

Abstract
Limited information is available regarding the frequency, spectrum, and clinical relevance of somatic mutations in the developing fetus. The goal of this study was to determine somatic mutant frequencies (Mfs) at the hypoxanthine phosphoribosyltransferase (HPRT) reporter gene in cord blood T lymphocytes from preterm infants to gain insight into in utero mutational events. Mf determinations were made by using the HPRT T cell cloning assay on cord blood samples from 52 preterm infants. Natural logarithm Mfs (lnMfs) from preterm infants were compared with results from our database for full-term infants. Our analysis revealed higher lnMfs in cord blood T lymphocytes from preterm compared with full-term infants (P = 0.008). In addition, preterm females had significantly higher lnMfs compared with full-term females (P < 0.001), whereas preterm males were found to have significantly lower lnMfs than preterm females (P = 0.005). Regression analyses also demonstrate a significant relationship between lnMf and gestational age for preterm females that does not exist for preterm males. These results demonstrate the gender-specific association between Mf and age in humans.
Little is known about the clinical effects of spontaneous somatic mutations during fetal development. During embryogenesis, somatic mutational events are influenced by a number of unique factors that are not operative in adults, specifically high rates of DNA replication associated with cellular proliferation, as well as variable expression of enzymes that metabolize genotoxic compounds, V(D)J recombinase and DNA-damage repair enzyme systems. Another unique aspect of embryogenesis is that there are a number of gender-specific developmental processes that occur in utero that appear to have clinical relevance. Specifically, gender-specific in utero developmental processes have been described for neural differentiation (1), growth (2), lung maturation (3), cardiac development (4), and glutathione-reductase expression (5). Investigations of genetic events that occur in utero are critical because mutations during this time have been directly associated with diseases in children, in particular cancer, and also may contribute to multigenetic events associated with adult-onset diseases such as diabetes, autoimmune diseases, and cancer. These studies also may provide insight into gender-specific differences seen with disease pathogenesis.
Previously we have used the hypoxanthine phosphorybosiltransferase (HPRT) T cell cloning reporter gene assay to determine the in vivo frequency and spectrum of somatic mutational events in healthy infants and children (6–8). We have demonstrated an age-specific increase in spontaneous mutant frequency (Mf) in children from birth to 17 years of age that is significantly different from that seen in adults (6, 9). We also have demonstrated an age-specific HPRT mutational spectrum in children characterized by “illegitimate” V(D)J recombinase-mediated large deletions, which predominates in children less than 5 yr old. Such illegitimate V(D)J recombinase-mediated rearrangements have been shown to have clinical significance because such genetic alterations are associated with pediatric hematopoietic malignancies (10–14). These studies have provided insight into the developmental association between spontaneous somatic mutational events and oncogenesis, as well as contributed a database for future investigations directed at studying the potential age-specific genetic susceptibility of children to genotoxic exposures.
In this study, we have expanded on our previous work and describe the background somatic Mf in cord blood T lymphocytes from 52 infants born prematurely (gestational age < 36 wk). We compared the Mf and unselected cloning efficiency (CE) in these infants to our previously reported data for full-term infants and found that for preterm infants a significantly higher Mf exists that is inversely related to gestational age (GA). The most intriguing observation is that this increase in Mf in preterm infants is solely the consequence of a gender-specific increase in Mf of preterm female infants.
MATERIALS AND METHODS
Study Population and Sample Collection.
Heparinized umbilical cord blood samples from 52 preterm infants (<36-wk gestation) and 63 full-term infants were obtained from the labor and delivery unit of Fletcher Allen Hospital of the University of Vermont College of Medicine. Only blood samples from infants showing no clinical evidence of perinatal infection, systemic illness, or congenital anomaly and whose mothers had no history of systemic or chronic disease, chronic use of medication, or significant genotoxic exposure including radiation were included in this study. Medical and socioeconomic history as well as informed consent were obtained, following the procedure approved by the Committee on Human Research at the University of Vermont. The 63 full-term infants included 60 subjects from our previous study (8) plus three additional infants. Among the preterm infants three subjects had been in our previous study but had gestational ages under 36 wk.
HPRT T Cell Cloning Assay.
The HPRT T cell cloning assay has been described in detail elsewhere (8, 15). Umbilical cord blood samples were diluted with basic RPMI medium 1640 (containing 25 mM Hepes, 25 mM l-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin sulfate), pH 7.2. Mononuclear cells (MNC) were isolated by histopaque density sedimentation (400 × g for 30 min at 20°C) and washed twice with saline G. The isolated MNC were inoculated at 1, 2, and 5 cells/well in 96-well round bottom microtiter plates for determining unselected cloning efficiency (CE). To select HPRT mutant isolates, 3 or 4 × 104 cells/well were plated in the presence of 10 μM 6-thioguanine. The medium used consisted of 55% RPMI medium 1640, 20% HL-1 medium, 5% calf bovine serum, 20% T cell growth factor (TCGF), 0.25 μg/ml of phytohemagglutinin, 100 units/ml of penicillin, and 100 μg/ml of streptomycin sulfate. TCGF is a supernatant medium derived from a lymphokine-activated killer cell therapy containing approximately 1,500 units/ml of recombinant interleukin 2. Each well contained 200 μl of the above medium and ≈104 γ-irradiated 36X4 accessory cells, a B lymphoblastoid cell line in which the HPRT gene is deleted. Microtiter plates were incubated in a humidified atmosphere with 5–6% CO2 at 37°C for 14–21 days and scored for positive colonies.
Statistical Analysis.
