

The fact that human cancers have preferential sites of distant spread indicates that metastasis is a highly specific and regulated process rather than a random event. Breast cancer is one of several cancers, including prostate, thyroid, and kidney, that display a very high frequency of metastasis to bone (1). The predilection for breast cancer to spread to bone was recognized more than a hundred years ago by Paget (2), who proposed that bone provides a favorable and specific “soil,” or microenvironment, for the growth of specific “seed,” or breast cancer cells.
As a densely mineralized tissue, bone represents a particularly harsh environment for successful tumor cell establishment and growth. Older ideas of pathogenesis favor the view that tumor cells themselves directly cause bone breakdown as a result of their general invasive properties. Such invasive capability is essential, of course, but there is little doubt that the most important specific property required of cancer cells to metastasize in bone is the ability to promote bone resorption, thereby providing a niche in which tumor cells can grow and expand in this otherwise hostile environment. The resorption by host osteoclasts is essential to provide space for tumor establishment and expansion within the mineralized bone matrix (3). Human breast cancer deposits in bone are surrounded by active osteoclasts (3, 4), and evidence obtained from animal models demonstrates active softening, resorption, and destruction of bony tissue (osteolysis) (4, 5) metastasis to bone (6). Furthermore, inhibition of osteoclast-mediated bone resorption is effective in reducing bone metastatic burden in clinical (7) and experimental studies (8). In this issue of the JCI, Gallwitz and colleagues (9) investigate the inhibition of osteolytic bone destruction associated with metastatic human breast cancer cells.
Osteoclast formation
The development of active osteoclasts requires intimate contact between osteoblastic stromal cells and osteoclast precursors of the monocyte/macrophage lineage. This process is influenced by several hormones and cytokines, including 1.25 dihydroxyvitamin D3, parathyroid hormone (PTH) and PTH-related protein (PTHrP) prostaglandin E2 IL-6, and IL-11, all of which enhance osteoclast formation (10). These factors are unable to mediate osteoclast differentiation in the absence of osteoblastic stromal cells, but they act directly upon the latter to control the production of regulators of osteoclast formation and activity. The receptor activator of NF-κB ligand (RANKL) is a member of the TNF ligand family, produced by osteoblastic stromal cells as well as T cells, and acts upon its receptor, RANK, in mononuclear cells to program osteoclast differentiation and maintain their activity (reviewed in ref. 10). Together with the soluble TNF receptor molecule osteoprotegerin (OPG), a decoy receptor that is a powerful inhibitor of RANKL/RANK interaction, RANKL interaction with RANK, modulated by OPG, constitutes an exquisitely regulated system that ensures controlled osteoclast formation from hemopoietic precursors, as well as the maintenance of active osteoclasts. These molecules are intimately involved in the increased osteoclast formation around breast cancer metastases in bone.
PTHrP: an osteolytic cancer product
The discovery of PTHrP as a likely cause of hypercalcemia in many patients with cancer provided new insights into the pathogenesis of the skeletal complications of malignancy. Studies have revealed PTHrP as a previously unrecognized hormone, important in fetal development and in the pathogenesis of hypercalcemia when produced in excess in certain cancers, but otherwise exerting essential paracrine actions in a number of fetal and adult tissues (11). The syndrome of the humoral hypercalcemia of malignancy is explained by the production of excess PTHrP, which acts generally upon the skeleton to increase bone resorption and on the kidney to reduce calcium excretion. PTHrP does so in a manner identical to that of PTH, by acting upon the receptor (PTH1R) that it shares with PTH. Neutralizing anti–PTHrP antibodies are effective in preventing and treating the syndrome in animal models (12, 13).
In addition to this role of excessive circulating PTHrP in several cancers, PTHrP is produced by two-thirds of primary breast cancers and 90% of bone metastases (14), leading to the concept that local PTHrP production by breast cancer cells that reach bone promotes the bone resorption process, thus favoring tumor establishment and expansion. The experimental model that has provided the most support for this is one in which PTHrP–producing human breast cancer cells have established themselves and grown as lytic deposits in bone after injection into the arterial circulation of immune-deficient mice (8, 15). This tumor establishment and growth can be prevented by inhibition of bone resorption, using bisphosphonates or neutralizing mAb’s against PTHrP (8, 15).
Anti–PTH-rP therapeutics?
