

Abstract
We have identified a human gene encoding a novel MBD2-interacting protein (MBDin) that contains an N-terminal GTP-binding site, a putative nuclear export signal (NES), and a C-terminal acidic region. MBDin cDNA was isolated through a two-hybrid interaction screening using the methyl-CpG-binding protein MBD2 as bait. The presence of the C-terminal 46-amino-acid region of MBD2 and both the presence of the acidic C-terminal 128-amino-acid region and the integrity of the GTP-binding site of MBDin were required for the interaction. Interaction between MBD2 and MBDin in mammalian cells was confirmed by immunoprecipitation experiments. Fluorescence imaging experiments demonstrated that MBDin mainly localizes in the cytoplasm but accumulates in the nucleus upon disruption of the NES or treatment with leptomycin B, an inhibitor of NES-mediated transport. We also found that MBDin partially colocalizes with MBD2 at foci of heavily methylated satellite DNA. An MBD2 deletion mutant lacking the C-terminal region maintained its subnuclear localization but failed to recruit MBDin at hypermethylated foci. Functional analyses demonstrated that MBDin relieves MBD2-mediated transcriptional repression both when Gal4 chimeric constructs and when in vitro-methylated promoter-reporter plasmids were used in transcriptional assays. Southern blotting and bisulfite analysis showed that transcriptional reactivation occurred without changes of the promoter methylation pattern. Our findings suggest the existence of factors that could be targeted on methylated DNA by methyl-CpG-binding proteins reactivating transcription even prior to demethylation.
Addition of methyl groups mostly at the CpG dinucleotide represents the major epigenetic modification of eukaryotic genomes heritable by somatic cells after cell division. DNA methylation plays an essential role in mammalian development (24) and in different biological processes such as tissue-specific gene expression (40), X-chromosome inactivation (17), genomic imprinting (2, 33), and repression of transposable elements (46). Abnormal methylation is a cause of human genetic diseases, including ICF syndrome (16), and is involved in carcinogenic processes primarily through aberrant hypermethylation of tumor suppressors' promoter regions (12, 21, 22).
Methylated DNA is generally associated with transcriptional silencing (6, 7, 32), but how DNA methylation silences gene expression is not well understood. Studies are now focusing on the different components of the DNA methylation system and particularly on the mechanisms by which methyl-CpG signal is targeted, read, and maintained. Recently, a family of five mammalian methyl-CpG-binding proteins (MeCP2, MBD1, MBD2, MBD3, and MBD4) has been found to be essential to interpret the methylation patterns and to mediate the biological consequences of DNA methylation (1, 15, 18). All methyl-binding proteins share a common structural stretch of 60 to 80 amino acids called the mCpG-binding domain (MBD) and, with the exception of MBD4 (19, 34), mediate transcriptional repression by changing the local chromatin structure, mainly through recruitment of histone deacetylases (HDACs) (1, 18, 29, 43).
MBD2 is a component of a large protein complex, MeCP1, which represses transcription from densely methylated genes. MeCP1 includes HDAC1, HDAC2, and RbAp46/48 proteins, so that MBD2 can target deacetylase activity at methylated sites (30). MBD2 can associate with different other proteins that could confer additional activities on MBD2. MBD2 associates with MBD3, which belongs to the Mi-2/NuRD corepressor complex (48), but although MBD3 is 70% similar to MBD2, it lacks an intrinsic ability to bind methylated DNA (18). By virtue of MBD2/MBD3 interaction, the Mi-2/NuRD complex can be recruited on methylated DNA to silence transcription. In addition, MBD2 and MBD3 form a complex with DNA methyltransferase 1 on hemimethylated DNA at replication foci and may help to establish or maintain the repressed state of chromatin (42). Recently, Sekimata et al. (39) identified a novel protein, MIZF, that interacts with MBD2 and functions as a negative regulator of transcription in an HDAC-dependent manner. The association of MIZF with MBD2 significantly enhances HDAC protein recruitment and activity (39).
Moreover, MBD2 has been also described as an enzyme capable of directly removing methyl groups from methylated CpG-containing DNA both in vitro and in vivo (5, 10). Such an activity of MBD2 would provide a means of active DNA demethylation not involving DNA replication. However, other groups failed to confirm the demethylase activity of MBD2 (8, 30, 44).
At present, it seems likely that the activity of MBD2 can depend on its association with different molecular partners and that additional undescribed roles could be found for this methyl-binding protein. The aim of this study was to identify, using yeast two-hybrid screening, molecular partners of MBD2. Here we describe a novel MBD2-interacting protein, MBDin, which is able to modulate the MBD2-mediated transcriptional functions.
MATERIALS AND METHODS
Construction of fusion genes and expression plasmids.
For the construction of Gal4 binding domain (Gal4BD) and Gal4 activation domain (Gal4AD) fusion genes, different fragments were amplified by PCR with pairs of primers linked to restriction sites and cloned in pBridge, pSG424 (35), and pGADGH (Clontech) plasmids: pBridge-MBD2 and pGal4-MBD2 were obtained by cloning the entire MBD2b coding sequence (786 bp) in pBridge and pSG424, respectively; pBridge-ΔMBD2, pBridge-MUT1, pBridge-MUT2, pBridge-MUT3, and pBridge-MUT4 are deletion mutants of pBridge-MBD2 and were obtained by cloning, respectively, 471-bp (amino acids 103 to 260), 177-bp (amino acids 103 to 162), 339-bp (amino acids 103 to 216), 153-bp (amino acids 209 to 260), and 180-bp (amino acids 156 to 216) fragments of the human MBD2 coding sequence in the pBridge plasmids; pGad-MBDinΔN30 and pGad-MBDinΔC128 are deletion mutants of the library plasmid pGadGH-MBDin and were obtained by cloning, respectively, a 1,032-bp (amino acids 30 to 374) and a 720-bp (amino acids 1 to 246) fragment of the human MBDin coding sequence in the pGADGH plasmids. Primers containing restriction sites (underlined) were as follows: for pBridge-MBD2, MBD2-1 (5′-AGTCGAATTCATGGATTGCCCGGCCCTCCCC-3′) and MBD2-2 (5′-AGTCGGATCCTTAGGCTTCATCTCCACTGTC-3′); for Gal4-MBD2, MBD2-1 and MBD2-3 (5′-AGTCGAATTCTTAGGCTTCATCTCCACTGTC-3′); for pBridge-ΔMBD2, MBD2-7 (5′-AGTCGAATTCAAACAACCGGTAACCAAAG-3′) and MBD2-2; for pBridge-MUT1, MBD2-7 and MBD2-9 (5′-AGTCGGATCCTTATCCTTGAAGACCTTTGGGTAG-3′); for pBridge-MUT2, MBD2-7 and MBD2-10 (5′-AGTCGGATCCTTAGACAATAAAAGCTTTGCAGAG-3′); for pBridge-MUT3, MBD2-11 (5′-AGTCGAATTCCTCTGCAAAGCTTTTATTGTC-3′) and MBD2-2; for pBridge-MUT4, MBD2-12 (5′-AGTCGAATTCCTACCCAAAGGTCTTCAAGGA-3′) and MBD2-10 (5′-AGTCGGATCCTTAGACAATAAAAGCTTTGCAGAG-3′); for pGadGHΔMBDin, MBDin-1 (5′-AGTCGGATCCAGGGAAAACCACTTTG-3′) and MBDin-2 (5′-AGTCGAATTCCAAGTGAAGTGTGCTAAAGTCTC-3′); and for pGad-MBDinΔC128, MBDin-3 (5′-AGTCGAATTCGATGGCGGCGTCCGCAGC-3′) and MBDin-5 (5′-AGTCGTCGACCTACACCCTGAGTGAGCTGTAAAACTC-3′). For the expression of red fluorescent protein (RFP) and green fluorescent protein (GFP) fused to MBD2 and MBDin, different fragments were amplified by PCR with pairs of primers linked to restriction sites and cloned in pDsRedN1 and pEGFP1 (Clontech): pRFPMBD2 was obtained by cloning the entire MBD2b coding sequence (786 bp) in pDsRedN1; pRFPΔMBD2 is a deletion mutant of pRFPMBD2 and was obtained by cloning a 627-bp fragment (amino acids 1 to 209) of the human MBD2b coding sequence in the pDsRedN1 plasmids; pRFPMBDin and pGFPMBDin were obtained by cloning the entire MBDin coding sequence (1,122 bp) in pDsRedN1 and pEGFP1, respectively; pGFPMBD2 was obtained by cloning the entire MBD2b coding sequence (786 bp) in pEGFP1. Primers containing restriction sites (underlined) were as follows: for RFPMBD2, MBD2-4 (5′-AGTCGAATTCAGCACAATGGATTGCCCGGCCCTCCCC-3′) and MBD2-14 (5′-AGTCGGATCCAAGGCTTCATCTCCACTGTCCAT-3′); for pRFPΔMBD2, MBD2-4 and MBD2-17 (5′-AGTCGGATCCGTGACAATAAAAGCTTTGCAGAG-3′); for pRFPMBDin, MBDin-9 (5′-AGTCGAATTCAGCACAATGGCGGCGTCCGCAGCTGC-3′) and MBDin-10 (5′-AGTCGTCGACGATTTATTGTTTGTCTTCCAGTATTG-3′); for pGFPMBDin, MBDin-12 (5′-AGTCAGATCTGCGGCGTCCGCAGCTGCCGCT-3′) and MBDin-7 (5′-AGTCGAATTCCAAGTGAAGTGTGCTAAAGTCTC-3′); for pGFPMBD2, MBD2-20 (5′-AGTCGAATTCGATGGATTGCCCGGCCCTCCC C-3′) and MBD2-2. To construct pMyc-MBD2 and pHA-MBDin expression vectors, PCR-generated full-length MBD2 and MBDin fragments were cloned into pcDNA3.1A (Invitrogen) and pCEFL-HA (11) plasmids, respectively. Primers containing restriction sites (underlined) were as follows: for pMyc-MBD2, MBD2-4 and MBD2-5 (5′-AGTCGGATCCGGCTTCATCTCCACTGTCCAT-3′), and for pHA-MBDin, MBDin-3 (5′-AGTCGAATTCATGGCGGCGTCCGCAGCTGC-3′) and MBDin-2. All PCR-derived products as well as subcloning junctions were verified by sequence analysis. Site-specific mutagenesis of MBDin was obtained by using a QuikChange kit (Stratagene). Mutagenic oligonucleotides were as follows: for pGFP-MBDinLV/AA, MU1 (5′-GGTACTGGATTAGATGAAGCCTTTGCGCAAGTTACCAGTGC-3′) and ML1 (5′-GCACTGGTAACTTGCGCAAAGGCTTCATCTAATCCAGTACC-3′); for pGad-MBDinΔGKT, MU2 (5′-GTGTTGGGAATGGCGGGATCCACTTTTGTACAGAGGCTCACAG-3′) and ML2 (5′-CTGTGAGCCTCTGTACAAAAGTGGATCCCGCCATTCCCAACAC-3′); and for pGad-MBDinK/N, MU3 (5′-GGCGGGATCCGGGAACACCACTTTTGTACAG-3′) and ML3 (5′-CTGTACAAAAGTGGTGTTCCCGGATCCCGCC-3′).
Yeast two-hybrid assay.
We performed yeast two-hybrid screens by yeast mating according to the manufacturer's instructions (Clontech). In the screens we used a human HeLa S3 cell line cDNA library pretransformed in Saccharomyces cerevisiae strain Y187 cloned downstream of the Gal4AD in pGADGH (Clontech). We cloned “bait” sequences downstream of the Gal4BD in the pBridge vector (Clontech), transformed the PJ692A S. cerevisiae strain by the lithium acetate method, and then treated it according to the manufacturer's instructions. The diploid strains were plated on high-stringency synthetic medium lacking histidine, adenine, tryptophan, and leucine. His+ Ade+ diploid colonies were patched on selective plates and assayed for β-galactosidase activity using a colony-lift filter assay (9). Total DNA was prepared from colonies displaying a His+ Ade+ LacZ+ phenotype, and pGADGH library plasmids were then rescued after transformation of a HB101 Leu− Escherichia coli strain. The isolated pGADGH library plasmids were tested for specificity by cotransformation into PJ692A either alone or in combination with the pBridgeMBD2 construct or in combination with plasmids containing different unrelated baits (syntenin, HMGA1, and galectin-1 cDNAs) fused to sequences encoding the Gal4BD. The cDNA inserts from specific clones were sequenced by the dideoxy termination method (38). For measurement of protein interaction, plasmids were cotransformed into yeast strain Y187 and plated on synthetic medium lacking tryptophan and leucine. After 4 days of growth, colonies were inoculated into liquid synthetic dextrose medium lacking Leu and Trp for 24 h, and a liquid assay for β-galactosidase activity was performed according to the manufacturer's instructions (Clontech).
Isolation of a human full-length MBDin cDNA clone.
A human breast carcinoma cDNA library in λgt11 was purchased from Clontech. A total of 2 × 106 recombinant clones were screened by an in situ plaque hybridization technique (37). The 543-bp PstI fragment of pGADGH-MBDin was labeled by a random priming procedure (13) and used as a probe. Hybridizations were carried out at 60°C, and filters were washed under low-stringency conditions (0.6× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]-0.4% sodium dodecyl sulfate at 40°C). Phage DNA was isolated by a small-scale purification procedure (37). Inserts from positive clones were sequenced by using the dideoxy termination method (38).
Northern blots.
A panel with poly(A)+ RNA from mouse embryo and human tissues was purchased from Clontech. A 543-bp PstI fragment of pGadGHMBDin was labeled by a random priming procedure (13) and used as a probe. A mouse β-actin probe was used to control equal RNA loading. Hybridizations were carried out as described previously (37).