CEs were calculated by using the Poisson distribution relationship, Po = e−x, where Po is the fraction of wells without colony growth and x is the average number of clonable cells per well. CE = −ln Po/N, where N is the average number of cells inoculated per well by limited dilution. The mean CE was determined from three different concentration (i.e., n = 1, 2, and 5 cells/well) of unselected cells. The Mf is the ratio of the mean CE in the presence (selected) and absence (unselected) of 6-thioguanine. Confidence intervals (95%) were calculated (Table 1) as described (16).
Table 1.
Summary of GA, sex, CE, Mf, and smoking exposure for preterm and full-term newborns
| Subject number | Sex | GA, weeks | CE | Mf × 10−6 | lnMf × 10−6 | 95% Confidence interval
|
Smoking group* | |
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | |||||||
| Preterm newborns | ||||||||
| PI 4 | F | 26 | 0.24 | 1.04 | 0.04 | −0.86 | 0.94 | I |
| PI 9 | F | 27 | 0.08 | 4.36 | 1.47 | 0.62 | 2.32 | I |
| PS 8 | F | 27 | 0.19 | 2.52 | 0.92 | 0.02 | 1.82 | III |
| PI 31 | F | 29 | 0.01 | 17.41 | 2.86 | 0.08 | 5.63 | III |
| PS 9 | F | 29 | 0.34 | 0.54 | −0.61 | −1.76 | 0.53 | II |
| PI 22 | F | 30 | 0.12 | 1.45 | 0.37 | −1.04 | 1.79 | I |
| PS 25 | F | 30 | 0.06 | 1.16 | 0.15 | −1.29 | 1.58 | I |
| PI 25 | F | 31 | 0.11 | 3.17 | 1.15 | 0.14 | 2.17 | I |
| PI 33 | F | 31 | 0.14 | 0.41 | −0.88 | −2.86 | 1.09 | III |
| PI 41 | F | 31 | 0.16 | 6.54 | 1.88 | −0.10 | 3.85 | I |
| PS 26a | F | 31 | 0.53 | 0.66 | −0.42 | −1.18 | 0.35 | I |
| PS 26c | F | 31 | 0.40 | 0.44 | −0.83 | −2.80 | 1.14 | I |
| PI 36 | F | 32 | 0.19 | 0.78 | −0.24 | −1.40 | 0.91 | I |
| MFS 28 | F | 33 | 0.03 | 1.93 | 0.66 | −1.44 | 2.76 | I |
| PI 3 | F | 33 | 0.10 | 1.74 | 0.55 | −0.86 | 1.97 | IV |
| PI 14 | F | 33 | 0.04 | 17.54 | 2.86 | 1.95 | 3.78 | I |
| PI 1 | F | 34 | 0.05 | 3.98 | 1.38 | 0.34 | 2.43 | II |
| PS 12b | F | 34 | 0.18 | 0.54 | −0.61 | −2.02 | 0.80 | I |
| PS 5 | F | 35 | 0.15 | 2.09 | 0.74 | 0.04 | 1.44 | III |
| PS 14 | F | 35 | 0.36 | 0.41 | −0.89 | −1.89 | 0.11 | IV |
| PS 21 | M | 26 | 0.37 | 1.21 | 0.19 | −0.64 | 1.02 | IV |
| PS 22 | M | 27 | 0.31 | 0.15 | −1.89 | −3.86 | 0.08 | I |
| PI 40 | M | 28 | 0.09 | 1.93 | 0.66 | −1.33 | 2.65 | I |
| PS 30 | M | 29 | 0.25 | 1.84 | 0.61 | −0.04 | 1.26 | I |
| PI 28 | M | 31 | 0.02 | 1.16 | 0.15 | −1.88 | 2.18 | II |
| PS 23 | M | 31 | 0.31 | 0.37 | −0.98 | −2.38 | 0.41 | IV |
| PS 26b | M | 31 | 0.48 | 0.31 | −1.17 | −2.32 | −0.02 | I |
| PS 29 | M | 31 | 0.46 | 0.76 | −0.28 | −1.09 | 0.54 | II |
| PI 8 | M | 32 | 0.17 | 0.91 | −0.09 | −1.10 | 0.92 | II |
| PI 18 | M | 32 | 0.11 | 2.26 | 0.82 | −0.10 | 1.74 | II |
| PI 43 | M | 32 | 0.11 | 1.58 | 0.46 | −1.52 | 2.44 | I |
| PS 18 | M | 32 | 0.16 | 0.49 | −0.71 | −1.87 | 0.44 | II |
| PS 19 | M | 32 | 0.32 | 0.57 | −0.56 | −1.32 | 0.21 | I |
| MFS 104 | M | 33 | 0.49 | 0.36 | −1.03 | −2.18 | 0.11 | I |
| PI 2 | M | 33 | 0.09 | 1.93 | 0.66 | −0.76 | 2.08 | IV |
| PI 12 | M | 33 | 0.17 | 1.54 | 0.43 | −0.75 | 1.61 | I |
| PI 34 | M | 33 | 0.15 | 0.77 | −0.26 | −2.23 | 1.72 | II |
| PI 44 | M | 33 | 0.14 | 1.42 | 0.35 | −0.66 | 1.36 | IV |
| PI 46 | M | 33 | 0.23 | 0.25 | −1.38 | −3.35 | 0.59 | II |
| PS 6 | M | 33 | 0.18 | 0.39 | −0.95 | −2.92 | 1.02 | I |
| PS 10 | M | 33 | 0.07 | 0.52 | −0.66 | −2.08 | 0.77 | II |
| PI 6 | M | 34 | 0.02 | 3.48 | 1.25 | −0.25 | 2.74 | I |
| PI 7 | M | 34 | 0.04 | 1.34 | 0.29 | −1.18 | 1.76 | I |
| PS 16 | M | 34 | 0.10 | 1.92 | 0.65 | −0.07 | 1.37 | IV |
