death. Recently, Marquez and Pietras have shown that administration of antibodies to ERα in nude mice blocked the growth of human breast cancer xenografts (23). This probably resulted from inhibition by the antibodies of membrane ER signaling to ERK and PI 3-kinase. Spatially, the receptor appears to be localized primarily but not exclusively to caveolar fractions of the PM (6, 13). In this confined area, ER potentially interact with a variety of signaling molecules that must localize to the PM for activation. The PM ER acts as a G protein-coupled receptor, directly (31, 50) or indirectly (10), leading to activation of multiple signaling pathways. This results in cAMP generation (4), PLC and inositol triphosphate (IP3) activation (16, 41), and the stimulation of cascades leading to enhanced activity of ERK, JNK, and p38 MAP kinases (23, 32, 33). The importance of these nongenomic mechanisms of estrogen action is analogous to that of the actions of steroids in plants. In Arabidopsis spp., brassinosteroids mediate plant cell developmental growth and fertility (22), and cell action results from steroid binding to a transmembrane, tyrosine kinase receptor protein (44). Thus, steroid action at the cell surface is an ancient function conserved from plants to humans, further indicating its importance.
One important issue with regard to the cell surface ER that we addressed here is the structural requirements for a population of ER to translocate to the PM. We found that ER are not posttranslationally lipidated, as occurs with other PM-localized proteins. Rather, we identified serine 522 as important for membrane translocation. Compared to expression of wt mouse ERα, mutation of this serine to alanine resulted in 62% fewer receptors expressed at the membrane, with little influence on receptor affinity for ligand. However, there was no appreciable effect on the nuclear receptor numbers, affinity, and function (transactivation of an ERE-luciferase reporter). Furthermore, expression of the S522A mutant receptor was markedly less efficient in supporting E2-induced ERK activation, cAMP generation, and stimulation of IP3 than wt receptor expression. Presumably, reduced signaling resulted from a decreased number of receptors available at the membrane. Supporting this, we did not find a loss of association at the membrane between S522A mutant ERα and signaling molecules, compared to that for wt ERα. However, this receptor can serve as a dominant-negative protein for wt ER when expressed in MCF-7 cells, and therefore additional mechanisms of abolishing signal transduction may be relevant (see below). Supporting the specificity of our results, we found that substitution of alanine for serine at residues 10 and 582 of the mouse ERα had no effect on either E2 binding to the membrane or signaling by E2, when these mutants were compared to wt ERα.
How does ERα localize to the membrane, and how does S522 contribute? It was recently determined that caveolin 1 protein facilitates the translocation of ER to the PM and that the two endogenous proteins physically bind in both the cytosol and the PM (31). The scaffolding domain of caveolin 1 (amino acids 82 to 101) is essential for this protein to move from the cytosol to the membrane (3, 27), and we determined that the scaffolding domain facilitates ER movement to the PM. An important question, then, is whether serine 522 is necessary for the association of ERα and caveolin 1. We report here that in the cytoplasm, the physical association between these two proteins was 60% decreased by the mutation of serine 522. In contrast, association of caveolin with S10- or S582-mutated ERα was comparable to that with wt ER (data not shown). It has recently been shown that residues 1 to 282 of ERα bind to caveolin 1 (37). However, Lu et al. recently showed that caveolin 1 associates with the androgen receptor through both N-terminal (A/B domain) and E domain elements (20). We found that an A/B domain deletion mutant ERα localizes to the membrane and supports E2 signaling to ERK equivalently to wt ERα. Thus, the interaction between caveolin and the N terminus of ER may not be functionally important for the membrane ER.
What supports the relevance of S522A for ER action at the membrane? Kousteni et al. showed that by targeting only the E domain of ERα to the PM but not to the nucleus, E2 could rescue cells from apoptotic death (14). It was recently demonstrated that the E domain (and here the full-length ER) is sufficient to convey robust ERK activation in response to E2 when targeted to the PM (31). These overall findings suggest that the E domain is generally important for ERα actions originating at the membrane. Identification of serine 522 provides a novel insight into the specific structural requirements for membrane localization, steroid action, and cell biological functions of E2. We suggest that similar examination of the role of the ligand binding domains of the progesterone, androgen, and other steroid receptors is warranted.
