

Abstract
VMA1-derived endonuclease (VDE), a site-specific endonuclease in Saccharomyces cerevisiae, enters the nucleus to generate a double-strand break in the VDE-negative allelic locus, mediating the self-propagating gene conversion called homing. Although VDE is excluded from the nucleus in mitotic cells, it relocalizes at premeiosis, becoming localized in both the nucleus and the cytoplasm in meiosis. The nuclear localization of VDE is induced by inactivation of TOR kinases, which constitute central regulators of cell differentiation in S. cerevisiae, and by nutrient depletion. A functional genomic approach revealed that at least two karyopherins, Srp1p and Kap142p, are required for the nuclear localization pattern. Genetic and physical interactions between Srp1p and VDE imply direct involvement of karyopherin-mediated nuclear transport in this process. Inactivation of TOR signaling or acquisition of an extra nuclear localization signal in the VDE coding region leads to artificial nuclear localization of VDE and thereby induces homing even during mitosis. These results serve as evidence that VDE utilizes the host systems of nutrient signal transduction and nucleocytoplasmic transport to ensure the propagation of its coding region.
Homing endonucleases are site-specific endonucleases encoded by introns and inteins (internal protein sequences) to promote homing of their genetic elements (homing endonuclease genes [HEG]) into allelic intronless and inteinless alleles, respectively (26). Homing endonuclease genes, widely found in Archaea, Bacteria, and Eukarya, are thought to be parasitic mobile genetic elements because they have no known host functions and their catalytic activity results in self-propagation.
In Saccharomyces cerevisiae, the VMA1 intein encodes a homing endonuclease (14) termed the VMA1-derived endonuclease (VDE) (also called PI-SceI [36]), which is produced by an autocatalytic protein splicing reaction (19, 25). When heterozygous VMA1(+)/VMA1(Δ) diploid cells undergo meiosis under nutrient-deficient conditions, VDE expressed from the intein-containing allele [referred to as VMA1(+)] specifically cleaves the VDE recognition sequence in the inteinless VMA1 allele [referred to as VMA1(Δ)] in the nuclear genome (4, 14) to make a double-strand break. The double-strand break is then repaired, with the VMA1(+) allele as a template. This leads to the unidirectional gene conversion of VMA1(Δ) to VMA1(+), which is called homing. Consequently, the VDE coding region exhibits super-Mendelian inheritance, allowing it to spread in yeast populations even though VDE confers no known benefit to the host yeast cells.
VDE-mediated homing has the following two characteristic features: VDE homing occurs only in meiosis and not in mitosis even if the VMA1(+) and VMA1(Δ) alleles coexist (14). In contrast, other homing endonucleases initiate homing in vitro and in vivo once the HEG(+) allele is accompanied by the HEG(−) allele (7, 29). Meiosis is assumed to be the best time for homing because of the homologue pairing and bias towards the interchromosomal repair pathway. Second, nuclear entry of VDE is a prerequisite for homing, since the recognition sequence of VDE resides only in the nuclear genome. These characteristics are likely attributed to spatial and temporal regulations of VDE activity. VDE is worth analyzing because the mechanism of homing in the nuclear genomes of simple eukaryotes remains completely unknown, in contrast to that of the T4 phage (29), mitochondrial, and chloroplast genomes.
As in other eukaryotic cells, the yeast nuclear envelope may serve as an intracellular barrier. It can prevent VDE newly synthesized in the cytoplasm from accessing the target recognition DNA site. Whereas small molecules can diffuse through nuclear pores in an energy-independent manner, macromolecules such as VDE must be actively imported into the nucleus with the aid of carrier molecules termed karyopherins and the nuclear pore complex (33). In Saccharomyces cerevisiae, one karyopherin of the α family and 14 members of the karyopherin β family have been identified (6, 47). Some of them have been shown to serve as receptors for import and/or export of diverse cargo molecules such as proteins and RNAs (15).
Each karyopherin mediates nucleocytoplasmic transport by binding to the targeting signal in cargo molecules. A classical nuclear targeting signal is simian virus 40 large T antigen nuclear localization signal (NLS), which is rich in basic amino acids (24, 27). This NLS is recognized by a heterodimer of karyopherin α and karyopherin β (Srp1p/Kap60p and Kap95p, respectively, in S. cerevisiae) (11, 15). In the cytoplasm, karyopherin α serves as an adaptor for direct binding to the NLS, whereas karyopherin β binds to karyopherin α. Subsequent to the discovery of this classical transport, karyopherin β proteins have been identified as mediators that accomplish the nuclear translocation of certain classes of proteins independently of karyopherin α (6, 20). In nucleocytoplasmic transport directly mediated by karyopherin β, a nonclassical targeting signal substitutes for the role of the NLS.
Karyopherin-mediated nucleocytoplasmic transport is involved in diverse cellular processes, such as gene expression, signal transduction, and cell cycle progression. Hence, many target proteins are regulated to enter the nucleus at appropriate times. For example, the nuclear localization of several nutrient-responsive transcription factors, including Gln3p and Msn2/4p, is regulated by highly conserved TOR protein kinases, the central controllers of cell growth (1). Inactivation of the TOR signaling pathway by nutrient depletion or by treatment with a specific inhibitor, rapamycin, promotes nuclear import of TOR-regulated transcription factors and triggers transcription of nutrient-responsive genes. VDE function in the nucleus is restricted to meiosis, which suggests the possibility that nucleocytoplasmic transport of VDE also regulates its function. However, the mechanism of nuclear import of VDE was not at all inferable, as VDE has neither a typical NLS nor any host functions such as the transcription factors described above.
In this study, we found that the nuclear import of VDE is driven by nutrients and the TOR signaling cascade. At least two karyopherins, Srp1p and Kap142p, are implicated in the import and export, respectively, of VDE. Furthermore, artificial VDE localization to the nucleus induces homing even in mitotic cells, indicating that nuclear import of VDE is required for self-propagation of the VDE coding region.
MATERIALS AND METHODS
Strains and plasmids.
The Escherichia coli strain SCS1 was used as the plasmid host. The S. cerevisiae strains used in this study are listed in Table 1. YOC2759 [VMA1(Δ)], NOY388 (SRP1), NOY612 (srp1-31), JK9-3Da (TOR1), JH11-1c (TOR1-1), and the deletion and temperature-sensitive karyopherin mutants have been described previously (18, 32, 45, 46).
TABLE 1.