Cell culture, transient transfections, and CAT assays.
293T, HeLa, and NIH 3T3 cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum (calf serum for NIH 3T3) (Life Technologies). Cells were plated at a density of about 250,000 per 60-mm petri dish 16 h before transfections. DNA transfections were carried out by calcium phosphate precipitation using Calphos (Clontech) or Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. For transcription assays, cultures were cotransfected with test plasmids (5 μg) and different amounts of effector plasmids as indicated below. For normalization of transfection efficiencies, a β-galactosidase expression plasmid, pSVβ-gal (Promega) (1 μg), was included in the cotransfection mixtures as an internal standard for transfection efficiency. Chloramphenicol acetyltransferase (CAT) assays were performed with different amounts of extracts to ensure linear conversion of the chloramphenicol with each extract, and results are presented as the means of at least three independent transfection experiments. CAT activity was quantified with the Molecular Dynamics PhosphorImager system. Total protein extracts (5 μg) were assayed for β-galactosidase activity as previously described (25).
Immunoprecipitation and immunoblotting.
293T and HeLa cell extracts were prepared in Nonidet P-40 lysis buffer (1% Nonidet P-40, 50 mM Tris-HCl [pH 8.0], 150 mM NaCl) with proteinase inhibitors (Roche Molecular Biochemicals). Immunoprecipitations were performed as described previously (11). The samples were immunoprecipitated with rabbit polyclonal antihemagglutinin (anti-HA) antibodies (Roche Molecular Biochemicals) and mouse monoclonal anti-Myc antibodies (Santa Cruz) and resolved by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis. Western blot analyses were performed by using rabbit polyclonal or mouse monoclonal anti-HA (Roche Molecular Biochemicals), anti-Myc, goat polyclonal anti-xeroderma pigmentosum group A protein (XPA), and goat polyclonal anti-MBD2a antibodies (Santa Cruz), diluted 1:500 in Tween-Tris-buffered saline (20 mM Tris-HCl [pH 7.6], 137 mM NaCl, 0.1% Tween 20), and the antigenic proteins were detected with an enhanced chemiluminescence detection system (Amersham Biosciences). Goat polyclonal anti-MBD2a antibodies were selective for the long form of MBD2 (MBD2a; molecular mass, 43.5 kDa). For transcriptional assays, immunoblotting experiments were performed by using mouse monoclonal anti-HA antibody and rabbit polyclonal anti-Gal4 (DNA binding domain [DBD]) (Santa Cruz) antibodies.
Methylation analysis.
pGAT1700 plasmid (50 μg) (36) was treated with 50 U of SssI methylase (New England Biolabs) at 37°C in the presence of 5 mM adenosylmethionine for 8 h. Complete methylation of treated plasmids was confirmed by HpaII restriction enzyme digestion. After transfections, cells were harvested and used for both transcription assays (see above) and methylation analysis. Total DNA was prepared from cells by a standard phenol-chloroform extraction method (37). DNA (20 μg) was digested with HpaII and MspI restriction endonucleases. All digestions were carried out overnight with a three- to fivefold excess of enzyme. Completeness of digestion was verified by adding 1 μg of phage λ DNA to the reaction mixture. Southern blot analysis was performed by a standard procedure (37). The pGAT1700 plasmid was digested with EcoRI, labeled by a random priming procedure (37), and used as a probe. Sodium bisulfite analysis was done essentially as described by Frommer et al. (14). An 8-μg sample of total DNA was digested with EcoRI and denatured in 0.3 M NaOH for 15 min at 37°C in a volume of 100 μl, and then 60 μl of 10 mM hydroquinone and 1.04 ml of 3.6 M sodium bisulfite (pH 5) were added. Reaction mixtures were incubated at 50°C for 16 h in the dark. The DNA was desalted and concentrated using Geneclean (Bio101), denatured with 0.3 M NaOH for 15 min at 37°C, neutralized with 3 M ammonium acetate (pH 7), and ethanol precipitated. An aliquot of DNA was amplified by using the modified primers MG2 (5′-GTGTTAGGATTTTGAGGGAGGGTTAGG-3′) and MG1 (5′-TCCTAAAACCTACTCCACCAACAATCAAAAAACTCC-3′). All PCRs were carried out in 100-μl volumes containing 10 mM Tris, 50 mM KCl, 1 mM MgCl2, 5% dimethyl sulfoxide, 0.2 mM deoxynucleotide triphosphates, 10 pmol of each primer, and 2 U of Taq polymerase (Stratagene). The amplification cycles were as described previously (14). The amplified fragments were cloned into the pCRII vector of the TA cloning system (Invitrogen), and then at least 20 independent clones for each fragment were sequenced by using the T7 primer to determine the methylation pattern of individual molecules.
Fluorescence imaging.
A 5-μg sample of each expression construct was transfected into NIH 3T3 cells by using Lipofectamine Plus reagents (Invitrogen) on a 2-cm2 coverslip of cells at 50% confluence according to the manufacturer's instructions. Leptomycin B (LMB) was a gift from Minoru Yoshida. At 48 h after transfection, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline for 20 min at 37°C and then washed twice in phosphate-buffered saline. Cells were then incubated in phosphate-buffered saline containing 5 μg of 4′,6-diamidino-2-phenylindole (DAPI) per ml for 30 min at 37°C and then washed twice in phosphate-buffered saline. Coverslips were mounted with 4.8% Mowiol in 50 mM Tris (pH 8.5). Images were obtained using a Zeiss Axioskop MC 100 fitted with a camera, and all the images were acquired with a 40× Plan-Neofluar objective. Images were subjected to scale adjustment by using image software (Photoshop; Adobe Systems, Inc., San Diego, Calif.).
RESULTS
Interaction screening of a human HeLa cDNA library with MBD2.
In order to identify genes capable of encoding proteins specifically interacting with MBD2 we carried out a yeast two-hybrid screening of a HeLa cDNA library. A total of 3 × 106 clones were tested using the short form of MBD2 cDNA, MBD2b (18), as bait. Approximately 120 independent cDNA clones exhibited His+ Ade+ reporter gene activation. A following step, consisting of the activation of the β-galactosidase reporter gene, reduced to 60 the number of positive clones. The majority of the positive clones contained a 1,791-bp insert that revealed the presence of an entirely open reading frame of 1,125 bp, leaving 15 and 651 nucleotides of 5′ and 3′ untranslated sequences, respectively. A consensus Kozak sequence (23) was found around the initiation codon. To be sure that we isolated the full-length cDNA of MBDin with its initiator ATG, we screened a full-length human breast carcinoma cDNA library to obtain larger cDNA clones. All the positive clones obtained revealed that the 5′ untranslated sequence contained 21 nucleotides, and no additional ATG codon was present. The open reading frame encoded a polypeptide of 374 amino acids with a predicted molecular mass of 41 kDa. We termed this protein MBDin (for “MBD2 interactor”). A search against the databases revealed that the cDNA of MBDin was identical to XAB1 (DDBJ/EMBL/GenBank accession no. AB044661), a GTPase previously identified as interacting partner of XPA (31). The N-terminal region of this protein contains the consensus sequence for a GTP-binding site (GMAGSGKT) within a large amino acid region conserved among GTPases (Fig. 1) (41). Moreover, we identified a potential nuclear export signal (NES) consensus sequence (Fig. 1), which is contained in several proteins that share the ability to shuttle between the nucleus and cytoplasm (28, 47). The C-terminal region of MBDin is characterized by an acidic domain containing a cluster of acidic amino acid residues (Fig. 1). MBDin mRNA is ubiquitously expressed in different adult organs as a major transcript of 2.1 kb and two minor transcripts of approximately 2.9 and 1.4 kb (data not shown). We investigated the expression of MBDin during development. Northern blot analysis of mouse embryos at different days postcoitus showed that MBDin was abundantly expressed as a single transcript of 2.1 kb at all developmental stages, with a peak at 11 days postcoitus (Fig. 2).