To establish the roles of the membrane ER in cell biology, several approaches could be taken. Targeting of ER to only one compartment in the cell may suggest a specific function for a pool of the endogenous receptor. Another approach is to devise specific agonists or antagonists for the membrane ER, reagents that do not enter the cell to bind the nuclear receptor. Several ER agonists have recently been described that dissociate some membrane signaling from transcriptional activity (14). However, ER signaling through the membrane receptor stimulates gene transcription (9, 46), and thus, these two functions may not always reflect membrane versus nuclear receptor action. A third approach is to express mutant sex steroid receptors that specifically interfere with endogenous ER actions at the membrane. We show here in MCF-7, ZR-75-1, and endothelial cells that ERα S522A is capable of significantly preventing E2 signaling from the endogenous membrane receptor. We propose that this could result from preventing endogenous ER localization at the membrane. Since the dimerization motifs for ERα do not involve serine 522, we reasoned that wt and mutant ER could heterodimerize and thus sequester wt ER from localizing fully at the PM. Supporting this, we provide evidence of heterodimerization between the mutant and wt ERα and a loss of membrane wt ERα when both receptors are coexpressed.
In MCF-7 or ZR-75-1 breast cancer cells, expression of ERα S522A interfered with endogenous ER function. Expression of ERα S522A inhibited E2-induced ERK activation, cyclin D1 production, cdk4 activity, and G1/S progression. Many of these actions of E2 require signaling from the membrane to kinases such as ERK. Furthermore, the utility of this approach was shown in a second cell type, where membrane E2-ER signaling to p38β MAP kinase (33) was significantly prevented by expression of ERα S522A. The strong inhibition of cyclin D1 protein in MCF-7 cells by ERα S522A expression and the linkage to modulation of ERK activity suggests an important therapeutic intervention in breast cancer. In vitro, E2 induction of cyclin D1 overcomes the tamoxifen-induced G1/S cell cycle block (47). Also, tamoxifen sensitivity can be restored through p27 function, resulting from ERK downregulation (8). In this respect, limiting endogenous membrane ER signaling to ERK (19) and cyclin D1 may be therapeutically desirable, as suggested by our use of the S522A mutant ERα. It has also been recently reported that specifically cyclin D1 is essential to the development of rodent breast cancer, resulting from Ras or Neu oncogene signaling (51). Cyclin D1 has several important functions, but arguably the most important is the regulation of the inactivating phosphorylation of the Rb protein by cdk4, allowing E2F release and the subsequent transcription of genes that drive cell cycle progression in breast cancer (39). Our demonstration that ERα S522A significantly limits these events both points out therapeutic targets and reveals the importance of E2 signaling from the membrane. The ultimate goal of hormone replacement after the menopause is to activate specific, desirable effects of sex steroids (osteoblast survival) without invoking unwanted actions (breast cancer proliferation). This strategy will be best served by defining the array of discrete actions of E2 that result from binding at membrane and nuclear ER in various target cells. Expression of ERα S522A may be very useful in this regard.
Acknowledgments
This work was supported by grants from the Research Service of the Department of Veterans Affairs, the Avon Products Breast Cancer Research Foundation, the Department of Defense Breast Cancer Research Program (grant BC990915), and the NIH (HL-59890) (to E.R.L.).
We thank M. Lisanti for caveolin plasmids.