Features of the strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| YPH501 | 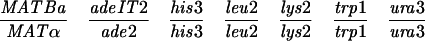 |
41 |
| YOC2759 | 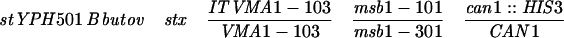 |
32 |
| YOC2816 | 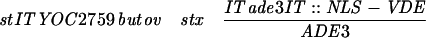 |
This study |
| YOC1705 | MATaade2 his3 leu2 lys2 trp1 ura3 can1::HIS3 msb1-101 | 32 |
| YOC2754 | MATα ade2 his3 leu2 lys2 trp1 ura3 VMA1-103 msb1-301 | 32 |
| YOC2758 |
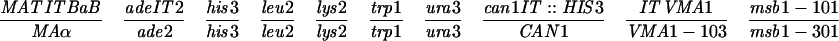 (YOC1705 × YOC2754) (YOC1705 × YOC2754) |
This study |
| NKY278 | 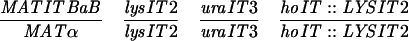 |
N. Kleckner |
| YOC2790 | 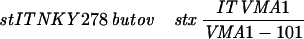 |
This study |
| NOY388 | MATaade2 his3 leu2 trp1 ura3 can1-100 | 46 |
| NOY612 | MATα ade2 his3 leu2 trp1 ura3 can1-100 srp1-31 | 46 |
| JK9-3da | MATaleu2 trp1 ura3 his4 rme1 HMLa | 18 |
| JH11-1c | JK9-3da but TOR1-1 | 18 |
| L40 | MATatrp1 leu2 his3 LYS2::lexA-HIS3 URA3::lexA-lacZ | 35 |
| BY4741 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | 45 |
| his3Δ | BY4741 but his3Δ | 45 |
| kap108Δ | BY4741 but kap108Δ | 45 |
| kap114Δ | BY4741 but kap114Δ | 45 |
| kap120Δ | BY4741 but kap120Δ | 45 |
| kap122Δ | BY4741 but kap122Δ | 45 |
| kap123Δ | BY4741 but kap123Δ | 45 |
| kap127Δ | BY4741 but kap127Δ | 45 |
| kap142Δ | BY4741 but kap142Δ | 45 |
YOC2758 is a diploid strain made by crossing YOC1705 and YOC2754 (32), while YOC2790 is a derivative of NKY278 with its VMA1 locus (VMA1-105/VMA1-105) converted to VMA1/VMA1-101. For description of the VMA1 loci, refer to Nogami et al. (32). Briefly, VMA1 contains the VDE coding region but lacks the VDE recognition sequence, whereas VMA1-101, VMA1-103, and VMA1-105 have complete deletions of the VDE coding region. Additionally, VMA1-103 and VMA1-105 carry nucleotide substitutions in the VDE recognition sequence.
For two-hybrid analyses, pBTM116-SRP1 was constructed by cloning the PCR-amplified SRP1 gene into the BamHI site of pBTM116. pACTII-HK-VDE was constructed by ligating the VDE coding region of pYO314-VMA1 into the BamHI-SmaI gap of pACTII-HK.
For expression of tagged Srp1p, YEp-Flag1-SRP1 and pYES2-SRP1 were constructed by cloning the PCR-amplified SRP1 gene into the BamHI site of YEp-Flag1 (Sigma) and pYES2 (Invitrogen), respectively.
For NLS-VDE (YCpTV-VDEC-NLS-B) construction, synthetic oligonucleotides NLS-BglII F (5′-GATCGGCCGAAGAAGAAGCGCAAGGTC-3′) and NLS-BglII R (5′-GATCGACCTTGCGCTTCTTCTTCGGCC-3′) were annealed for 2 min at 72°C, incubated on ice, and cloned into the BglII site of YCpTV-VDEC, which carries the full-length VDE coding region on pYO314. YIpTV-VDEC-NLS-B was generated by insertion of the BamHI-SalI fragment from pRS314-VDEC-NLS-B into YIpTade3S. YOC2816 was made by transformation of YOC2759 with the SacI-AvrII fragment of YIpTV-VDEC-NLS-B.
Media, growth conditions, and DNA manipulations.
Yeast cells were grown in either a rich medium, YPD (1% Bacto yeast extract [Difco], 2% Bactopeptone [Difco], and 2% glucose [Wako Chemicals]), or a synthetic growth medium, SD (0.67% yeast nitrogen base without amino acids [Difco] and 2% glucose) supplemented appropriately. For selection without uracil or tryptophan, 0.5% Casamino Acids (Difco) was added to SD. Yeast growth, tetrad analysis, mating type determinations, and yeast transformation were performed as described previously (23). Standard procedures were used for all DNA manipulations and E. coli transformation (39).
Synchronous sporulation and nutrient depletion.
Synchronous sporulation was performed according to Ohta et al. (34) with modifications. A single colony on a YPD plate was inoculated into 2 ml of presporulation medium (SPS), consisting of 0.5% Bacto yeast extract, 1% polypeptone (Nihon Seiyaku), 0.17% yeast nitrogen base without ammonium sulfate and amino acids (Difco), 0.05 M potassium phthalate, 1% potassium acetate, and 0.5% ammonium sulfate, with the pH adjusted to 5.0, plus nutritional supplements. After overnight culturing at 30°C, a small amount of preculture suspension was inoculated into 0.5 liter of SPS with appropriate amino acids and 0.001% silicone oil SH-200 (Nakarai Tesque), and cells were cultured at 30°C to a density of 1 × 107 to 2 × 107 cells per ml. The cells were harvested by centrifugation and washed once in sterile water, and portions were taken as premeiotic samples. The rest was inoculated into 0.5 liter of sporulation medium (SPM; 1% potassium acetate and 0.001% polypropyleneglycol [Nakarai Tesque]), with appropriate amino acids, and cultured at 30°C for 2, 4, 6, and 24 h with vigorous aeration.
For nitrogen source starvation, carbon source starvation, and glucose depletion, cells in log phase were washed with sterile water and suspended in SC-N medium (0.17% yeast nitrogen base without amino acids and ammonium sulfate and 2% glucose), YP (1% yeast extract, 2% Bactopeptone), and YPGly (1% yeast extract, 2% Bactopeptone, 3% glycerol), respectively. For rapamycin treatment, log-phase cells were washed with sterile water to which rapamycin (Sigma) was added to a final concentration of 0.2 μg/ml with a stock solution of 20 μg/ml in 90% ethanol and 10% Triton X-100. All experiments were performed at 30°C except for specified cases.