| PS 20 | M | 34 | 0.25 | 0.42 | −0.87 | −2.27 | 0.53 | I |
| MFS 7 | M | 35 | 0.68 | 0.57 | −0.56 | −1.56 | 0.43 | IV |
| PI 10 | M | 35 | 0.08 | 2.18 | 0.78 | −0.65 | 2.21 | I |
| PI 20 | M | 35 | 0.11 | 0.79 | −0.24 | −2.21 | 1.74 | I |
| PS 2 | M | 35 | 0.19 | 0.26 | −1.34 | −3.31 | 0.63 | I |
| PS 7 | M | 35 | 0.44 | 0.37 | −1.00 | −1.47 | −0.54 | I |
| PS 13 | M | 35 | 0.25 | 0.11 | −2.18 | −4.15 | −0.21 | I |
| PS 15 | M | 35 | 0.38 | 0.57 | −0.56 | −1.45 | 0.34 | II |
| Full-term newborns | ||||||||
| MFS 61a | F | 36 | 0.45 | 0.32 | −1.14 | −2.08 | −0.20 | IV |
| PS 32 | F | 36 | 0.07 | 2.69 | 0.99 | 0.08 | 1.89 | II |
| MFS 66 | F | 37 | 0.25 | 0.70 | −0.36 | −2.33 | 1.61 | IV |
| MFS 77 | F | 38 | 0.10 | 0.87 | −0.14 | −1.56 | 1.28 | I |
| MFS 94 | F | 38 | 0.47 | 0.14 | −1.97 | −3.38 | 0.56 | IV |
| MFS 111 | F | 38 | 0.36 | 1.30 | 0.26 | −0.88 | 1.41 | II |
| MFS 8 | F | 39 | 0.57 | 0.50 | −0.69 | −1.58 | 0.20 | I |
| MFS 29 | F | 39 | 0.29 | 0.40 | −0.92 | −2.89 | 1.06 | II |
| MFS 78 | F | 39 | 0.29 | 0.60 | −0.51 | −2.06 | 1.04 | IV |
| MFS 85 | F | 39 | 0.35 | 0.66 | −0.42 | −1.31 | 0.48 | III |
| MFS 105 | F | 39 | 0.18 | 0.18 | −1.71 | −3.69 | 0.26 | III |
| MFS 6 | F | 40 | 0.59 | 0.29 | −1.24 | −2.38 | −0.10 | II |
| MFS 15 | F | 40 | 0.20 | 0.24 | −1.43 | −2.83 | −0.03 | I |
| MFS 24 | F | 40 | 0.22 | 0.79 | −0.24 | −1.14 | 0.66 | IV |
| MFS 41 | F | 40 | 0.36 | 0.10 | −2.30 | −4.27 | −0.33 | I |
| MFS 53 | F | 40 | 0.25 | 0.56 | −0.58 | −1.98 | 0.82 | III |
| MFS 54 | F | 40 | 0.15 | 0.71 | −0.34 | −1.37 | 0.69 | III |
| MFS 73 | F | 40 | 0.07 | 1.81 | 0.59 | −0.83 | 2.02 | II |
| MFS 79 | F | 40 | 0.20 | 0.58 | −0.54 | −1.70 | 0.61 | IV |
| MFS 108 | F | 40 | 0.06 | 1.16 | 0.15 | −1.84 | 2.14 | I |
| MFS 63 | F | 40 | 0.32 | 0.54 | −0.62 | −1.76 | 0.53 | I |
| MFS 9 | F | 41 | 0.58 | 0.10 | −2.30 | −4.27 | −0.33 | IV |
| MFS 11 | F | 41 | 0.48 | 1.20 | 0.18 | −0.46 | 0.82 | II |
| MFS 26 | F | 41 | 0.22 | 0.95 | −0.05 | −1.20 | 1.10 | I |
| MFS 42 | F | 42 | 0.16 | 0.18 | −1.71 | −3.83 | 0.40 | II |
| MFS 62 | F | 42 | 0.33 | 0.47 | −0.76 | −2.15 | 0.64 | II |
| PS 24 | M | 36 | 0.68 | 0.82 | −0.19 | −0.72 | 0.33 | III |
| MFS 74 | M | 36 | 0.61 | 0.87 | −0.14 | −1.28 | 1.00 | II |
| MFS 61b | M | 36 | 0.50 | 0.23 | −1.47 | −2.87 | −0.07 | IV |
| MFS 31 | M | 37 | 0.04 | 0.96 | −0.04 | −2.07 | 1.98 | III |
| MFS 36 | M | 37 | 0.60 | 0.47 | −0.76 | −1.47 | −0.04 | I |
| MFS 16 | M | 38 | 0.33 | 1.06 | 0.06 | −1.09 | 1.20 | I |
| MFS 27 | M | 38 | 0.22 | 0.23 | −1.47 | −2.62 | −0.32 | I |
| MFS 45 | M | 38 | 0.13 | 0.67 | −0.40 | −2.38 | 1.58 | I |
| MFS 103 | M | 38 | 0.27 | 0.24 | −1.43 | −2.58 | −0.28 | IV |
| MFS 65 | M | 38 | 0.15 | 6.37 | 1.85 | 1.34 | 2.36 | IV |
| MFS 82 | M | 38 | 0.18 | 0.64 | −0.45 | −1.60 | 0.071 | II |
| MFS 1 | M | 39 | 0.30 | 1.16 | 0.15 | −0.85 | 1.15 | IV |
| MFS 5 | M | 39 | 0.19 | 1.80 | 0.59 | −0.42 | 1.60 | III |
| MFS 30 | M | 39 | 0.09 | 3.60 | 1.28 | 0.25 | 2.31 | IV |
| MFS 43 | M | 39 | 0.43 | 0.47 | −0.76 | −1.51 | 0.00 | IV |
| MFS 58 | M | 39 | 0.36 | 0.58 | −0.54 | −1.37 | 0.28 | III |
| MFS 59 | M | 39 | 0.17 | 0.85 | −0.16 | −1.07 | 0.75 | IV |
| MFS 64 | M | 39 | 0.13 | 2.93 | 1.26 | 0.23 | 1.92 | IV |
| MFS 71 | M | 39 | 0.20 | 0.75 | −0.29 | −1.06 | 0.49 | II |
| MFS 72 | M | 39 | 0.47 | 0.85 | −0.16 | −0.88 | 0.55 | III |
| MFS 87 | M | 39 | 0.47 | 0.45 | −0.80 | −1.94 | 0.35 | I |
| MFS 95 | M | 39 | 0.43 | 0.88 | −0.13 | −0.89 | 0.63 | IV |