REFERENCES
- 1.Albanese, C., J. Johnson, G. Watanabe, N. Eklund, D. Vu, A. Arnold, and R. G. Pestell. 1995. Transforming p21 ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 270:23589-23597. [DOI] [PubMed] [Google Scholar]
- 2.Alton, G., M. Hasilik, R. Niehues, K. Paneerselvam, J. R. Etchison, F. Fana, and H. H. Freeze. 1998. Direct utilization of mannose for mammalian glycoprotein biosynthesis. Glycobiology 8:285-295. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, R. G. 1998. The caveolae membrane system. Annu. Rev. Biochem. 67:199-225. [DOI] [PubMed] [Google Scholar]
- 4.Aronica, S. M., W. L. Kraus, and B. S. Katzenellenbogen. 1994. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 91:8517-8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catoria, G., M. V. Barone, M. Di Domenico, A. Bilancio, D. Ametrano, A. Migliaccio, and F. Auricchio. 1999. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J. 18:2500-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambliss, K. L., I. S. Yuhanna, C. Mineo, P. Liu, Z. German, T. S. Sherman, M. E. Mendelsohn, R. G. W. Anderson, and P. W. Shaul. 2000. Estrogen receptor α and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ. Res. 87:e44-e52. [DOI] [PubMed] [Google Scholar]
- 7.Cooper, L. F., J. C. Tiffee, J. P. Griffin, H. Hamano, and Z. Guo. 2000. Estrogen-induced resistance to osteoblast apoptosis is associated with increased hsp27 expression. J. Cell. Physiol. 185:401-407. [DOI] [PubMed] [Google Scholar]
- 8.Donovan, J. C. H., A. Milic, and J. M. Slingerland. 2001. Constitutive MEK/MAPK activation leads to p27kip1 deregulation and antiestrogen resistance in human breast cancer cells. J. Biol. Chem. 276:40888-40895. [DOI] [PubMed] [Google Scholar]
- 9.Duan, R., W. Xie, and S. Safe. 2001. Estrogen receptor-mediated activation of the serum response element in MCF-7 cells through MAPK-dependent phosphorylation of Elk-1. J. Biol. Chem. 276:11590-11598. [DOI] [PubMed] [Google Scholar]
- 10.Filardo, E. J., J. A. Quinn, K. I. Bland, and R. A. Frackelton, Jr. 2000. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, gpr30, and occurs via transactivation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 14:1649-1660. [DOI] [PubMed] [Google Scholar]
- 11.Greene, G., N. Sobel, W. King, and E. Jensen. 1984. Immunochemical studies of estrogen receptors. J. Steroid Biochem. 20:51-56. [DOI] [PubMed] [Google Scholar]
- 12.Holt, A., G. Alton, C. H. Scaman, G. Loppnow, A. Szpacenko, I. Svendsen, and M. M. Palcic. 1998. Identification of the quinone growth cofactor in mammalian semicarbazide-sensitive amine oxidase. Biochemistry 37:4946-4957. [DOI] [PubMed] [Google Scholar]
- 13.Kim, H. P., J. Y. Lee, J. K. Jeong, S. W. Bae, H. K. Lee, and I. Jo. 1999. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochem. Biophys. Res. Commun. 263:257-262. [DOI] [PubMed] [Google Scholar]
- 14.Kousteni, S., T. Bellido, L. I. Plotkin, C. A. O'Brien, D. L. Bodenner, L. Han, K. Han, G. B. DiGregorio, J. A. Katzenellenbogen, B. S. Katzenellenbogen, P. K. Roberson, R. S. Weinstein, R. L. Jilka, and S. C. Manolagas. 2001. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 104:719-730. [PubMed] [Google Scholar]
- 15.Lavoie, J. N., G. L'Allemain, A. Brunet, R. Muller, and J. Pouyssegur. 1996. Cyclin D1 expression is regulated by the p42/p44 MAPK and negatively by the p38/HOG MAPK pathway. J. Biol. Chem. 271:20608-20616. [DOI] [PubMed] [Google Scholar]
- 16.Le Mellay, V., B. Grosse, and M. Lieberherr. 1997. Phospholipase C beta and membrane action of calcitriol and estradiol. J. Biol. Chem. 272:11902-11907. [DOI] [PubMed] [Google Scholar]