Fluorescence microscopy.
Exponentially growing cells were fixed in the growing medium by adding 1 M potassium acetate (pH 6.5) and formaldehyde (1:10 dilution) for 30 min at 30°C with continuous shaking. The fixed cells were harvested by centrifugation and resuspended in 30 ml of formaldehyde solution (10% formaldehyde and 0.1 M potassium acetate [pH 6.5]) for 45 min at 25°C with gentle shaking.
The procedures for nuclear staining and immunofluorescence microscopy are based on a previously published method (37). For VDE staining, a rabbit polyclonal antibody against VDE was used as the first antibody at a 1:1,000 dilution, and a rhodamine-conjugated anti-rabbit immunoglobulin antibody was used as the second antibody at a 1:100 dilution. Stained cells were observed under the Leica DMRE microscope. All the images presented in this paper were captured with a charge-coupled device camera and Metamorph Imaging software and subsequently processed with Adobe Photoshop software.
Semiquantitative PCR for homing detection.
To normalize the initial amounts of genomic DNA, the PCR product of the chromosomal RHO1 gene was used as the internal control for each sample. The internal standard was amplified with ExTaq polymerase with primers RHO1-1 (5′-GGGCATATGTCACAACAAGTTGGTAACAGT-3′) and RHO1-2 (5′-ATTAACCCTCACTAAAGAGATCTCTATAACAAGACACACTT-3′). A dilution series of the reaction mixture for amplification of the internal standard was prepared. The final reaction mixture (25 μl) contained diluted yeast DNA solution and 10 pmol each of the primers. The reaction was taken through 30 cycles, each of which consisted of 94°C, 50°C, and 72°C, all for 1 min. The PCR products were visualized in 0.7% agarose gels stained with ethidium bromide after electrophoresis. The intensity of the ethidium bromide staining of each band was measured with a charge-coupled device imaging system.
After normalization of the amounts of genomic DNA, the target was amplified with ExTaq with primers LEU2atg (5′-CACCTGTAGCATCGATAGCA-3′) and CgURA3taa (5′-CTGATTCAAGACATATCC-3′). To load equal amounts of DNA samples, we concurrently ran a PCR for RHO1. The final reaction mixture (50 μl) contained 10 pmol each of the primers and 1 μl of yeast DNA solution. To identify the point just prior to saturation, we subjected replicates of the reaction solution through 25, 27, 29, and 30 cycles, each consisting of the same thermocycle as above. The PCR products were visualized as described above.
Two-hybrid analysis.
Yeast strain L40 was cotransformed with pBTM116 or pBTM116-VDE and pACTII-HK or pACTII-HK-SRP1. Transformants were cultured in SD medium lacking tryptophan and leucine. β-Galactosidase activity was measured according to the o-nitrophenyl-β-d-galactopyranoside (ONPG) method (19).
Expression of FLAG fusion protein and in vitro binding assays.
S. cerevisiae cells were transformed with YEp-FLAG1-SRP1 or YEp-FLAG1-BAP (Sigma), and the transformants were cultured for 2 to 3 days to saturation. Supernatants from cell suspensions were prepared, to which 1/10 volume of 10× lysis buffer (final concentrations: 50 mM Tris-HCl [pH 7.5], 7.5 mM NaCl, 1 mM EDTA, 1% Triton X-100) and protease inhibitors (1 mM phenylmethylsulfonyl fluoride and 25 μg/ml each of tosylphenylalanine chloromethyl ketone, tosyllysine chloromethyl ketone, leupeptin, pepstatin A, antipain, and aprotinin) were added.
FLAG-Srp1p and FLAG-BAP were purified by incubating the supernatants with anti-FLAG M2 agarose gel (Sigma) overnight at 4°C and washing four times with wash buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl). The FLAG fusions bound to the anti-FLAG M2 agarose gel were incubated for 2 h at 4°C with cell lysates (50 μg of protein) prepared in lysis buffer plus protease inhibitors with glass beads and were then washed four times with wash buffer. The FLAG fusion proteins and bound materials were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis on a 10% gel, the proteins were electroblotted onto polyvinylidene difluoride membranes (Amersham Pharmacia Biotech), probed with an anti-VDE antibody or an anti-FLAG M2 antibody (Sigma), and developed with the ECL detection kit (Amersham Pharmacia Biotech) with an anti-rabbit immunoglobulin-horseradish peroxidase antibody in the former case and an anti-mouse immunoglobulin-horseradish peroxidase antibody in the latter.
Immunoprecipitation.
For immunoprecipitations, whole-cell extracts were prepared from S. cerevisiae cells functionally expressing Xpress-Srp1p and either untreated or treated with rapamycin for an hour. Log-phase S. cerevisiae cells were harvested and lysed in lysis buffer by vortexing with glass beads. Then 120 μg of total protein from each sample was diluted in lysis buffer plus the protease inhibitors and immunoprecipitated for 2 h at 4°C by addition of an anti-Xpress antibody (Invitrogen) and protein A-Sepharose (Amersham Pharmacia Biotech). The immunoprecipitates formed were washed five times with lysis buffer, and the proteins were eluted and subjected to SDS-PAGE. After electrophoresis in 10% gels, the proteins were electroblotted onto polyvinylidene difluoride membranes and probed with the anti-VDE antibody as described above and the anti-Xpress antibody, followed by development with the anti-mouse immunoglobulin-horseradish peroxidase antibody and the ECL detection kit.
RESULTS
VDE is excluded from the nucleus in exponentially growing mitotic cells.
To examine the cellular localization of VDE in both mitotic and meiotic S. cerevisiae cells, indirect immunofluorescence microscopy was performed with an anti-VDE antibody that specifically recognizes the 50-kDa VDE protein in yeast cell extracts (Fig. 1A). We found that VDE was excluded from the nucleus in mitotic cells. Most of the signals were detected evenly in the cytoplasm, frequently forming dot-like structures (Fig. 1B, log phase).
FIG. 1.