Reasoning that tumor cell production of PTHrP in bone is an important factor in helping the cancer to flourish there, Gallwitz et al. (9) looked for small molecule inhibitors of PTHrP production by screening for inhibitors of transcription through the PTHrP gene promoter. The two molecules found to be active in the appropriate in vitro and in vivo assays, 6-thioguanine and 6-thioguanosine, were discovered by a screen carried out on a number of known anticancer drugs. Therapeutics directed at transcriptional controls would be expected to have problems of specificity, but a further screen of a relatively small library of compounds (less than 10,000) yielded no other positives, and in another test of specificity, the two candidate molecules had no effect on the activity of five unrelated gene promoter-reporters. Both of the compounds were effective in combatting humoral hypercalcemia and bone tumor growth in experimental murine models, and the authors propose that modulation of PTHrP production with this approach could provide a new therapeutic pathway for the treatment of the skeletal complications associated with cancer. If drug discovery is embarked upon using the promotor-based assay as a screening method, then it would clearly be useful to cast the net more widely and make use of a high throughput screen with libraries of at least several hundred thousand compounds.
There may be additional roles for PTHrP in the pathophysiology of cancer, especially those suggested in the “vicious cycle” hypothesis of Mundy (5). This theory proposes that, following initiation, the bone metastatic process is amplified because the microenvironment (with enhanced production of TGF-β [ref. 16] and perhaps other growth factors) increases PTHrP production, thereby favoring osteoclast formation and furthering tumor growth (Figure 1). Candidate drugs discovered during a PTHrP–promoter screen should also be evaluated for their ability to inhibit TGF-β stimulation, since TGF-β’s effect on PTHrP production is transcriptional as well as posttranslational (17, 18). Indeed, a protein kinase C inhibitor, calphostin C, has been shown to inhibit activity of the PTHrP promoter; thus protein kinase C inhibitors might constitute another class of compounds worth investigating as inhibitors of PTHrP production (18).
Figure 1.
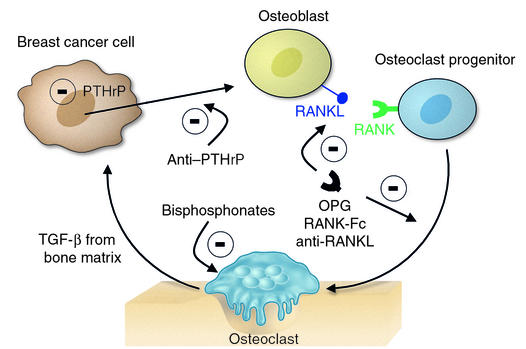
Local factors acting either directly or indirectly on hemopoietic precursors regulating osteoclast formation and activity. Locally generated TGFβ promotes PTHrP production by tumor cells (16). Therapeutic aims could be to reduce PTHrP production or prevent its action. Some other current approaches to reducing osteoclast formation or activity are shown (reviewed in refs. 5 and 21), with a minus sign indicating inhibition: OPG as an inhibitor of RANKL stimulation; RANK-Fc as a competitive inhibitor of RANKL binding to RANK; anti-RANKL; and bisphosphonates acting upon osteoclasts to reduce their activity and enhance apoptosis.(7). Not shown here are several targets for therapeutic intervention within the osteoclast itself (21).
The frequency of PTHrP production in breast cancer, its ability to promote bone resorption, and the efficacy of resorption inhibition in limiting bone metastasis growth all suggest that reduction of PTHrP production or action could be of benefit in late stages of cancer. Reduction of PTHrP availability or action could be addressed with humanized anti-PTHrP antibodies, one of which is currently in clinical trial (reported in ref. 5), or with receptor antagonists if they are suitably effective and specific, or by aiming at pharmacological reduction of tumor PTHrP production, as proposed by Gallwitz et al. (9). Figure 1 illustrates these approaches to inhibiting cancer cell interaction with bone and also identifies targets that are of current interest for use in the therapeutic reduction of osteoclast formation and activity.
Use of PTHrP reduction as a therapeutic mechanism does, however, raise other questions. In the only prospective clinical study to date, women with PTHrP–positive primary breast cancers had a significantly better prognosis than did those with PTHrP–negative tumors, as evidenced by reduced metastases at all sites, including bone (19). This raises the possibility that PTHrP production at an early stage might confer a less invasive phenotype upon the breast cancer cell. The underlying mechanisms remain speculative, but PTHrp is a multifunctional protein (11) that may yet offer some surprises. Attention was drawn recently to an antiangiogenic effect of N-terminal PTHrP (20). The prospect of PTHrP being “protective” at one stage of cancer and having a deleterious role at another is an intriguing one. The two are by no means inconsistent, since resorption stimulation does not reflect a general invasive property but a specific function that comes into play in complementing the general invasive properties of the cancer cells. It is enhanced by factors in the bone microenvironment, and it specifically favors growth in bone.
The factors involved in the generation and maintenance of osteoclast activity are receiving much attention, especially since the discovery of the controlling influence of the TNF ligand and TNF receptor family members. Therapeutic efforts directed against the skeletal complications of cancer embrace those targets (OPG, RANKL and RANK) (Figure 1), as well as PTHrP as a proximal cause. It should also be recognized that there are other breast cancer products that might profoundly influence bone metastasis establishment — prostaglandins, IL-6, and IL-11, to mention a few (10). PTHrP has been the focus of much more intensive study than these, but their participation is plausible, and their contribution to the skeletal complications of cancer will need to be fully evaluated and put into perspective.