| MFS 99 | M | 39 | 0.23 | 0.43 | −0.84 | −2.81 | 1.13 | II |
| MFS 2 | M | 40 | 0.75 | 1.22 | 0.20 | −0.37 | 0.77 | IV |
| MFS 25 | M | 40 | 0.20 | 0.87 | −0.14 | −1.29 | 1.01 | I |
| MFS 39 | M | 40 | 0.69 | 0.13 | −2.04 | −3.44 | −0.64 | I |
| MFS 40 | M | 40 | 0.42 | 0.55 | −0.60 | −1.49 | 0.30 | IV |
| MFS 44 | M | 40 | 0.24 | 0.16 | −1.83 | −3.81 | 0.14 | I |
| MFS 56 | M | 40 | 0.14 | 1.56 | 0.44 | −0.47 | 1.36 | I |
| MFS 60 | M | 40 | 0.13 | 0.45 | −0.80 | −2.21 | 0.61 | I |
| MFS 84 | M | 40 | 0.61 | 0.35 | −1.05 | −2.05 | −0.05 | I |
| MFS 97 | M | 40 | 0.50 | 0.14 | −1.97 | −3.36 | −0.57 | II |
| MFS 57 | M | 41 | 0.09 | 2.77 | 1.02 | 0.09 | 1.95 | IV |
| MFS 68 | M | 41 | 0.40 | 0.64 | −0.45 | −1.21 | 0.32 | IV |
| MFS 75 | M | 41 | 0.31 | 1.61 | 0.48 | −0.42 | 1.37 | IV |
| MFS 86 | M | 41 | 0.42 | 1.11 | 0.10 | −0.54 | 0.75 | I |
| MFS 88 | M | 42 | 0.45 | 1.21 | 0.19 | −0.57 | 0.95 | I |
Group I, preterm newborns whose mothers had no history of active or passive cigarette exposure during the pregnancy; group II, preterm newborns whose mothers actively smoked cigarettes throughout the pregnancy; group III, preterm newborns whose mothers actively smoked cigarettes during first trimester only; and group IV, preterm newborns whose mothers were exposed only to passive cigarette smoke.
Because the distribution of Mf is skewed, statistical analyses were based on the natural logarithms of Mf × 10−6 (lnMf × 10−6). Adjusted lnMfs × 10−6 were calculated as the difference between the observed lnMf × 10−6 and a correction factor representing the lnMf × 10−6 expected for that CE. The correction factor was derived by regressing lnMf × 10−6 on CE, group (preterm or full term) and an interaction term to allow the effects of CE to differ in the two groups.
Preterm and full-term infants were compared by using t tests to assess differences in CE, Mf × 10−6, and lnMf × 10−6. Gender differences also were analyzed by t tests. Regression analyses were used to examine the relationships between lnMf × 10−6 and GA.
To examine the potentially confounding effect of transplacental tobacco exposure on Mf and CE, subjects were divided into four groups based on maternal history of tobacco exposure as described (8) (see Table 1). Groups were compared by ANOVA using the Student-Newman-Kuels procedure to adjust for multiple pairwise comparisons. Regression analyses, including smoking exposure group variables, also were done to examine the significance of the relationship between GA and lnMf × 10−6 after controlling for tobacco exposure.
RESULTS
A summary of the GA, gender, CE, Mf, and transplacental cigarette smoke exposure for the preterm and full-term cord blood samples used in this study is shown in Table 1. A total of 52 preterm subjects (32 male and 20 female) and 63 full-term infants (37 male and 26 female) were enrolled.
Comparison of Preterm and Full-Term Infants.
A significantly lower CE was seen for premature infants compared with full-term infants, P = 0.001, (Table 2). In addition, the mean lnMf for preterm infants was significantly higher than the mean lnMf of full-term infants, P = 0.008.
Table 2.
Comparison of CE, Mf, and lnMf in preterm and full-term infants
| N | CE* | Mf × 10−6* | lnMf × 10−6* | |
|---|---|---|---|---|
| Preterm | 52 | 0.21 | 1.95 | 0.02 |
| (0.15) | (3.37) | (1.05) | ||
| Full-term | 63 | 0.32 | 0.92 | −0.46 |
| (0.18) | (0.99) | (0.88) | ||
| P value | 0.001 | 0.008 |
Mean (SD).