VDE localizes in the nucleus in meiosis, when homing takes place. (A) Immunoblot analysis with the anti-VDE antibody. Whole-cell lysates (25 μg of protein) from log-phase culture of NNY311 [VDE(−)] or YPH501 [VDE(+)] were used. (B) Time course of VDE localization in a culture synchronously entering meiosis. YOC2790 cells were cultured in YPD to a density of 4 × 106 cells/ml (labeled log phase). The cells were harvested, washed, and transferred to presporulation medium, SPS. After incubation in SPS for 24 h (corresponding to 0 h in synchronous meiosis), cells were harvested, washed, and transferred to sporulation medium, SPM. The upper panels show localization of VDE, while the lower panels show the location of the nuclei stained with 4′,6′-diamidino-2-phenylindole (DAPI) in those cells. Note that the antibody staining accumulates in the nucleus as time passes. DAPI staining at 6 h shows the image of the meiotic cells. (C) Semiquantitative PCR to detect homing. The VDE recognition sequence (indicated by an open triangle) was artificially inserted in the MSB1 locus (recipient), and the donor and the recipient chromosomes were originally marked with the URA3 (black box) and LEU2 (gray box) genes, respectively. Homing will generate a tight linkage of the LEU2 and URA3 genes. PCRs with the primers indicated by arrows were used to detect the homing product. (D) Detection of meiosis-specific homing. Yeast genomic DNA was prepared from a log-phase culture of YOC2758 and a culture undergoing synchronized meiosis and subjected to semiquantitative PCR. The upper panel shows amplified DNA derived from the homing product. The lower panel shows the amplified RHO1 gene as an internal standard to normalize the initial amount of template DNA in each sample.
Synchronous meiosis was then induced first by transferring cells from the rich medium (YPD) to the presporulation medium (SPS), containing fewer nutrients than YPD, and then to the sporulation medium (SPM). After a 24-h incubation of cells under nutrient-limited condition in SPS, some VDE was localized in the nucleus (Fig. 1B, 0 h). This period during which the cells are competent for entering meiosis is defined as the premeiotic phase in cells of the SK1 background (34). The nuclear localization of VDE was maintained for the duration of observation after the transfer to SPM for meiosis induction. After 6 h of incubation in SPM, the signal intensities from the nucleus and the cytoplasm became nearly identical, with the only unstained regions corresponding to vacuoles (Fig. 1B, 6 h). The intensities of the signals did not increase further. These results implied that VDE migration is already complete during the premeiotic phase, in advance of meiosis.
FIG. 6.

srp1-31 does not respond to rapamycin, implying that the yeast karyopherin α, Srp1p, is required for nuclear import of VDE. (A) The temperature-sensitive srp1-31 mutant cells show defects in nuclear transport of VDE. NOY388 (SRP1) or NOY612 (srp1-31) cells were cultured to log phase in YPD at 25°C, shifted to 37°C for 3 h, and either treated with 0.2 μg of rapamycin per ml for an hour or left untreated. The panels are presented as in Fig. 4A. Note that rapamycin-treated wild-type cells exhibit nuclear import of VDE (indicated by yellow in the nuclear zones), whereas, in contrast to other karyopherin mutations, rapamycin-treated srp1-31 cells do not. (B) The numbers of cells with nuclear fluorescence were counted, and the fractions were calculated as in Fig. 4B.
To examine the correlation between the intracellular localization of VDE and its function, we constructed a tester diploid strain, YOC2758, containing msb1-101 (recipient)/msb1-301 (donor) and detected homing of URA3 in several conditions (Fig. 1C). In this strain, the donor and recipient chromosomes were originally marked with URA3 and LEU2, respectively, and the VDE recognition sequence was artificially inserted in the recipient chromosome. Homing of URA3 mediated by authentic VDE was detected by semiquantitative PCR analyses with primers corresponding to the sequences of the URA3 and LEU2 genes, which are tightly linked after homing (32). We found that homing products were hardly detectable in the mitotic cells but became highly visible during meiosis (Fig. 1D), as expected from the previous report (14). Thus, the nuclear entry of VDE coincides with homing in meiosis.
VDE import into the nucleus after nutrient deprivation.
To examine whether nutrient limitation in premeiosis causes the alteration of VDE localization, we observed its cellular localization in the early, mid-, and late log and stationary phases, quantifying the population of cells with VDE residing in the nucleus. As shown in Fig. 2A and B, the number of fluorescent cells in the nucleus increased after nutrient limitation: the number of cells with nucleus-localized VDE were few in early log phase, increased to 60% in late log phase, and rose further in stationary phase. These results suggested that VDE migrates to the nucleus in response to nutrient deprivation.
FIG. 2.

VDE import into the nucleus after nutrient deprivation. (A) Growth properties of YPH501 in YPD medium. (B) Nuclear import of VDE induced by starvation. Cells were sampled at the time points indicated as a to f in A. Cellular localization of VDE was observed by indirect immunofluorescence with the anti-VDE antibody, and cells with signals in the nucleus were counted. Approximately 100 cells were counted at each time point. Note that VDE quickly enters the nucleus upon the diauxic shift (between c and d).
Nuclear import of VDE is induced by depletion of nitrogen and fermentable carbon sources.
To identify the nutrients that prevented VDE translocation into the nucleus, VDE localization was examined under various conditions, including glucose depletion (YPGly medium), depletion of fermentable carbon sources (YP medium), and depletion of nitrogen sources (SC-N medium) and in sporulation-inducing medium (SPM medium, an acetate-based, nutrient-deprived medium). In YP, SC-N, and SPM media, most cells exhibited fluorescent signals from the nucleus within 2 h (Fig. 3). On the other hand, cells growing in the glycerol-containing medium YPGly exhibited only partial migration into the nucleus (Fig. 3). These results suggested that depletion of both nitrogen and fermentable carbon sources resulted in the translocation of VDE into the nucleus.
FIG. 3.

Effects of nutrient depletion on cellular localization of VDE. (A) Time course of VDE transport to the nucleus induced by depletion of nitrogen or fermentable carbon sources. Strain YPH501 was grown to early log phase in YPD, harvested by centrifugation, washed once with sterile water, and resuspended in YPD (solid squares), YPGly (solid circles), YP (open triangles), SC-N (open squares), or SPM (open circles). Nuclear import of VDE was examined by indirect immunofluorescence with the anti-VDE antibody. More than 100 cells were examined in each of two independent experiments at each time point. (B) Typical localization of VDE. At each time point, the left panels show VDE localization, while the right panels show DAPI-stained nuclei.
TOR signaling pathway is involved in subcellular localization of VDE.