Acknowledgments
The author thanks Matthew T. Gillespie for helpful comments.
Footnotes
See the related article beginning on page 1559.
Conflict of interest: The laboratory of the author receives funding from the Chugai Pharmaceutical Company.
Nonstandard abbreviations used: parathyroid hormone (PTH); parathyroid hormone–related protein (PTHrP); receptor activator of NF-κB ligand (RANKL); receptor activator of NF-κB (RANK).
References
- 1.Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer. 1987;55:61–66. doi: 10.1038/bjc.1987.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 3.Mundy GR, Martin TJ. Pathophysiology of skeletal complications of cancer. In Handbook of experimental pharmacology. Physiology and pharmacology of bone.G.R. Mundy and T.J. Martin, editors. Springer-Verlag. Berlin, Germany. 1993;107:641–671. [Google Scholar]
- 4.Clohisy DR, Perkins SL, Ranmaraine ML. Review of cellular mechanisms of tumor osteolysis. Clin Orthop. 2000;373:104–114. doi: 10.1097/00003086-200004000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 6.Lelelakis M, et al. A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis. 1999;17:163–170. doi: 10.1023/a:1006689719505. [DOI] [PubMed] [Google Scholar]
- 7.Berenson JR, Lipton A. Bisphosphonates in the treatment of malignant bone disease. Annu Rev Med. 1999;50:237–248. doi: 10.1146/annurev.med.50.1.237. [DOI] [PubMed] [Google Scholar]
- 8.Yoneda T, et al. Inhibition of osteolytic bone metastasis of breast cancer by combined treatment with the bisphosphonate ibandronate and tissue inhibitor of the matrix metalloproteinase-2. J Clin Invest. 1997;99:2509–2517. doi: 10.1172/JCI119435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallwitz WE, Guise TA, Mundy GR. Guanosine nucleotides inhibit different syndromes of PTHrP excess caused by human cancers in vivo. J Clin Invest. 2002;110:1559–1572. doi:10.1172/JCI200211936. doi: 10.1172/JCI11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suda T, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 11.Martin, T.J., Lam, M.H.C., Kartsogiannis, V., and Gillespie, M.T. 2000. Parathyroid hormone-related protein. In Skeletal growth factors. E. Canalis, editor. Lippincott Williams & Wilkins. Philadelphia, Pennsylvania, USA. 335–354.
- 12.Kukreja SC, et al. Tumor resection and antibodies to parathyroid hormone-related protein cause similar changes on bone histomorphometry in hypercalcemia of cancer. Endocrinology. 1990;127:305–310. doi: 10.1210/endo-127-1-305. [DOI] [PubMed] [Google Scholar]
- 13.Iguchi H, Onuma E, Sato K, Ogata E. Involvement of parathyroid hormone-related protein in experimental cachexia induced by a human lung cancer-derived cell line established from a bone metastasis. Int J Cancer. 2001;20:24–27. doi: 10.1002/ijc.1425. [DOI] [PubMed] [Google Scholar]
- 14.Powell GJ, et al. Localization of parathyroid hormone-related protein in breast cancer metastases: increased incidence in bone compared with other sites. Cancer Res. 1990;50:7710–7716. [PubMed] [Google Scholar]
- 15.Guise TA, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin JJ, et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiriyama T, et al. TGFβ stimulation of parathyroid hormone-related protein (PTHrP): a paracrine regulator? Mol Cell Endocrinol. 1993;92:55–62. doi: 10.1016/0303-7207(93)90074-t. [DOI] [PubMed] [Google Scholar]
- 18.Lindemann RK, Ballschmieter P, Nordheim A, Dittmer J. Transforming growth factor β regulates parathyroid hormone-related protein expression in MDA-MB-231 breast cancer cells through a novel Smad/Ets synergism. J Biol Chem. 2001;276:46661–46670. doi: 10.1074/jbc.M105816200. [DOI] [PubMed] [Google Scholar]
- 19.Henderson MA, et al. Parathyroid hormone-related protein production by breast cancers, improved survival and reduced bone metastases. J Natl Cancer Inst. 2001;93:234–237. doi: 10.1093/jnci/93.3.234. [DOI] [PubMed] [Google Scholar]
- 20.Bakre MM, et al. Parathyroid hormone-related peptide is a naturally occurring, protein kinaseA-dependent angiogenesis inhibitor. Nat Med. 2002;9:995–1003. doi: 10.1038/nm753. [DOI] [PubMed] [Google Scholar]
- 21.Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]