FIG. 1.

Characteristics of the MBDin protein. The complete predicted amino acid sequence of the human MBDin protein is shown; the complete human cDNA sequence is available under DDBJ/EMBL/GenBank accession number AB044661. The gray shading represents the region conserved among GTP-binding proteins, the N-terminal consensus sequence for the GTP-binding site (amino acids 26 to 33) and the NES motif (amino acids 247 to 256) are boxed, and the C-terminal acidic region is underlined.
FIG. 2.

Expression of the MBDin gene in mouse embryos. The Northern blot of poly(A)+ RNA (2 μg) from mouse embryos at different days postcoitus was hybridized with the MBDin cDNA probe. The relative abundance of the RNA samples was determined by hybridization with β-actin probe (β-act). The mobility of RNA size standards is shown on the left of the blot.
Interaction of MBD2 deletion mutants with MBDin.
To narrow the regions of MBD2 and MBDin that promoted the interaction between these two proteins, portions were cloned into the yeast two-hybrid vectors and tested for interaction. As shown in Fig. 3, deletion of the N-terminal 103 amino acids (pBridge-ΔMBD2) of MBD2, comprehending the MBD, did not affect the ability of MBD2 to associate with MBDin, demonstrating that the MBD is dispensable for the interaction. The further removal of 100 or 46 amino acids at the C terminus (pBridge-MUT1 and pBridge-MUT2, respectively) abolished the capacity of MBD2 to bind MBDin, showing that the C-terminal region is necessary for the interaction. An MBD2 deletion mutant lacking both N-terminal and C-terminal amino acids (pBridge-MUT4) was also unable to associate with MBDin. The ability of pBridge-Mut3 to associate with MBDin indicated that the C-terminal 53-amino-acid region of MBD2 is sufficient for the interaction with MBDin.
FIG. 3.

Mapping of MBD2 and MBDin binding region. (A) Schematic representation of structural and functional domains of MBD2 and MBDin proteins. For MBD2, the MBD is indicated as a black box; the putative coiled-coil domain is indicated as a gray box. For MBDin, the black box indicates the GTP-binding site, the dotted box indicates the region of homology with GTPases, the gray box indicates the NES motif, and the hatched box indicates the C-terminal region containing a cluster of acidic amino acid residues. (B) Mapping of the MBDin-binding region of MBD2 by yeast two-hybrid experiments. Full-length MBD2 and deletion mutants were fused to the Gal4 DBD (bait), and full-length MBDin was fused to the Gal4AD (prey). The interaction between the bait proteins and prey was tested on the basis of the His and Ade reporter gene activation and by quantitative liquid β-galactosidase assay. (C) Mapping of the MBD2-binding region of MBDin by yeast two-hybrid experiments. Full-length MBDin and mutants were fused to the Gal4 DNA activation domain, and full-length MBD2 was fused to the Gal4BD. The interaction between the bait proteins and prey was tested as described above.
We also investigated the ability of different MBDin deletion mutants to interact with MBD2 (Fig. 3C). Deletion of the MBDin acidic C-terminal 128-amino-acid region (pGad-MBDinΔC128), as well as deletion of the N-terminal 30 amino acids (pGad-MBDinΔN30), abolished the ability of MBDin to associate with MBD2, demonstrating that both N-terminal and C-terminal regions are necessary for the interaction. Because the N-terminal region includes a GTP-binding site, we investigated whether changes of amino acid residues critical for the GTP binding (27) were able to abolish the interaction. Results showed that both deletion of GKT (amino acids 31 to 33) (pGad-MBDinΔGKT) and alteration of lysine 32 to asparagine (pGad-MBDinK/N) inhibited the binding to full-length MBD2. These results show that both the integrity of the GTP-binding site and the presence of the C-terminal region of MBDin are necessary for the interaction.
Immunoprecipitation experiments.
Coimmunoprecipitation experiments were performed to investigate whether MBD2 and MBDin form a complex in mammalian cells. 293T cells were cotransfected with expression vectors containing the Myc-tagged full-length MBD2 cDNA (pMyc-MBD2) and the HA-tagged full-length MBDin cDNA (pHA-MBDin). Subsequently, cell lysates were immunoprecipitated with anti-Myc and anti-HA antibodies, respectively. The immunoprecipitated fractions were then immunoblotted and hybridized with both anti-HA and anti-Myc antibodies. As shown in Fig. 4A, MBDin and MBD2 coimmunoprecipitated when both expression plasmids were transfected. Empty vectors were also cotransfected as negative controls. Moreover, because HeLa cells express at high levels the long form of MBD2 (MBD2a) (reference 18 and data not shown), we also checked whether the endogenous protein could associate with MBDin. We found that in HeLa cells, endogenous MBD2a coimmunoprecipitates with HA-MBDin (Fig. 4B). These results indicate that MBD2 and MBDin form a complex in mammalian cells.
FIG. 4.