To further examine the relationship between GA on Mf, a regression analysis was performed by using GA as a continuous variable to predict lnMf × 10−6. Both preterm and full-term infants were included in this analysis and the following regression equation was obtained:
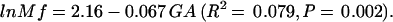 |
This analysis demonstrates that there is a significant correlation between lnMf and GA in which lnMf decreases as GA increases.
Gender-Specific Effects of GA.
When analyses were performed separately for male and female infants, only preterm female subjects demonstrate a significantly higher lnMf (P < 0.001) as compared with full-term female subjects (Tables 3 and 4). In contrast, a comparison of lnMf between preterm males and full-term males was not significant (P = 0.98) (Tables 3 and 4).
Table 3.
Summary of CE, Mf, and gender in preterm and full-term newborns
| Male
|
Female
|
|||
|---|---|---|---|---|
| Preterm | Full-term | Preterm | Full-term | |
| N | 32 | 37 | 20 | 26 |
| CE* | 0.23 | 0.34 | 0.17 | 0.29 |
| (0.16) | (0.19) | (0.14) | (0.16) | |
| Mf × 10−6* | 1.02 | 1.08 | 3.43 | 0.69 |
| (0.80) | (1.18) | (5.06) | (0.58) | |
| lnMf × 10−6* | −0.30 | −0.31 | 0.53 | −0.68 |
| (0.86) | (0.88) | (1.15) | (0.85) | |
Mean (SD).
Table 4.
Statistical comparisons of gender for CE and lnMf in preterm and full-term infants
|
P values
|
||
|---|---|---|
| CE | lnMf × 10−6 | |
| Preterm vs full-term | ||
| Female | 0.012 | <0.001 |
| Male | 0.012 | 0.946 |
| Female vs male | ||
| Preterm | 0.244 | 0.005 |
| Full-term | 0.315 | 0.098 |
The relationship between lnMf and GA was analyzed further by separate regression analysis for each gender, revealing the following gender relationships: female, lnMf = 4.55 − 0.131 GA (R2 = 0.280, P < 0.001); and male, lnMf = −0.22 − 0.002 GA (R2 = 0.00, P = 0.933).
This regression analyses demonstrate a highly significant gender-specific relationship between lnMf and GA for females that does not exist for males. As a result, the inverse relationship between GA and lnMf seen for infants described in the previous section is the consequence of a gender-specific increase in Mf for female infants. The relationship between lnMf and GA is graphically depicted in Fig. 1 for both genders.
Figure 1.

Regression analysis comparing lnMf at the HPRT locus and GA for preterm females (Left; P < 0.001) and preterm males (Right; P = 0.933).
As a consequence of the differing effects of GA in males and females, gender differences in lnMf were reversed for preterm and full-term infants. For preterm infants, females had a significantly higher lnMf compared with males (P = 0.005), whereas for full-term infants, females had a lower lnMf compared with males that is not statistically different (P = 0.098) (Tables 3 and 4).
Adjustment of Differences in CE.
Previous studies have demonstrated an inverse relationship between lnMf and CE in normal control populations. The question then can be raised about whether differences in lnMf could be caused at least in part by differences in CE. We examined this possibility by adjusting lnMf for CE, by using a correction factor based on data from both preterm and full-term infants. The results were very similar to the unadjusted results, reinforcing our findings of gender differences. In fact, the comparison of full-term males vs. full-term females, which was not significant for the unadjusted analyses, demonstrated a significant difference for the adjusted analysis, with the females having a lower adjusted lnMf. For the regression analyses, the relationships with GA were stronger than for the unadjusted lnMf. These results indicated that the significant differences and relationships observed for lnMf are not attributable to differences in CE.
Effect of Transplacental Tobacco Exposure.
We were concerned that the significant differences seen in lnMf and lnMf for premature infants could be influenced by transplacental exposure to cigarette smoke because there was a significant difference in the distribution of maternal cigarette exposure between full-term and preterm infants. Previously we reported that transplacental exposure to cigarette smoke from smoking mothers and mothers exposed only to passive cigarette smoke showed no significant differences in lnMf for full-term infants compared with controls (8). In this study, lnMf in preterm infants also showed no significant differences between the smoking groups shown in Table 1 (P = 0.071). The relationship between GA and lnMf remained significant when smoking exposure groups were included in the regression analysis (P = 0.001). An additional regression using smoking exposure, gender, group (full or preterm), and the interaction between group and gender as predictors confirmed that the gender-specific differences between full-term and preterm infants were not the result of differences in smoking exposure. LnMf was significantly related to gender (P = 0.004), group (P < 0.001), and their interaction (P = 0.003) but was not related to smoking exposure in this particular study.
DISCUSSION
It has been estimated that about half of all in vivo somatic mutations arise during development, with about half of these occurring in utero (17). As a result, in utero somatic mutational events most likely play a major role in the development of pediatric diseases, in particular cancer. Recently, in utero leukemogenic rearrangements involving t(4;11)(q21;q23) translocations (resulting in genomic fusions between the MLL-AF4 loci) and t(12;21)(p13;q22) translocations (TEL-AML1 genomic fusions) have been identified in children diagnosed with acute lymphocytic leukemia (ALL) (18, 19). In addition, identical leukemic clones with rearrangement of MLL gene on chromosome 11q23 were found in twins with infant acute B cell lymphoblastic leukemia (20–22), and T cell ALL (23), further suggesting the importance of in utero leukemogenesis. These observations illustrate that it is critical to determine the in utero genetic effects of exogenous environmental exposures as well as endogenous developmental processes that may influence the initiation of in utero leukemogenic events.