Nutrient limitation has been reported to cause inactivation of the TOR signaling pathway (38, 40). Therefore, we examined the effect of rapamycin, an inhibitor of TOR kinases (38, 40), on the nuclear migration of VDE. As shown in Fig. 4, VDE became equally localized in the cytoplasm and in the nucleus after a 1-h incubation with rapamycin. In the presence of rapamycin, few cells formed buds, consistent with the fact that rapamycin is effective in arresting S. cerevisiae cells at the early G1 phase (18). In a rapamycin-resistant TOR1 mutant (18), nuclear import of VDE was not observed, confirming that the rapamycin-induced VDE translocation is dependent on TOR inactivation (Fig. 4). Thus, the TOR signaling pathway appears to prevent the translocation of VDE into the nucleus.
FIG. 4.

Effects of TOR signaling pathway and rapamycin treatment on subcellular localization of VDE. (A) Rapamycin treatment leads to nuclear import of VDE except in the TOR1-1 strain, a rapamycin-resistant TOR1 mutant. Each strain was grown to early log phase in YPD and either left untreated or treated with 0.2 μg of rapamycin per ml for an hour. The leftmost panels show localization of VDE visual-ized by indirect immunofluorescence with the anti-VDE antibody, while the middle panels show nuclei stained with DAPI. The right panels are the merged images of VDE (red) and nucleus (green). Note that rapamycin-treated haploid TOR1 cells exhibit nuclear import of VDE (indicated by yellow in the nuclear zones) like diploid wild-type cells (YPH501), whereas rapamycin-treated TOR1-1 cells do not. (B) The numbers of cells with nuclear fluorescence were counted, and the fractions were calculated. The sample sizes were all in excess of 200.
Subcellular localization of VDE in karyopherin mutants.
Genomic sequence analysis revealed that a total of 14 karyopherin βs and one karyopherin α exist in the S. cerevisiae genome (6, 47). We used a functional genomic approach to identify karyopherins that affect the nuclear localization of VDE. We screened available S. cerevisiae karyopherin mutants by indirect immunofluorescence staining of VDE (Fig. 5 and 6 ) and individually examined the subcellular localization of VDE in those mutants in the presence and absence of rapamycin. All the mutants except the srp1-31(Ts) and kap142Δ mutants showed a VDE localization pattern similar to that of the wild-type strain: VDE was excluded from the nucleus in the absence of rapamycin, while it became evenly distributed in the nucleus and the cytoplasm in its presence (Fig. 5 and 6). In the srp1-31(Ts) mutant (46), almost all the VDE appeared to remain in the cytoplasm even in the presence of rapamycin at the nonpermissive temperature (Fig. 6).
FIG. 5.

Functional genomic strategy to identify Kap142p as a nuclear transport factor. The kap142 mutation allows VDE to enter the nucleus, implying that Kap142p is a nuclear transport factor for VDE; none of the other six karyopherin β mutants exhibited VDE localization in the nucleus except when treated with rapamycin. (A) Effects of several karyopherin mutations on cellular distribution of VDE. Exponentially growing cells of each mutant were either left untreated or treated with 0.2 μg of rapamycin per ml for an hour. The localization of VDE was examined by indirect immunofluorescence after staining with the anti-VDE antibody. Yeast nuclei were stained with DAPI. (B) The numbers of cells with nuclear fluorescence were counted, and the fractions were calculated as in Fig. 4B.
Control experiments with the srp1-31(Ts) mutant at the permissive temperature (data not shown) and the wild-type strain (Fig. 6) showed that we could observe the nuclear localization of VDE in the presence of rapamycin. We also found a certain amount of VDE localized in the nucleus prior to nutrient starvation in the kap142Δ mutant (Fig. 5). This result implies that the mutant is defective in either exit from the nucleus or preventing entry into the nucleus. Taken together, the data obtained suggested that Srp1p and Kap142p mediate nuclear localization of VDE.
Srp1p interacts with VDE in vitro and in vivo.
S. cerevisiae has a single karyopherin α, termed Srp1p, which directly interacts with cargo proteins. The finding that nuclear import of VDE is mediated by Srp1p, described above, gives rise to the hypothesis that Srp1p binds to VDE. We therefore investigated possible protein-protein interactions between Srp1p and VDE. The actual interaction between the two proteins was first indicated by an experiment with the yeast two-hybrid system (Fig. 7A). Cells were cultured in SC liquid medium without leucine and tryptophan, in which two-hybrid interactions were detected as β-galactosidase activity (23). Cloning vectors pBTM116 and pACTII-HK were used as negative controls in combination with pBTM116-SRP1 and pACTII-HK-VDE. Both controls were negative in the β-galactosidase assay, suggesting that the coexistence of Srp1p and VDE is required for their interaction.
FIG. 7.

Srp1p interacts with VDE. (A) Interaction between Srp1p and VDE detected by two-hybrid analysis. S. cerevisiae strain L40 was cotransformed with a pBTM116-derived plasmid and a pACTII-HK-derived plasmid. The transformants were cultured in SD medium lacking tryptophan and leucine, and β-galactosidase activity was measured. The values are the averages of data from more than three transformants. (B) VDE binds Srp1p in vitro. S. cerevisiae whole-cell extracts were incubated with either FLAG-Srp1p or FLAG-BAP bound to an anti-FLAG M2-agarose gel. FLAG fusion proteins and bound materials were separated by SDS-PAGE and detected by Western blotting with the anti-VDE antibody or the anti-FLAG M2 antibody. The leftmost lane represents 10% of the total VDE used to bind to FLAG-Srp1p or FLAG-BAP. (C) Enhanced interaction between Srp1p and VDE after rapamycin treatment in vivo. Whole-cell extracts were prepared from S. cerevisiae cells expressing Xpress-Srp1p or the vector alone with or without rapamycin treatment for an hour. Each protein sample was immunoprecipitated by addition of the anti-Xpress antibody and protein A-Sepharose. The immunoprecipitates were subjected to SDS-PAGE and detected by Western blotting with the anti-VDE antibody or the anti-Xpress antibody. The total lysates were used as controls.
The interaction was then biochemically analyzed. We incubated yeast cell extracts with either secreted FLAG-bacterial alkaline phosphatase (FLAG-BAP) or FLAG-Srp1p from S. cerevisiae cells. Proteins that interacted with the FLAG-tagged proteins were separated by SDS-PAGE and identified with the anti-VDE antibody. We found that VDE specifically bound to FLAG-Srp1p but not to FLAG-BAP (Fig. 7B). Furthermore, we carried out coimmunoprecipitation experiments with cell extracts expressing Xpress-Srp1p and VDE to verify the finding mentioned previously, Extracts were prepared from untreated and rapamycin-treated cells expressing the Xpress-tagged Srp1 protein. Xpress-Srp1p was precipitated with the anti-Xpress monoclonal antibody, and the precipitates were subjected to SDS-PAGE, followed by VDE analysis by Western blotting. As shown in Fig. 7C, VDE and Xpress-Srp1p coimmunoprecipitated, and this interaction was enhanced by rapamycin treatment. These results clearly demonstrated that Srp1p-dependent nuclear localization of VDE after treatment with rapamycin (Fig. 5) is caused by physical interaction of Srp1p and VDE.