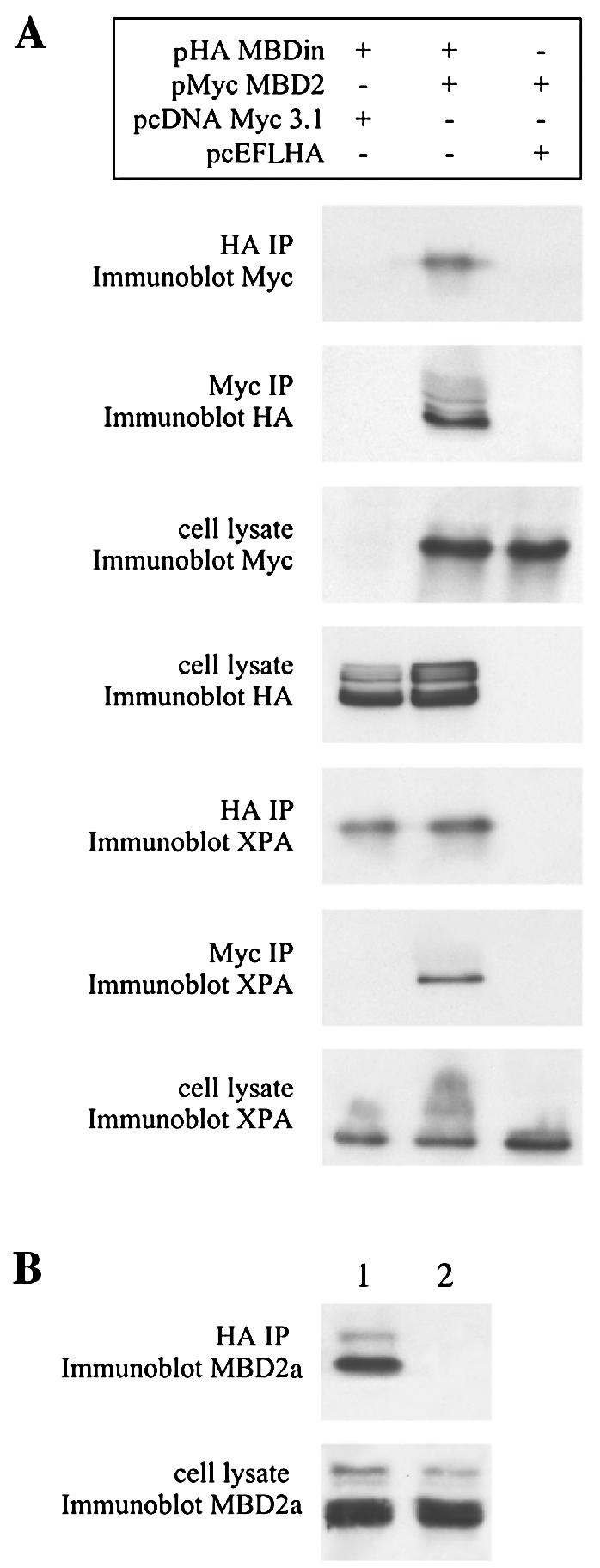
In vivo association of MBD2 with MBDin and XPA. (A) pHA-MBDin and pMyc-MBD2 and control empty vectors were transfected in 293T cells in the indicated combinations. After 48 h in culture, whole-cell extracts were prepared and subjected to immunoprecipitation with rabbit polyclonal anti-HA and mouse monoclonal anti-Myc antibody. Immunoprecipitated (IP) proteins were analyzed by immunoblotting with mouse monoclonal anti-Myc antibody, rabbit polyclonal anti-HA antibody, or anti-XPA antibody. Cell lysates (5%) were subjected to immunoblotting prior to immunoprecipitation. (B) HeLa cells were transfected with pHA-MBDin (lane 1) and pCEFL-HA empty vector (lane 2). Immunoprecipitations were carried out with rabbit polyclonal anti-HA antibody. Immunoprecipitated proteins and cell lysates (5%) were analyzed by immunoblotting with goat polyclonal anti-MBD2a antibody.
Because MBDin (XAB1) has been recently found to associate with XPA (31), we also tested the possibility that XPA belongs to the same complex as MBD2. Since XPA is expressed at high levels in 293T cells (Fig. 4), we investigated the presence of XPA in the immunoprecipitated fractions described above. We found that when all the three proteins were present in the extracts, XPA was detected when MBD2 (Myc) or MBDin (HA) was immunoprecipitated, while XPA was not detected when only MBD2 was transfected (Fig. 4). These results indicate that MBDin, MBD2, and XPA belong to same complex and that MBDin might function as a bridge.
Subcellular localization of MBDin and MBD2.
Subcellular localization of MBDin was studied by transfecting NIH 3T3 cells with a mammalian expression vector encoding the GFP-MBDin fusion protein (pGFPMBDin). GFP-MBDin was detected mainly in the cytoplasm, but nuclear staining was also observed (Fig. 5A). These data are in agreement with previously reported immunofluorescence and immunoblotting experiments on the endogenous XAB1 protein in HeLa cells (31).
FIG. 5.

Subcellular localization of MBD2 and MBDin. NIH 3T3 cells were transiently transfected with GFP-MBDin in the absence (A) or presence (B) of LMB and with GFP-MBDinLV/AA (C). (D) Subcellular localization of RFP-MBD2 (left) and DAPI staining (right) of the same nuclei. (E) NIH 3T3 cells were cotransfected with RFP-MBD2 and GFP-MBDin; RFP and GFP fluorescence indicate the location of MBD2 and MBDin fusion proteins (right and left, respectively). (F) NIH 3T3 cells were cotransfected with RFPΔ-MBD2 and GFP-MBDin. RFP and GFP fluorescence indicate the location of MBD2Δ and MBDin fusion proteins (right and left, respectively).
From the analysis of the MBDin amino acid sequence we found the presence of a potential NES at amino acids 247 to 256 (Fig. 1). The NES is a conserved stretch of amino acids, displayed by a number of proteins that share the ability to shuttle between the nuclear and cytoplasmic compartments (28, 47). NES-mediated transport is inhibited by LMB (47). Thus, we investigated the localization of GFP-MBDin in the presence of LMB. Our results show that 24 h after addition of 20 nM LMB, GFP-MBDin accumulated in the nucleus (Fig. 5B), indicating the NES-dependent nuclear export of MBDin and a possible role of MBDin in the nucleus. To confirm the role of the putative NES, two point mutations converting two critical hydrophobic residues (45), leucine 254 and valine 256, of MBDin to alanines were introduced in the GFP-MBDin fusion protein (plasmid pGFP-MBDinLV/AA). The GFP-MBDinLV/AA mutant accumulated in the nucleus (Fig. 5C), showing that nuclear export was significantly impaired.
We then investigated the possible colocalization of MBDin and MBD2. First, we transfected NIH 3T3 cells with an RFP-MBD2 fusion protein expression vector (pRFPMBD2). In agreement with previous studies (18), RFP-MBD2 colocalized with foci of heavily methylated satellite DNA, which in mouse cells corresponds to regions of the nucleus that stain brightly with DAPI (Fig. 5D). When GFP-MBDin and RFP-MBD2 were cotransfected, both proteins localized mainly with major satellite DNA, suggesting that MBDin is recruited on hypermethylated DNA regions when MBD2 is abundantly expressed (Fig. 5E). Similar results were obtained when we used an opposite combination (RFP-MBDin and GFP-MBD2) of fusion proteins (data not shown). In order to confirm that amino acids 216 to 262 of MBD2 are essential for the interaction with MBDin in vivo (Fig. 3), we prepared an RFP-ΔMBD2 expression vector, lacking the MBDin interacting region and still containing the MBD (pRFPΔMBD2). pRFPΔMBD2 was transfected alone or together with pGFPMBDin. RFP-ΔMBD2 localized in nuclear hypermethylated regions (Fig. 5F, left), but this truncated protein failed to recruit the overexpressed MBDin, as shown by the diffuse localization of MBDin in double transfectants (Fig. 5F, right).