It is of interest that prematurity itself is associated with cancer risk. Specifically, several epidemiologic studies conclude that children born prematurely are at an increase risk for hepatoblastomas (24, 25), malignant germ-cell tumors (26), and breast cancer as adults (27), as well as a reduced risk of prostate cancer (28). These studies infer that transplacental exposures as well as other gender-specific endogenous in utero mechanisms may result in specific genetic alterations associated with malignant transformation in children. There are currently no biologic data that can provide insight into these observations except that gender-specific expression of sex hormones likely plays some role.
In this study we determined that the mean nonselected CE of preterm infants (male and female) is significantly lower than the mean nonselected CE of full-term infants. The reason for this difference is unclear. The proportion of T lymphocytes found in the mononuclear fraction of cord blood samples is lower compared with children and adults (29). This decrease in the proportion of T lymphocytes in cord blood samples could result in a smaller fraction of clonable T lymphocytes for our assay system and be reflected as lower nonselected CE, because the nonselected CE is based on total mononuclear cell content.
The most interesting observation in this study is that there is a gender-specific difference in the effect of GA on the frequency of somatic mutations. Specifically, the lnMf for female preterm infants is significantly higher than for preterm males and that it decreases with increasing GA whereas for male preterm infants the lnMf is unrelated to GA. An inverse relationship between lnMf and CE has been observed in healthy populations, raising the question of whether these differences in lnMf could be caused at least in part by differences in CE. We examined this possibility by adjusting lnMf for CE, using a correction factor based on data from both the preterm and full-term infants. The results were very similar to the unadjusted results, reinforcing our findings of gender differences. In fact, the comparison of full-term males vs. full-term females, which was not significant for the unadjusted analyses, demonstrated a significant difference for the adjusted analysis, with the females having a lower adjusted lnMf. It is also important to consider that this gender difference in Mf is not the result of in vivo clonal expansion of HPRT mutant clones, resulting in a falsely elevated Mf. Preliminary molecular analysis of HPRT mutant isolates from both female and male preterm infants have demonstrated no significant in vivo clonal expansion; therefore, in this study, Mf can be considered a reflection of in vivo Mf.
The reasons for gender-specific differences for in utero somatic mutational events are unclear. Gender-specific in utero developmental processes in humans have been described for neural differentiation (1), growth (2), lung maturation (3), cardiac development (4), and glutathione-reductase expression (5). With respect to molecular differences, adult females have been found to have gender-specific differences in the spectrum of p53 mutations and levels of DNA adducts in lung cancer tumors (30, 31) as well as a more active immune response (32). In this respect, higher Mfs have been previously demonstrated in adults with increased lymphocyte proliferation associated with autoimmune diseases (33, 34). If in utero, the immune system in females is more active and mature than for males, then the higher Mf seen for female preterm infants may be the consequence of increased in utero lymphocyte proliferation. Such increased lymphocyte proliferation and immune activity has been associated with premature births in response to maternal bacterial vaginosis and/or chorioamnionitis (35).
The gender-specific decrease in Mf with increasing GA in female preterm infants is a significant observation. This change in Mf with GA may be the result of a number of factors. The first factor to consider is the rate at which mutations are being generated. In the absence of a known genotoxic exposure, or DNA damage and repair system deficiency, the mutation rate is mainly a consequence of cell proliferation and division. The second factor that will influence changes in Mf with GA is the removal of mutant isolates. It is intriguing to speculate that such a removal may be associated with an in utero-mediated immunological maturation event that selectively eliminates autoreactive T cells via apoptosis. Interestingly, a high number of autoimmune T lymphocytes has been observed in full-term cord blood samples (36). Elimination of these “autoimmune” T lymphocytes during fetal development may be reflected in the decreasing number of total T cells that have developed proliferation-related HPRT mutations.
An alternative possibility for such a decrease in Mf may be explained by the change in selective pressure against HPRT− T lymphocytes during the observed gestational period. It has been known that Mf of cord blood T lymphocytes is extremely low, despite the high rate of lymphocyte proliferation evidenced by high lymphocyte count in cord blood (29). This paradox may be explained by the selective pressure against HPRT mutant isolates that lack the purine salvage pathway (37). Evidence for such selective pressure against HPRT mutant isolates has been reported in female heterozygous for Lesch-Nyhan syndrome mutations (38). The fraction of HPRT− T lymphocytes in these heterozygous females is substantially lower than the expected 50% (38–40). Recently, one study demonstrated that only 8.3% of T lymphocytes carried HPRT mutations in a cord blood sample of a Lesch-Nyhan heterozygous female characterized prenatally (P. O’Neill, personal communication). The selection against HPRT mutant isolates is thought to take place in bone marrow, where purine synthesis is limited to the salvage pathway (41, 42). Interestingly, during the life span of HPRT heterozygous female mice, the decrease of HPRT mutant isolates in the juvenile period was much more rapid compared with the decrease in adult stage (43). Such biphasic decrease was attributed to the initial high selection pressure against HPRT mutant isolates in bone marrow and later selection with lower selective pressure in peripheral lymphatic organs (43). In addition, primary hematopoietic site shifts from liver to bone marrow during the late stage of human fetal development (44); therefore, the decrease in apparent Mf in female infants may be the result of the shift of hematopoietic site along with an increased proliferation rate in the lymphocyte population. For male preterm infants, the lack of an association between Mf and GA may be the result of a lower proliferation rate in males and/or a distinctive switch compared with females in hematopoietic site development. This hypothesis is supported by the observation that a higher number of CD3+ cells occurs in female cord blood samples compared with those from males (45).