VDE mislocalization in mitotic cells induces enforced homing.
To determine the functional relevance of VDE localization in the nucleus, we measured homing frequencies in the mitotic cells whose VDE localization was artificially altered. We found that rapamycin-treated mitotic cells in which VDE is also mislocalized in the nucleus (Fig. 4) exhibited an increased homing frequency. While no homing was observed in untreated cells, homing products were clearly detectable by semiquantitative PCR in rapamycin-treated mitotic cells (Fig. 8A). The amount of homing products in the rapamycin-treated cells was almost the same as in the premeiotic cells (data not shown). Mitotic cells expressing recombinant VDE attached to the nuclear localization signal (NLS) showed similar results (Fig. 8B). VDE tagged with the simian virus 40 large T NLS (NLS-VDE), consisting of a short lysine-rich sequence (31), accumulated heavily in the nucleus in mitotic cells (Fig. 8B). NLS-VDE expressed in mitotic cells normally functioned in the nucleus and promoted homing (Fig. 8C). Thus, nuclear localization of VDE in mitotic cells is sufficient to induce homing, which is an essential step for the self-propagation of its coding region.
FIG. 8.

Artificial mislocalization of VDE causes homing even in mitotically growing cells. (A) Detection of homing products in rapamycin-treated mitotic cells. S. cerevisiae genomic DNA was prepared from a log-phase culture of YOC2758 without rapamycin treatment (left) or with rapamycin treatment for an hour (right) and subjected to semiquantitative PCR analysis. The upper and lower panels show amplified DNA derived from the homing product and the amplified RHO1, respectively, as in Fig. 1C. (B) Immunolocalization of NLS-tagged VDE in mitosis. YOC2758 and YOC2816 (expressing NLS-VDE) were grown to early log phase in YPD. The localization of VDE was examined by indirect immunofluorescence staining with the anti-VDE antibody. S. cerevisiae nuclei were stained with DAPI. (C) Effects of NLS on homing frequency. S. cerevisiae genomic DNA was prepared from log-phase cultures of YOC2758 (left) and YOC2816 (right) and subjected to semiquantitative PCR analysis. Data are presented as in Fig. 1C.
DISCUSSION
Previous studies of homing endonucleases have focused mainly on their structure and biochemical functions in vitro (13, 21), and thus the spatial and temporal regulation of homing endonuclease activity remained unresolved. We found that starvation induces nuclear translocation of VDE, providing a vital clue to understanding the in vivo regulation of this endonuclease activity. Translocation of VDE to the nucleus is regulated by the TOR signaling pathway, which is inactivated after nutrient depletion. Nuclear entry of VDE is mediated by one of the karyopherins, Srp1p, through physical interaction (Fig. 9).
FIG. 9.

Model of nucleocytoplasmic transport of VDE mediated by the TOR signaling pathway and karyopherins.
Nucleocytoplasmic transport mechanism of VDE.
The TOR signaling pathway broadly controls nutrient metabolism by sequestering several transcription factors in the cytoplasm (38, 40). Several known cases regulated by this pathway include the nucleocytoplasmic transport of nutrient-responsive transcription factors, such as Gln3p (42) and Msn2/4p (16). Nutrient deprivation or rapamycin treatment causes rapid nuclear accumulation of Gln3p and Msn2/4p to induce transcription activation of their downstream genes (1).
The molecular mechanism of VDE bears a similarity to that of Gln3p. Both Gln3p and VDE enter the nucleus after nutrient deprivation or rapamycin treatment, dependent on the TOR signaling pathway. In addition, both Gln3p and VDE interact with Srp1p to enter the nucleus. Thus, it is likely that VDE uses the same nuclear import mechanism as Gln3p for its import. Another supporting evidence for this is the involvement of Ure2p in the nuclear import of Gln3p and VDE. Ure2p is likely involved in cytoplasmic retention of VDE, like that of Gln3p, since 58% of exponentially growing Δure2 cells showed nuclear localization of VDE (data not shown).
Physical interaction of VDE with Srp1p may occur during nuclear import of VDE. Although Srp1p is known to bind to the NLS of cargo proteins, VDE lacks such typical NLS sequences, unlike Gln3p. Our plausible explanation is that VDE interacts with Srp1p through some adapter proteins that possesses a typical NLS. An alternative possibility is that domain I of VDE, which contains sequences considerably rich in basic residues, may function as a “fake” NLS to interact with Srp1p and promote its nuclear import. Since this signal presumably functions less strongly and less directly than the conventional NLS, VDE cannot accumulate in the nucleus without the help of another NLS fused to itself. Identification of the region that binds to Srp1p will contribute to understanding the more detailed mechanisms of karyopherin-mediated nucleocytoplasmic transport of VDE.
Our functional genomic analyses of karyopherins showed that Kap142p is also required for the controlled localization of VDE. Kap142p, which belongs to the karyopherin β family, is a candidate protein involved in the nuclear export or maybe inhibition of the nuclear import of VDE. While several proteins are known to accumulate in the nucleus of kap142Δ cells (3, 10, 22, 28), VDE is evenly distributed in both the nucleus and the cytoplasm in these mutant cells. This observation suggests that other karyopherins or unknown molecular mechanisms may prevent VDE accumulation in the nucleus. Our results revealed a new aspect of homing endonucleases: host machineries such as that of the TOR signaling pathway and that of karyopherin-mediated nuclear transport are required for the self-propagation of homing endonucleases, although they are not known to have any host function, unlike Gln3p.
Difference in VDE localization patterns in mitosis and in meiosis gives rise to meiosis-specific homing mediated by VDE.
One of the most intriguing features of VDE is that it does not initiate homing during mitosis, although other homing endonucleases start homing once intron/intein and intronless/inteinless sequences are both present (14). VDE, as a nuclear genomic parasite, assumes the strategy of the meiosis-specific function for self-propagation for the following two reasons. First, homologous chromosomes get paired at meiosis, ensuring that repair will take place with its own coding region as the template. Second, the risk that VDE will create a lethal double-strand break is low because the host repair system in meiosis is more active than it is in mitosis. We found that nuclear localization of VDE during mitosis leads to homing, implying that VDE could mediate homing whenever it enters the nucleus.