MBDin reactivates MBD2-repressed promoters.
To explore whether MBDin could affect the transcriptional repression potential of MBD2, we performed transient-transfection assays in human 293T cells using the G5-Myc-XDN and the pG5-simian virus 40 promoter-reporter constructs, bearing five Gal4 DNA binding sites upstream from the c-Myc P2 and simian virus 40 minimal promoters, respectively (11, 26). The Gal4-MBD2 fusion protein repressed the c-Myc promoter activity in a dose-dependent manner (Fig. 6 and data not shown), in agreement with previously reported data on the β-globin promoter (18). To determine whether MBDin could affect the MBD2 repression potential, cotransfections of pGal4-MBD2 along with increasing amounts of pHA-MBDin expression vector were performed. The results presented in Fig. 6 show that MBDin is able to relieve MBD2-mediated repression even at a low concentration. At higher MBDin concentrations, the promoter activity was significantly higher than basal levels, indicating that MBDin behaves as a transcriptional activator. Such activity was specifically related to the presence of MBD2, because no significant changes of reporter expression were observed in the absence of Gal4-MBD2. The above findings could not be explained by a decreased cellular concentration of MBD2, because immunoblotting analysis of cell extracts showed that increasing amount of MBDin did not alter MBD2 concentration (Fig. 6). Similar results were obtained when a pG5-SV40 promoter-reporter construct was employed (data not shown). These results indicate that MBDin can relieve MBD2-mediated repression.
FIG. 6.

MBDin reactivates transcription from MBD2-repressed c-Myc promoter. The pG5-XΔN reporter plasmid (5 μg) was cotransfected into 293T cells with Gal4-MBD2 (5 μg) and/or HA-MBDin expression plasmids in the indicated amounts. The level of transcription is given as a percentage of the CAT activity seen in the absence of expression plasmids. CAT activity was quantitated as described in Materials and Methods. The results are averages of at least three independent experiments plus standard deviations. A 20-μg sample of each cell extract was resolved on a 12% polyacrylamide gel under denaturing conditions and subjected to immunoblotting with anti-Gal4 (DBD) and anti-HA antibodies.
MBD2 and MBDin activity on methylated promoters.
Next we questioned whether MBDin could reactivate transcription from methylated promoters. For this purpose we used a plasmid construct containing 1,700 bp of the mouse galectin-1 promoter upstream of a CAT reporter gene (pGAT1700) (36). It was previously demonstrated that galectin-1 promoter constructs are repressed by in vitro DNA methylation (4). Thus, we performed transcription assays after transfection of MBD2 and MBDin expression vectors along with either unmethylated or in vitro-methylated pGAT1700 in 293T cells. The unmethylated pGAT1700 was highly active, while the methylated form was almost completely repressed (Fig. 7). The overexpression of MBD2 caused a further repression of the methylated pGAT1700 activity but did not influence the activity of the unmethylated plasmid. When MBDin was overexpressed, no significant changes in the reporter expression, either from methylated or unmethylated constructs, was observed. In contrast, when both MBD2 and MBDin were transfected, MBDin clearly reactivated, in a dose-dependent manner, the methylated pGAT1700, indicating that MBDin can be recruited by MBD2 on the methylated promoter and is able to reactivate transcription.
FIG. 7.

Influence of MBD2 and MBDin overexpression on nonmethylated and methylated promoter activity. 293T cells were transfected with 5 μg of nonmethylated (A) or in vitro-methylated (B) pGAT1700 reporter plasmid. Cotransfected expression plasmids were pCMVβ (1 μg) and, where indicated, pMyc-MBD2 (3 μg) and pHA-MBDin in different amounts. The level of transcription is given as a percentage of the CAT activity seen in the presence of unmethylated pGAT1700 reporter plasmid alone. Percent relative activity of the promoters is expressed as the ratio of CAT to β-galactosidase activity. The results are averages of multiple experiments plus standard deviations. Each bar represents the mean of four independent transfections.
Because MBD2 has been described to function as a transcriptional repressor (30) or as a demethylase (5), we investigated whether the MBDin-mediated reactivation was accompanied by demethylation of the reporter construct, implying that MBDin triggered the MBD2 demethylation activity, or whether the reactivation occurred without changes of the promoter methylation pattern. For this purpose we analyzed the general state of methylation of the reporter plasmid by performing a Southern blot analysis in parallel with the transcription assays. Total DNA was extracted from 293T cells 48 h after transfections and digested with either methylation-sensitive HpaII or methylation-insensitive MspI. Comparison of hybridization signals obtained by probing digested DNA indicated that pGAT1700 did not undergo demethylation at CCGG sites even when both MBD2 and MBDin were present (data not shown), a condition in which transcription of the CAT reporter gene was reactivated (Fig. 7).
Because we previously demonstrated that changes in the methylation pattern of a small region around the transcriptional start site account for the galectin-1 promoter activity, we analyzed at high resolution the methylation pattern of a 188-bp region of the transfected mouse galectin-1 promoter covering the region from −177 to +11. This region contains seven methylatable CpG sites. Sodium bisulfite analysis using mouse-specific primers demonstrated that the seven CpG sites are almost fully methylated 48 h after transfection and no significant changes in their methylation state occurred when MBD2 alone or in combination with MBDin was transfected (Fig. 8).
FIG. 8.

Percentage of DNA methylation at individual CpG sites in the transfected galectin-1 promoter. 293T cells were transfected with in vitro-methylated pGAT1700 alone (black bars), together with pMyc-MBD2 (gray bars), or together with pMyc-MBD2 and pHA-MBDin (hatched bars). Total DNA from each sample was extracted 48 h after transfections, treated with sodium bisulfite, and amplified with the MG2 and MG1 primers (positions −206 to −177 and +11 to +45 relative to the galectin-1 transcription initiation site, respectively). Amplified fragments were cloned into the pCRII vector. Data obtained from sequencing with the T7 primer of at least 20 plasmid clones for each sample were compiled by individual CpG dinucleotides and expressed as the ratio of methyl-C to the number of clones. Positions of CpG sites relative to transcription initiation site are given below the bars.
These results show that MBDin, in the presence of MBD2, can reactivate transcription from a methylated promoter without altering the methylation pattern.
DISCUSSION
In this paper we describe a novel GTPase, MBDin, that binds to MBD2 and reverses MBD2-mediated transcriptional repression from methylated promoters without altering the promoter methylation pattern.
Several recent studies have revealed that MBD2 represses transcription through its ability to tether corepressor complexes on methylated DNA (1, 30, 43, 48). Two partially overlapping domains have been identified at the N terminus of MBD2, a methyl-CpG binding domain and a transcriptional repressor domain that recruits HDAC activity to bring about transcriptional silencing (8, 18, 30, 48). All the MBD2 partners described so far interact with the N-terminal region of MBD2 and mediate MBD2 transcriptional repression (30, 39, 48). Conversely, the novel MBD2 partner described in this paper, MBDin, associates with the extreme C-terminal region of MBD2 and relieves transcriptional repression. Interestingly, the MBD2 C-terminal region is unique among the known methyl-binding proteins and is the only region that clearly differs between MBD2 and the very similar MBD3 (18).