The overall increase in in utero Mf for females and the decrease of Mf with increasing GA, which may be caused by higher proliferation rate, also may have direct clinical implications. Most notably there is a female predominance for infant leukemia where the MLL gene rearrangements also predominate (46). MLL gene rearrangements associated with infant leukemia have been shown to be the consequence of “illegitimate” V(D)J recombinase-mediated chromosomal rearrangements as well as splice site mutations. In addition, there is a significant female predominance for pediatric autoimmune diseases such as juvenile rheumatic arthritis, juvenile psoriatic arthritis, systemic lupus erythematosus, and fybromyalgia (47).
Acknowledgments
We thank Holly Pasackow for her clinical assistance for obtaining cord blood samples and performing questionnaire interviews and C.J. Rairikar for his assistance with the statistical analysis. We also thank Richard Albertini for reviewing this manuscript. Research was supported by Grants 1K11HD01010 and 1R29HD35309–01A1 from the National Institute of Child Health, a Wyeth Pediatrics Neonatology Fellowship Research Grant, Grant RO1HDKA33648–01A1 from the National Institute of Child Health and Human Development, and a grant from the Children’s Miracle Network.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: HPRT, hypoxanthine phosphoribosyltransferase; Mf, mutant frequency; lnMf, logarithm Mf; GA, gestational age; CE, cloning efficiency.
References
- 1.Hutchison J B. Cell Mol Neurobiol. 1997;17:603–626. doi: 10.1023/A:1022581902880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smulian J C, Campbell W A, Rodis J F, Feeney L D, Fabbri E L, Vintzileos A M. Am J Obstet Gynecol. 1995;173:1195–1201. doi: 10.1016/0002-9378(95)91352-1. [DOI] [PubMed] [Google Scholar]
- 3.Hanley K, Rassner U, Jiang Y, Vansomphone D, Crumrine D, Komuves L, Elias P M, Feingold K R, Williams M L. J Clin Invest. 1996;97:2576–2584. doi: 10.1172/JCI118706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleisher L A, Dipietro J A, Johnson T R, Pincus S. Clin Sci. 1997;92:345–349. doi: 10.1042/cs0920345. [DOI] [PubMed] [Google Scholar]
- 5.Lovoie J C, Chessex P. Free Radical Biol Med. 1997;23:648–657. doi: 10.1016/s0891-5849(97)00011-7. [DOI] [PubMed] [Google Scholar]
- 6.Finette B A, Sullivan L M, O’Neil J P, Nicklas J A, Vacek P M, Albertini R J. Mutat Res. 1994;308:223–231. doi: 10.1016/0027-5107(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 7.Finette B A, Poseno T, Albertini R J. Cancer Res. 1996;56:1405–1412. [PubMed] [Google Scholar]
- 8.Finette B A, Poseno T, Vacek P M, Albertini R J. Mutat Res. 1997;377:115–123. doi: 10.1016/s0027-5107(97)00069-9. [DOI] [PubMed] [Google Scholar]
- 9.Robinson D, Goodall K, Albertini R J, O’Neill J P, Finette B A, Sala-Trepat M, Tates A D, Beare D, Green M H L, Cole J. Mutat Res. 1994;313:227–247. doi: 10.1016/0165-1161(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 10.Aplan P D, Lombardi D P, Ginsberg A M, Bertness V, Cossman J, Kirsch I R. Science. 1990;250:1426–1429. doi: 10.1126/science.2255914. [DOI] [PubMed] [Google Scholar]
- 11.Gu Y, Cimino G, Alder H, Nakamura T, Prasad R, Canaani O, Moir D T, Jones C, Nowell P C, Croce C M, Canaani E. Proc Natl Acad Sci USA. 1992;89:10464–10468. doi: 10.1073/pnas.89.21.10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cline M J. N Engl J Med. 1994;330:328–336. doi: 10.1056/NEJM199402033300507. [DOI] [PubMed] [Google Scholar]
- 13.Breit T M, Mol E J, Wovers-Tettero I L M, Ludwig W-D, Wering E R V. J Exp Med. 1993;177:965–977. doi: 10.1084/jem.177.4.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt C A, Przybylski G, Vogel D, Ludwig W D, Oettle H, Neubauer A, Siegert W. Leukemia. 1994;8:102–105. [PubMed] [Google Scholar]
- 15.O’Neill, J. P., McGinniss, M. J., Berman, J. K., Sullivan, L. M., Nicklas, J. A. & Albertini, R. J. (1987) Mutagenesis 87–94. [DOI] [PubMed]
- 16.Branda R F, Sullivan L M, O’Neil J P, Falta M T, Nicklas J A, Hirsch B, Vacek P M, Albertini R J. Mutat Res. 1993;285:267–279. doi: 10.1016/0027-5107(93)90115-v. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X B, Urlando C, Tao K S, Heddle J A. Mutat Res. 1995;338:189–201. doi: 10.1016/0921-8734(95)00024-z. [DOI] [PubMed] [Google Scholar]