Nuclear import of VDE occurs prior to meiosis under nutrient-depleted conditions. Considering that starvation of essential nutrients serves as a trigger of meiosis (9, 12), VDE may monitor extracellular environments at phases preceding meiosis in order for homing to occur precisely in meiosis. In contrast, meiosis does not seem to be a prerequisite for homing of I-SceI and I-SceII, the S. cerevisiae mitochondrial homing endonucleases encoded by the group I intron (8, 43). Upon conjugation, they home immediately after mitochondrial fusion, during which mitochondrial DNA frequently recombines (30) so that they bind their homing sites [HEG(−) alleles] in the matrix without any additional steps such as nuclear entry of VDE.
Although the accessibility of VDE to its target gene is regulated through its subcellular localization, nuclear localization alone may not be sufficient to fully activate VDE, because the generation of a double-strand break by VDE can be regulated by additional factors. Chromatin structures or proteins interacting with the VDE recognition sequence might regulate cleavage of the VDE recognition sequence in chromosomes. This idea is supported by the fact that a VDE-mediated double-strand break is repressed or strikingly delayed by blocking premeiotic DNA replication (unpublished data). Thus, at least two steps exist in the regulation of VDE activity: (i) karyopherin-mediated nuclear import of VDE under conditions conducive to entering meiosis and (ii) DNA cleavage after premeiotic DNA replication. Therefore, a complicated regulatory mechanism by the host yeast cells is required for controlling meiosis-specific homing. Elucidating the mechanism of VDE-mediated homing in detail will contribute to understanding how mobile insertion elements have developed strategies to ensure the long-term survival of endonucleases in natural populations.
Acknowledgments
We are grateful to M. Nomura for providing the srp1-31 mutant, to M. Hall for the TOR1-1 strain, to N. Kleckner for the NKY278 strain, and to K. Tanaka for the plasmids used for the two-hybrid analyses. We also thank K. Homma for critically reading the manuscript and members of the Laboratory of Signal Transduction for helpful discussions.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Science, Sports and Culture of Japan.
REFERENCES
- 1.Beck, T., and M. N. Hall. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402:689-692. [DOI] [PubMed] [Google Scholar]
- 2.Bertram, P. G., J. H. Choi, J. Carvalho, W. Ai, T.-F. Chan, and X. F. Zheng. 2000. Tripartite regulation of Gln3p by TOR, Ure2p, and phosphatases. J. Biol. Chem. 275:35727-35733. [DOI] [PubMed] [Google Scholar]
- 3.Blondel, M., P. M. Alepuz, S. Huang, G. Ammerer, and M. Peter. 1999. Nuclear export of Far1p in response to pheromones requires the export receptor Msn5p/Ste21p. Genes Dev. 13:2284-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bremer, M. C., F. S. Gimble, J. Thorner, and C. L. Smith. 1992. VDE endonuclease cleaves Saccharomyces cerevisiae genomic DNA at a single site: physical mapping of the VMA1 gene. Nucleic Acids Res. 20:5484.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carvalho, J., P. G. Bertram, S. R. Wente, and X. F. S. Zheng. 2001. Phosphorylation regulates the interaction between Gln3p and the nuclear import factor Srp1p. J. Biol. Chem. 276:25359-25365. [DOI] [PubMed] [Google Scholar]
- 6.Chook, Y. M., and G. Blobel. 2001. Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 11:703-715. [DOI] [PubMed] [Google Scholar]
- 7.Clyman, J., and M. Belfort. 1996. Trans and cis requirements for intron mobility in prokaryotic system. Genes Dev. 6:1269-1279. [DOI] [PubMed] [Google Scholar]
- 8.Colleaux, L., L. d'Auriol, M. Betermier, G. Cottarel, A. Jacquier, F. Galibert, and B. Dujon. 1986. Universal code equivalent of a yeast mitochondrial intron reading frame is expressed into E. coli as a specific double strand endonuclease. Cell 44:521-533. [DOI] [PubMed] [Google Scholar]
- 9.Colomina, N., E. Garí, C. Gallego, E. Herrero, and M. Aldea. 1999. G1 cyclins block the Ime1 pathway to make mitosis and meiosis incompatible in budding yeast. EMBO J. 18:320-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Vit, M. J., and M. Johnson. 1999. The nuclear exportin Msn5p is required for nuclear export of the Mig1 glucose repressor of Saccharomyces cerevisiae. Curr. Biol. 9:1231-1241. [DOI] [PubMed] [Google Scholar]
- 11.Enenkel, C., G. Blobel, and M. Rexach. 1995. Identification of a yeast karyopherin heterodimer that targets import substrate to mammalian nuclear pore complexes. J. Biol. Chem. 270:16499-16502. [DOI] [PubMed] [Google Scholar]
- 12.Freese, E. B., M. I. Chu, and E. Freese. 1982. Initiation of yeast sporulation by partial carbon, nitrogen, or phosphate deprivation. J. Bacteriol. 149:840-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gimble, F. S. 2000. Invasion of a multitude of genetic niches by mobile endonuclease. FEMS Microbiol. Lett. 185:99-107. [DOI] [PubMed] [Google Scholar]
- 14.Gimble, F. S., and J. Thorner. 1992. Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature 357:301-306. [DOI] [PubMed] [Google Scholar]
- 15.Gorlich, D., and U. Kutay. 1999. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:607-660. [DOI] [PubMed] [Google Scholar]
- 16.Görner, W., E. Durchschlag, M. T. Martinez-Pastor, F. Estruch, G. Ammerer, B. Hamilton, H. Luis, and C. Schüller. 1998. Nuclear localization of C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12:586-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guarente, L. 1983. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101:181-191. [DOI] [PubMed] [Google Scholar]
- 18.Heitman, J., N. R. Movva, and M. N. Hall. 1991. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905-909. [DOI] [PubMed]
- 19.Hirata, R., Y. Ohsumi, A. Nakano, H. Kawasaki, K. Suzuki, and Y. Anraku. 1990. Molecular structure of a gene, VMA1, encoding the catalytic subunit of H+-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 265:6726-6733. [PubMed] [Google Scholar]