MBDin and MBD2 are both expressed from early stages during development and ubiquitously in human adult tissues (reference 18 and this paper), suggesting their role in fundamental cell functions. MBDin is a nucleocytoplasmic GTPase, while MBD2 displays a distinct subnuclear localization (18). Although these two proteins localize mainly in distinct cell compartments, they partially colocalize at foci of heavily methylated satellite DNA, demonstrating a possible cooperation in interpreting the DNA methylation signal. It is worth noting that MBDin contains a putative NES (Fig. 1) and that our data showed that MBDin accumulates in the nucleus upon treatment with LMB, a drug that inhibits NES-mediated transport (47). In addition, we found that replacement of two critical hydrophobic residues in the NES consensus sequence of MBDin impaired the export of MBDin from the nucleus (Fig. 5C), as has also been demonstrated for other NES-containing proteins (45). These observations indicate that MBDin contains a functional NES and is subject to regulated nucleocytoplasmic shuttling and reinforce the hypothesis that, under certain conditions, MBDin may regulate the MBD2 functions in the nuclear compartment.
It was recently reported that MBDin (XAB1) associates with XPA (31), a nuclear protein that plays a central role in nucleotide excision repair (NER). In this case, the deletion of five amino acids required for nuclear localization of XPA abolishes interaction with XAB1, suggesting that this protein is involved in nuclear transport of XPA. The interaction of XPA with MBDin, which in turn associates with MBD2, and the observation that these three proteins belong to the same complex (Fig. 4) raise the intriguing possibility that NER and DNA methylation systems are linked. Interestingly, another methyl-binding protein, MBD4, interacts with MLH1 and is directly involved in the DNA mismatch repair system (3, 34). The possibility of a role for MBDin and MBD2 in NER deserves further investigation.
Although additional functions for MBDin can be possible, our data clearly show that this protein affects the transcriptional function of MBD2. In particular, the novelty of our findings is that MBDin reactivates methylated promoters on episomal DNA and such reactivation occurs only in the presence of MBD2. Whether this mechanism is operative in the regulation of endogenous methylated chromosomal genes remains to be investigated.
According to the current models, two possible mechanisms account for the reactivation of previously silent methylated genes: (i) the promoter can be demethylated (5); (ii) the methyl-binding proteins, and, as a consequence, the associated repressor complex, can be displaced from methylated DNA (20). MBD2 is involved in both mechanisms. In fact, MBD2 has been described to actively demethylate DNA (5), and, very recently, Hutchins et al. (20) demonstrated that MBD2 can be displaced from a methylated promoter by the GATA-3 transactivator. In the latter case, the transactivator displaces the repressor complex, including MBD2, from methyl-CpG sites, leading to transcriptional activation even prior to demethylation. In the case of MBDin-mediated reactivation, as well as for GATA-3, transcriptional reactivation occurs prior to demethylation. However, we found that MBDin reactivates transcription much better when MBD2 is coexpressed. These data suggest that MBDin is recruited by MBD2 on methylated DNA and possibly acts by interfering with the ability of MBD2 to associate with the repressor complex rather than through displacement of MBD2 from methyl-CpG sites. Our findings, showing that MBDin can relieve MBD2-mediated repression also when MBD2 is artificially tethered to the promoter (Fig. 6), further support this hypothesis.
Although further investigation will be necessary to define the precise mechanisms by which MBDin-mediated reactivation occurs, our data suggest the possible existence of novel factors that, through association with methyl-binding proteins, can lead to the transcriptional control of methylated genes. According to our findings, such control would be exerted not only in a repressive manner, as widely documented, but also by reactivating genes even prior to demethylation. This would confer on methyl-binding proteins a role as real interpreters of DNA methylation patterns rather than simple mediators of transcriptional repression.
Acknowledgments
We thank M. Yoshida (University of Tokyo, Tokyo, Japan) for the kind gift of LMB.
This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC), from the Consiglio Nazionale delle Ricerche (CNR), from the Ministero dell'Università e della Ricerca Scientifica e Tecnologica (MIUR), and from the Ministero della Salute.
REFERENCES
- 1.Ballestar, E., and A. P. Wolffe. 2001. Methyl-CpG-binding proteins: targeting specific gene repression. Eur. J. Biochem. 268:1-6. [DOI] [PubMed] [Google Scholar]
- 2.Bartolomei, M. S., and S. M. Tilghman. 1997. Genomic imprinting in mammals. Annu. Rev. Genet. 31:493-525. [DOI] [PubMed] [Google Scholar]
- 3.Bellacosa, A., L. Cicchillitti, F. Schepis, A. Riccio, A. T. Yeung, Y. Matsumoto, E. A. Golemis, M. Genuardi, and G. Neri. 1999. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc. Natl. Acad. Sci. USA 96:3969-3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benvenuto, G., M. L. Carpentieri, P. Salvatore, L. Cindolo, C. B. Bruni, and L. Chiariotti. 1996. Cell-specific transcriptional regulation and reactivation of galectin-1 gene expression are controlled by DNA methylation of the promoter region. Mol. Cell. Biol. 16:2736-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharya, S. K., S. Ramchandani, N. Cervoni, and M. Szyf. 1999. A mammalian protein with specific demethylase activity for mCpG DNA. Nature 397:579-583. [DOI] [PubMed] [Google Scholar]
- 6.Bird, A. 1992. The essentials of DNA methylation. Cell 70:5-8. [DOI] [PubMed] [Google Scholar]
- 7.Bird, A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6-21. [DOI] [PubMed] [Google Scholar]
- 8.Boeke, J., O. Ammerpohl, S. Kegel, U. Moehren, and R. Renkawitz. 2000. The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J. Biol. Chem. 275:34963-34967. [DOI] [PubMed] [Google Scholar]
- 9.Breeden, L., and K. Nasmyth. 1985. Regulation of the yeast HO gene. Cold Spring Harbor Symp. Quant. Biol. 50:643-650. [DOI] [PubMed] [Google Scholar]
- 10.Detich, N., J. Theberge, and M. Szyf. 2002. Promoter-specific activation and demethylation by MBD2/demethylase. J. Biol. Chem. 277:35791-35794. [DOI] [PubMed] [Google Scholar]
- 11.Fedele, M., G. Benvenuto, R. Pero, B. Majello, S. Battista, F. Lembo, E. Vollono, P. M. Day, M. Santoro, L. Lania, C. B. Bruni, A. Fusco, and L. Chiariotti. 2000. A novel member of the BTB/POZ family, PATZ, associates with the RNF4 RING finger protein and acts as a transcriptional repressor. J. Biol. Chem. 275:7894-7901. [DOI] [PubMed] [Google Scholar]