- 18.Gale K B, Ford A M, Repp R, Borkhardt A, Keller C, Eden O B, Greaves M F. Proc Natl Acad Sci USA. 1997;94:13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ford A M, Bennett C A, Price C M, Bruin M C A, Wering E R V, Greaves M. Proc Natl Acad Sci USA. 1998;95:4584–4588. doi: 10.1073/pnas.95.8.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ford A M, Ridge S A, Cabrera M E, Mahmoud H, Steel C M, Chan L C, Greaves M. Nature (London) 1993;363:358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- 21.Mahmoud H H, Ridge S A, Behm F G, Pui C-H, Ford A M, Raimondi S C, Greaves M F. Med Pediatr Oncol. 1995;24:77–81. doi: 10.1002/mpo.2950240203. [DOI] [PubMed] [Google Scholar]
- 22.Gill-Super H J, Rothberg P G, Kobayashi H, Freeman A I, Diaz M O, Rowley J D. Blood. 1994;83:641–644. [PubMed] [Google Scholar]
- 23.Ford A M, Pombo-de-Oliveira M S, McCarthy K P, MacLean J M, Carrico K C, Vincent R F, Greaves M. Blood. 1997;89:281–285. [PubMed] [Google Scholar]
- 24.Ikeda H, Matsuyama S, Tanimura M. J Pediatr. 1997;130:557–560. doi: 10.1016/s0022-3476(97)70239-7. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda H, Hachitanda Y, Tanimura M, Maruyama K, Koizumi T, Tsuchida Y. Cancer. 1998;82:1789–1796. [PubMed] [Google Scholar]
- 26.Shu X O, Nesbit M E, Buckley J D, Krailo M D, Robinson L L. Cancer Causes Controls. 1995;6:187–198. doi: 10.1007/BF00051790. [DOI] [PubMed] [Google Scholar]
- 27.Ekbom A, Hsieh C-C, Lipworth L, Adami H-O, Trichopoulos D. J Natl Cancer Instit. 1997;89:71–76. doi: 10.1093/jnci/89.1.71. [DOI] [PubMed] [Google Scholar]
- 28.Ekbom A, Hsieh C-C, Lipworth L, Wolk A, Ponten J, Adami H-O, Trichopoulos D. Br Med J. 1996;313:337–341. doi: 10.1136/bmj.313.7053.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hannet I, Yuksel F E, Lydyard P, Deneys V, DeBruyere M. Immunol Today. 1992;13:215–218. doi: 10.1016/0167-5699(92)90157-3. [DOI] [PubMed] [Google Scholar]
- 30.Guinee D G, Travis W D, Trivers G E, Benedetti V M D, Cawley H, Welsh J A, Bennett W P, Jett J, Colby T V, Tazelaar H, et al. Carcinogenesis. 1995;16:993–1002. doi: 10.1093/carcin/16.5.993. [DOI] [PubMed] [Google Scholar]
- 31.Kure E H, Ryberg D, Hewer A, Phillips D H, Skaug V, Baera R, Haugen A. Carcinogenesis. 1996;17:2201–2205. doi: 10.1093/carcin/17.10.2201. [DOI] [PubMed] [Google Scholar]
- 32.Chao T C. Chang Keng I Hsueh. 1996;19:95–106. [PubMed] [Google Scholar]
- 33.Ansari A A, Mayne A, Sundstrum J B, Gravanis M B, Kanter K, Sell K W, Villinger F, Siu C O, Herskowitz A. Circulation. 1995;92:862–874. doi: 10.1161/01.cir.92.4.862. [DOI] [PubMed] [Google Scholar]
- 34.Allegretta M, Nicklas J A, Subramanian S, Albertini R J. Science. 1990;247:718–721. doi: 10.1126/science.1689076. [DOI] [PubMed] [Google Scholar]
- 35.Goldberg R L, Thom E, Moawad A H, Johnson F, Roberts J, Caritis S N. Obstetr Gynecol. 1996;87:656–660. doi: 10.1016/0029-7844(96)00034-8. [DOI] [PubMed] [Google Scholar]
- 36.Fredrikson S, Sun J-B, Huang W-X, Li B-L, Olsson T, Link H. J Immunol. 1993;151:2217–2224. [PubMed] [Google Scholar]
- 37.Cole J, Skopek T R. Mutat Res. 1994;304:33–105. doi: 10.1016/0027-5107(94)90320-4. [DOI] [PubMed] [Google Scholar]
- 38.Albertini R J, DeMars R. Biochem Genet. 1974;11:397–411. doi: 10.1007/BF00486413. [DOI] [PubMed] [Google Scholar]
- 39.Dempsey J L, Morley A A, Seshadri R S, Emmerson B T, Gordon R, Bhagat C I. Hum Genet. 1983;64:288–290. doi: 10.1007/BF00279414. [DOI] [PubMed] [Google Scholar]
- 40.Kamatani N, Yamanaka H, Nishioka K, Nakamura T, Nakano K, Tanimoto K, Mizuno T, Nishida Y. Blood. 1984;63:912–916. [PubMed] [Google Scholar]
- 41.McKeran R O, Howell A, Andrews T M, Watts R W E, Arlett C F. J Neurol Sci. 1974;22:183–195. doi: 10.1016/0022-510x(74)90245-7. [DOI] [PubMed] [Google Scholar]
- 42.Lajtha L G, Vane J R. Nature (London) 1958;182:191–192. doi: 10.1038/182191a0. [DOI] [PubMed] [Google Scholar]
- 43.Deubel W, Bassuka I D, Schlereth W, Lorenz R, Hempel K. Mutat Res. 1996;351:67–77. doi: 10.1016/0027-5107(95)00214-6. [DOI] [PubMed] [Google Scholar]
- 44.Lukens J N. In: Wintrobe’s Clinical Hematology. Lee G R, Bithell T C, Foerster J, Athens J W, Lukens J N, editors. Vol. 1. Philadelphia: Lea and Febiger; 1993. pp. 79–80. [Google Scholar]
- 45.Motley D, Meyer M P, King R A, Naus G J. Hematopathology. 1996;105:38–43. doi: 10.1093/ajcp/105.1.38. [DOI] [PubMed] [Google Scholar]
- 46.Ross J A, Robison L L. Leuk Res. 1997;21:793–795. doi: 10.1016/s0145-2126(97)00051-9. [DOI] [PubMed] [Google Scholar]
- 47.Malleson P N, Fung M Y, Rosenberg A M. J Rheumatol. 1996;23:1981–1987. [PubMed] [Google Scholar]