- 20.Hood, J. K., and P. A. Silver. 1999. In or out? Regulating nuclear transport. Curr. Opin. Cell Biol. 11:241-247. [DOI] [PubMed] [Google Scholar]
- 21.Jurica, M. S., and B. L. Stoddard. 1999. Homing endonuclease: structure function and evolution. Cell. Mol. Life Sci. 55:1304-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaffman, A., N. M. Rank, E. M. O'Neill, L. S. Huang, and E. K. O'Shea. 1998. The receptor Msn5 exports the phosphorylated transcription factor Pho4 out of the nucleus. Nature 396:482-486. [DOI] [PubMed] [Google Scholar]
- 23.Kaiser, C., S. Michaelis, and A. Mitchell. 1994. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 24.Kalderon, D., B. L. Roberts, W. D. Rychardson, and A. E. Smith. 1984. A short amino acid sequence able to specify nuclear location. Cell 39:499-509. [DOI] [PubMed] [Google Scholar]
- 25.Kane, P. M., C. T. Yamashiro, D. F. Wolczyk, N. Neff, M. Goebl, and T. H. Stevens. 1990. Protein splicing converts the yeast TFP1 gene product to the 69-kDa subunit of the vacuolar H+-adenosine triphosphatase. Science 250:651-657. [DOI] [PubMed] [Google Scholar]
- 26.Lambowitz, A. M., and M. Belfort. 1993. Introns as mobile genetic elements. Annu. Rev. Biochem. 62:587-622. [DOI] [PubMed] [Google Scholar]
- 27.Lanford, R. E., and J. S. Butel. 1985. Replicative functions of the si,iam virus 40(cT)-3 mutant defective for nuclear transport of T antigen. Virology 147:72-80. [DOI] [PubMed] [Google Scholar]
- 28.Mahanty, S. K., Y. Wang, F. W. Farley, and E. A. Elion. 1999. Nuclear shuttling of yeast scaffold Ste5 is required for its recruitment to the plasma membrane and activation of the mating MAPK cascade. Cell 98:501-512. [DOI] [PubMed] [Google Scholar]
- 29.Mueller, J. E., J. Clyman, Y. Huang, M. M. Parker, and M. Belfort. 1996. Intron mobility in phase T4 occurs in the context of recombination-dependent DNA replication by way of multiple pathways. Genes Dev. 10:351-364. [DOI] [PubMed] [Google Scholar]
- 30.Nakagawa, K., N. Morishima, and T. Shibata. 1992. An endonuclease with multiple cutting sites, endo.SceI, initiates genetic recombination at its cutting site in yeast mitochondria. EMBO J. 11:2707-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson, M., and P. Silver. 1989. Context affects nuclear protein localization in Saccharomyces cerevisiae. Mol. Cell. Biol. 9:384-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nogami, S., T. Fukuda, Y. Nagai, S. Yabe, M. Sugiura, R. Mizutani, Y. Satow, Y. Anraku, and Y. Ohya. 2002. Homing at an extragenic locus mediated by VDE (PI-SceI) in Saccharomyces cerevisiae. Yeast 19:773-782. [DOI] [PubMed] [Google Scholar]
- 33.Ohno, M., M. Fornerod, and I. W. Mattaj. 1998. Nucleocytoplasmic transport: the last 200 nanometers. Cell 92:327-336. [DOI] [PubMed] [Google Scholar]
- 34.Ohta, K., A. Nicolas, M. Furuse, A. Nabetani, H. Ogawa, and T. Shibata. 1998. Mutations in the MRE11, RAD50, XRS2, and MRE2 genes alter chromatin configuration at meiotic DNA double-stranded break sites in premeiotic and meiotic cells. Proc. Natl. Acad. Sci. USA 95:646-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozaki, K., K. Tanaka, H. Imamura, T. Hihara, T. Kameyama, H. Nonaka, H. Hirano, Y. Matsuura, and Y. Takai. 1996. Rom1p and Rom2p are GDP/GTP exchange proteins (GEPs) for the Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J. 15:2196-2207. [PMC free article] [PubMed] [Google Scholar]
- 36.Perler, F. B., E. O. Davis, G. E. Dean, F. S. Gimble, W. E. Jack, N. Neff, C. J. Noren, J. Thorner, and M. Belfort. 1994. Protein splicing elements: inteins and exteins-a definition of terms and recommended nomenclature. Nucleic Acids Res. 22:1125-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pringle, J., R. A. Preston, A. E. Adams, T. Stearns, D. G. Drubin, B. K. Haarer, and E. W. Jones. 1989. Fluorescence microscopy methods in yeast. Methods Cell Biol. 31:357-435. [DOI] [PubMed] [Google Scholar]
- 38.Rohde, J., J. Heitman, and M. E. Cardenas. 2001. The Tor kinases link nutrient sensing to cell growth. J. Biol. Chem. 276:9583-9586. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 40.Schmelzle, T., and M. N. Hall. 2000. TOR, a central controller of cell growth. Cell 103:253-262. [DOI] [PubMed] [Google Scholar]
- 41.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vector and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanbrough, M., D. W. Rowen, and B. Magasanik. 1995. Role of the GATA factors Gln3p and Nil1p of Saccharomyces cerevisiae in the expression of nitrogen-regulated genes. Proc. Natl. Acad. Sci. USA 92:9450-9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wenzlau, J. M., R. J. Saldanha, R. A. Butow, and P. S. Perlman. 1989. A latent intron-encoded maturase is also an endonuclease needed for intron mobility. Cell. 56:421-430. [DOI] [PubMed] [Google Scholar]
- 44.Wiickner, R. B. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264:566-569. [DOI] [PubMed] [Google Scholar]
- 45.Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B., Andre, R., Bangham, R., Benito, J. D. Boeke, H. Bussey, A. M. Chu, C. Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury, S. H. Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M. Laub, H. Liao, R. W. Davis, et al. 1999. Functional characterization of the Saccharomyces cerevisiae genome by gene disruption and parallel analysis. Science 285:793-972. [DOI] [PubMed] [Google Scholar]
- 46.Yano, R., M. L. Oakes, M. M. Tabb, and M. Nomura. 1994. Yeast Srp1p has homology to armadillo/plakoglobin/B-catenin and participates in apparently multiple functions including the maintenance of nucleolar structure. Proc. Natl. Acad. Sci. USA 91:6880-6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yano, R., M. L. Oakes, M. Yamaghishi, J. A. Dodd, and M. Nomura. 1992. Cloning and characterization of SRP1, a suppressor of temperature-sensitive RNA polymerase I mutations in Saccharomyces cerevisiae. Mol. Cell. Biol. 12:5640-5651. [DOI] [PMC free article] [PubMed] [Google Scholar]