- 12.Feinberg, A. P. 2001. Cancer epigenetics takes center stage. Proc. Natl. Acad. Sci. USA 98:392-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6-13. [DOI] [PubMed] [Google Scholar]
- 14.Frommer, M., L. E. McDonald, D. S. Millar, C. M. Collis, F. Wat, G. W. Grigg, P. L. Molloy, and C. L. Paul. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 89:1827-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guy, J., B. Hendrich, M. Holmes, J. E. Martin, and A. Bird. 2001. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27:322-326. [DOI] [PubMed] [Google Scholar]
- 16.Hansen, R. S., C. Wijmenga, P. Luo, A. M. Stanek, T. K. Canfield, C. M. Weemaes, and S. M. Gartler. 1999. The Dnmt3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 96:14412-14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heard, E., P. Clerc, and P. Avner. 1997. X-chromosome inactivation in mammals. Annu. Rev. Genet. 31:571-610. [DOI] [PubMed] [Google Scholar]
- 18.Hendrich, B., and A. Bird. 1998. Identification and characterization of a family of mammalian methyl-CpG-binding proteins. Mol. Cell. Biol. 18:6538-6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendrich, B., U. Hardeland, H. H. Ng, J. Jiricny, and A. Bird. 1999. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 401:301-304. [DOI] [PubMed] [Google Scholar]
- 20.Hutchins, A. S., A. C. Mullen, H. W. Lee, K. J. Sykes, F. A. High, B. D. Hendrich, A. P. Bird, and S. L. Reiner. 2002. Gene silencing quantitatively controls the function of a developmental trans-activator. Mol. Cell 10:81-91. [DOI] [PubMed] [Google Scholar]
- 21.Jones, P. A., and P. W. Laird. 1999. Cancer—epigenetics comes of age. Nat. Genet. 21:163-167. [DOI] [PubMed] [Google Scholar]
- 22.Jones, P. A., and S. B. Baylin. 2002. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 6:415-428. [DOI] [PubMed] [Google Scholar]
- 23.Kozak, M. 1987. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15:8125-8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, E., T. H. Bestor, and R. Jaenisch. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915-926. [DOI] [PubMed] [Google Scholar]
- 25.Majello, B., P. De Luca, G. Hagen, G. Suske, and L. Lania. 1994. Different members of the Sp1 multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res. 22:4914-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majello, B., P. De Luca, G. Suske, and L. Lania. 1995. Differential transcriptional regulation of c-myc promoter through the same DNA binding sites targeted by Sp1-like proteins. Oncogene 10:1841-1848. [PubMed] [Google Scholar]
- 27.Marks, B., M. H. B. Stowell, Y. Vallis, I. G. Mills, A. Gibson, C. R. Hopkins, and H. T. McMahon. 2001. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410:231-234. [DOI] [PubMed] [Google Scholar]
- 28.Moroianu, J. 1999. Nuclear import and export pathways. J. Cell. Biochem. Suppl. 32-33:76-83. [DOI] [PubMed] [Google Scholar]
- 29.Ng, H. H., and A. Bird. 1999. DNA methylation and chromatin modification. Curr. Opin. Genet. Dev. 9:158-163. [DOI] [PubMed] [Google Scholar]
- 30.Ng, H. H., Y. Zhang, B. Hendrich, C. A. Johnson, B. M. Turner, H. Erdjument-Bromage, P. Tempst, D. Reinberg, and A. Bird. 1999. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 23:58-61. [DOI] [PubMed] [Google Scholar]
- 31.Nitta, M., M. Saijo, N. Kodo, T. Matsuda, Y. Nakatsu, H. Tamai, and K. Tanaka. 2000. A novel cytoplasmatic GTPase XAB1 interacts with DNA repair protein XPA. Nucleic Acids Res. 28:4212-4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Razin, A. 1998. CpG methylation, chromatin structure and gene silencing—a three-way connection. EMBO J. 17:4905-4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Razin, A., and H. Cedar. 1994. DNA methylation and genomic imprinting. Cell 77:473-476. [DOI] [PubMed] [Google Scholar]
- 34.Riccio, A., L. A. Aaltonen, A. K. Godwin, A. Loukola, A. Percesepe, R. Salovaara, V. Masciullo, M. Genuardi, M. Paravatou-Petsotas, D. E. Bassi, B. A. Ruggeri, A. J. Klein-Szanto, J. R. Testa, G. Neri, and A. Bellacosa. 1999. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 23:266-268. [DOI] [PubMed] [Google Scholar]
- 35.Sadovsky, Y., P. Webb, G. Lopez, J. D. Baxter, P. M. Fitzpatrick, E. Gizang-Ginsberg, V. Cavailles, M. G. Parker, and P. J. Kushner. 1995. Transcriptional activators differ in their responses to overexpression of TATA-box-binding protein. Mol. Cell. Biol. 15:1554-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salvatore, P., C. Contursi, G. Benvenuto, C. B. Bruni, and L. Chiariotti. 1995. Characterization and functional dissection of the galectin-1 gene promoter. FEBS Lett. 373:159-163. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 38.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sekimata, M., A. Takahashi, A. Murakami-Sekimata, and Y. Homma. 2001. Involvement of a novel zinc finger protein, MIZF, in transcriptional repression by interacting with a methyl-CpG-binding protein, MBD2. J. Biol. Chem. 276:42632-42638. [DOI] [PubMed] [Google Scholar]
- 40.Siegfried, Z., and H. Cedar. 1997. DNA methylation: a molecular lock. Curr. Biol. 7:305-307. [DOI] [PubMed] [Google Scholar]
- 41.Sprang, S. R. 1997. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66:639-678. [DOI] [PubMed] [Google Scholar]
- 42.Tatemat, K. I., T. Yamazaki, and F. Ishikawa. 2000. MBD2-MBD3 complex binds to hemi-methylated DNA and forms a complex containing DNMT1 at the replication foci in late S phase. Genes Cells 5:677-688. [DOI] [PubMed] [Google Scholar]
- 43.Wade, P. A. 2001. Methyl CpG-binding proteins and transcriptional repression. Bioessays 23:1131-1137. [DOI] [PubMed] [Google Scholar]
- 44.Wade, P. A., A. Gegonne, P. L. Jones, E. Ballestar, F. Aubry, and A. P. Wolffe. 1999. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet. 23:62-66. [DOI] [PubMed] [Google Scholar]
- 45.Wen, W., J. L. Meinkoth, R. Y. Tsien, and S. S. Taylor. 1995. Identification of a signal for rapid export of proteins from the nucleus. Cell 82:463-473. [DOI] [PubMed] [Google Scholar]
- 46.Yolder, J. A., C. P. Walsh, and T. H. Bestor. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13:335-340. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida, M., and S. Horinouchi. 1999. Trichostatin and leptomycin. Inhibition of histone deacetylation and signal-dependent nuclear export. Ann. N. Y. Acad. Sci. 886:23-36. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, Y., H. H. Ng, H. Erdjument-Bromage, P. Tempst, and A. Bird. 1999. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13:1924-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]