- 17.Levin, E. R. 1999. Cellular functions of the plasma membrane estrogen receptor. Trends Endocrinol. Metab. 10:374-376. [DOI] [PubMed] [Google Scholar]
- 18.Lieberherr, M., B. Grosse, M. Kachkache, and S. Balsan. 1993. Cell signaling and estrogens in female rat osteoblasts: a possible involvement of unconventional non-nuclear receptors. J. Bone Min. Res. 8:1365-1376. [DOI] [PubMed] [Google Scholar]
- 19.Lobenhofer, E. K., G. Huper, J. D. Iglehart, and J. R. Marks. 2000. Inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase activity in MCF-7 cells prevents estrogen-induced mitogenesis. Cell Growth Differ. 11:99-110. [PubMed] [Google Scholar]
- 20.Lu, M. L., M. C. Schneider, Y. Zhang, and J. P. Richie. 2001. Caveolin-1 interacts with androgen receptor: a positive modulator of androgen receptor mediated transactivation. J. Biol. Chem. 276:13442-13451. [DOI] [PubMed] [Google Scholar]
- 21.Lublin, D. 1992. Glucosyl-phosphatidylinositol anchoring of membrane proteins. Curr. Top. Microbiol. Immunol. 178:142-168. [DOI] [PubMed] [Google Scholar]
- 22.Mandava, M. B. 1988. Plant growth-promoting brassinosteroids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 39:23-52. [Google Scholar]
- 23.Marquez, D. C., and R. J. Pietras. 2001. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene 20:5420-5430. [DOI] [PubMed] [Google Scholar]
- 24.Migliaccio, A., M. Di Domenico, G. Castoria, A. de Falco, P. Bontempo, E. Nola, and F. Auricchio. 1996. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 15:1292-1300. [PMC free article] [PubMed] [Google Scholar]
- 25.Morey, A. K., A. Pedram, M. Razandi, B. A. Prins, R.-M. Hu, E. Biesiada, and E. R. Levin. 1997. Estrogen and progesterone inhibit human vascular smooth muscle proliferation. Endocrinology 138:3330-3339. [DOI] [PubMed] [Google Scholar]
- 26.Norfleet, A. M., M. L. Thomas, B. Gametchu, and C. S. Watson. 1999. Estrogen receptor-alpha detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary tumor cells by enzyme-linked immunocytochemistry. Endocrinology 140:3805-3814. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto, T., A. Schlegel, P. E. Scherer, and M. P. Lisanti. 1998. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J. Biol. Chem. 273:5419-5422. [DOI] [PubMed] [Google Scholar]
- 28.Pedram, A., M. Razandi, and E. R. Levin. 1998. Extracellular regulated kinase/jun kinase cross-talk underlies vascular endothelial cell growth factor-induced endothelial cell proliferation. J. Biol. Chem. 273:26722-26728. [DOI] [PubMed] [Google Scholar]
- 29.Pettersson, K., F. Delaunay, and J. A. Gustafsson. 2000. Estrogen receptor beta acts as a dominant regulator of estrogen signaling. Oncogene 19:4970-4978. [DOI] [PubMed] [Google Scholar]
- 30.Pietras, R., and C. M. Szego. 1977. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature 265:69-72. [DOI] [PubMed] [Google Scholar]
- 31.Razandi, M., P. Oh, A. Pedram, J. Schnitzer, and E. R. Levin. 2002. Estrogen receptors associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol. Endocrinol. 16:100-115. [DOI] [PubMed] [Google Scholar]
- 32.Razandi, M., A. Pedram, G. L. Greene, and E. R. Levin. 1999. Cell membrane and nuclear estrogen receptors derive from a single transcript: studies of ERα and ERβ expressed in CHO cells. Mol. Endocrinol. 13:307-319. [DOI] [PubMed] [Google Scholar]
- 33.Razandi, M., A. Pedram, and E. R. Levin. 2000. Estrogen signals to preservation of endothelial cell form and function. J. Biol. Chem. 275:38540-38546. [DOI] [PubMed] [Google Scholar]
- 34.Razandi, M., A. Pedram, and E. R. Levin. 2000. Plasma membrane estrogen receptors signal to anti-apoptosis in breast cancer. Mol. Endocrinol. 14:1434-1447. [DOI] [PubMed] [Google Scholar]
- 35.Resh, M. D. 1996. Regulation of cellular signalling by fatty acid acylation and prenylation of signal transduction proteins. Cell. Signal. 8:403-412. [DOI] [PubMed] [Google Scholar]
- 36.Russell, K. S., M. P. Haynes, D. Sinha, E. Clerisme, and J. R. Bender. 2000. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc. Natl. Acad. Sci. USA 97:5930-5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlegel, A., C. Wang, R. G. Pestell, and M. P. Lisanti. 2001. Ligand-independent activation of oestrogen receptor α by caveolin-1. Biochem. J. 359:203-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnitzer, J. E., D. P. McIntosh, A. M. Dvorak, J. Liu, and P. Oh. 1995. Separation of caveolae from associated microdomains of GPI-anchored proteins. Science 269:1435-1439. [DOI] [PubMed] [Google Scholar]
- 39.Sherr, C. J. 1993. Mammalian G1 cyclins. Cell 73:1059-1065. [DOI] [PubMed] [Google Scholar]
- 40.Simoncini, T., A. Hafezi-Moghadam, D. P. Brazil, K. Ley, W. W. Chin, and J. K. Liao. 2000. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407:538-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tesarik, J., and C. Mendoza. 1995. Nongenomic effects of 17β-estradiol on maturing human oocytes: relationship to oocyte developmental potential. J. Clin. Endocrinol. Metab. 80:1438-1443. [DOI] [PubMed] [Google Scholar]
- 42.Truss, M., and M. Beato. 1993. Steroid hormone receptors: interaction with deoxyribonucleic acid and transcription factors. Endocrine Rev. 14:459-479. [DOI] [PubMed] [Google Scholar]
- 43.Valentine, J. E., E. Kalkhoven, R. White, S. Hoare, and M. G. Parker. 2000. Mutations in the estrogen receptor ligand binding domain discriminate between hormone-dependent transactivation and transrepression. J. Biol. Chem. 275:25322-25329. [DOI] [PubMed] [Google Scholar]
- 44.Wang, Z.-Y., H. Seto, S. Fujioka, S. Yoshida, and J. Chory. 2001. BRl1 is a critical component of a plasma-membrane receptor for plant steroids. Nature 410:380-383. [DOI] [PubMed] [Google Scholar]
- 45.Watters, J. J., J. S. Campbell, M. J. Cunningham, E. G. Krebs, and D. M. Dorsa. 1997. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology 138:4030-4033. [DOI] [PubMed] [Google Scholar]
- 46.Watters, J. J., T. Y. Chun, Y. N. Kim, P. J. Bertics, and J. Gorski. 2000. Estrogen modulation of prolactin gene expression requires an intact mitogen-activated protein kinase signal transduction pathway in cultured rat pituitary cells. Mol. Endocrinol. 14:1872-1881. [DOI] [PubMed] [Google Scholar]
- 47.Watts, C. K., K. J. Sweeney, A. Warlters, E. A. Musgrove, and R. L. Sutherland. 1994. Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Canc. Res. Treat. 31:95-105. [DOI] [PubMed] [Google Scholar]
- 48.White, R., J. A. Lees, M. Needham, J. Ham, and M. Parker. 1987. Structural organization and expression of the mouse estrogen receptor. Mol. Endocrinol. 1:735-744. [DOI] [PubMed] [Google Scholar]
- 49.Wise, P. M., D. B. Dubal, M. E. Wislon, S. W. Rau, and M. Bottner. 2001. Neuroprotective effects of estrogen—new insights into mechanisms of action. Endocrinology 142:969-973. [DOI] [PubMed] [Google Scholar]
- 50.Wyckoff, M. H., K. L. Chambliss, C. Mineo, I. S. Yuhanna, M. E. Mendelsohn, S. M. Mumby, and P. W. Shaul. 2001. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Gαi. J. Biol. Chem. 276:27071-27076. [DOI] [PubMed] [Google Scholar]
- 51.Yu, Q., Y. Geng, and P. Sicinski. 2001. Specific protection against breast cancers by cyclin D1 ablation. Nature 411:1017-1021. [DOI] [PubMed] [Google Scholar]