

Abstract
Employing nonequilibrium molecular dynamics simulations, a comprehensive computational study of the photoinduced conformational dynamics of a photoswitchable bicyclic azobenzene octapeptide is presented. The calculation of time-dependent probability distributions along various global and local reaction coordinates reveals that the conformational rearrangement of the peptide is rather complex and occurs on at least four timescales: 1) After photoexcitation, the azobenzene unit of the molecule undergoes nonadiabatic photoisomerization within 0.2 ps. 2) On the picosecond timescale, the cooling (13 ps) and the stretching (14 ps) of the photoexcited peptide is observed. 3) Most reaction coordinates exhibit a 50–100 ps component reflecting a fast conformational rearrangement. 4) The 500–1000 ps component observed in the simulation accounts for the slow diffusion-controlled conformational equilibration of the system. The simulation of the photoinduced molecular processes is in remarkable agreement with time-resolved optical and infrared experiments, although the calculated cooling as well as the initial conformational rearrangements of the peptide appear to be somewhat too slow. Based on an ab initio parameterized vibrational Hamiltonian, the time-dependent amide I frequency shift is calculated. Both intramolecular and solvent-induced contributions to the frequency shift were found to change by 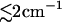 , in reasonable agreement with experiment. The potential of transient infrared spectra to characterize the conformational dynamics of peptides is discussed in some detail.
, in reasonable agreement with experiment. The potential of transient infrared spectra to characterize the conformational dynamics of peptides is discussed in some detail.
INTRODUCTION
The conformational dynamics of biomolecules may be crucial to such biological functions as ligand binding or enzymatic activity (1–3). Recent advances in experimental techniques allow for a real-time observation of these conformational motions on various timescales. Beautiful examples include atomic force microscopy techniques (4), laser-induced pH and temperature-jump experiments (5,6), and single molecule fluorescence spectroscopy (7,8). Furthermore, there have been various suggestions to include a molecular photoswitch into biomolecules (9–16). For example, Moroder and co-workers have synthesized various peptides, in which an azobenzene unit was incorporated directly in the backbone (11–14). This guarantees that the light-induced structural changes of the chromophore upon photoisomerization around the central N=N double bond are directly transferred into the peptide chain. By photoexciting the system by an ultrashort laser pulse, the subsequent conformational dynamics of the peptide is investigated by optical (17,18) or infrared (19,20) spectroscopy. These types of experiments, especially in combination with two-dimensional infrared probing (21), provide a new and promising way to study the folding and unfolding of peptides in unprecedented detail.
As an experimentally particularly well-characterized molecular system (11,17–20), we consider the octapeptide fragment H-Ala-Cys-Ala-Thr-Cys-Asp-Gly-Phe-OH which was connected head to tail via (4-aminomethyl)-phenylazobenzoic acid as well as by a disulfide bridge (see Fig. 1). According to nuclear magnetic resonance (NMR) experiments on this bicyclic azobenzene peptide (bcAMPB) in Renner et al. (11), in equilibrium, the trans azopeptide is predominantly in a single conformational state, while there are many conformations of similar energy in the equilibrium cis state of the peptide. To have a spectroscopically well-characterized final state, most time-resolved experiments have therefore studied the cis→trans photoisomerization of bcAMPB (17–20). These studies have revealed that the photoinduced response of the peptide is quite complex and occurs on various timescales.
FIGURE 1.

Structure and amino-acid labeling of the bicyclic azobenzene peptide bcAMPB.
For example, using transient infrared spectroscopy, Bredenbeck et al. (19) found two main features: 1) A red-shifted transient absorption band, which occurs immediately after photoswitching and decays on a timescale of 4 ps. This feature was attributed to the cooling of the molecule in the solvent. 2) A transient blue-shifted signature which is formed on a timescale of 6 ps. After 20 ps, the intensity and peak position of this feature are almost equivalent to the stationary FTIR difference spectrum. Since the blue shift of the amide I band can be directly related to the change of backbone structure (22,23), it was concluded that the main conformational changes associated with the stretching of the peptide are completed within only 20 ps.
Transient two-dimensional infrared spectroscopy (20), on the other hand, revealed substantial changes of the spectra for times up to 1 ns. Although two-dimensional infrared spectra in principle allow us to distinguish between homogeneous and inhomogeneous broadening, their interpretation is not straightforward for nonequilibrium processes. Optical pump probe spectroscopy (18) of bcAMPB also yielded various timescales: 1) 280 fs for the initial cis→trans photoisomerization. 2) A 5.4 ps constant, presumably reflecting a cooling process. 3) Two kinetic components with time constants of 100 and 1000 ps, reflecting conformational dynamics.
A first-principles theoretical description of the dynamics and spectroscopy of photoswitchable peptides represents a considerable challenge. First, we have recently performed extensive replica-exchange MD simulation (24) to study the equilibrium structure and conformational dynamics in the stable cis and trans states of bcAMPB. We were able to reproduce the NMR data, and also confirmed the general picture drawn by Renner et al. (11) that the trans-isomer of bcAMPB exhibits a well-defined structure, whereas the cis-isomer represents a conformational heterogeneous system comprising an ensemble of structures. Similar results were obtained by Carstens et al. (25) employing high temperature simulations. Second, one wants to describe the laser-induced conformational dynamics of biomolecules, starting from the experimentally achieved nonequilibrium preparation of the system (17,26–31). With this end in mind, we have developed a simple and practical computational strategy that allows us to extend well-established MD simulation techniques to the description of photoinduced dynamics in peptides (32). Employing a simple model of the photoisomerizationprocess, we generate a nonstationary phase-space distribution that mimics the laser-induced initial state of the molecule. Sampling this distribution by an ensemble of trajectories (typically some hundreds), the time evolution of the subsequent conformational dynamics is described via nonequilibrium MD simulations. Finally, to calculate time- and frequency-resolved optical and infrared spectra (33), a quantum-classical approach is employed that combines the classical MD description of the conformational dynamics with a quantum-mechanical calculation of the spectroscopic signals (34–37). In particular, there has been considerable effort to model the amide I vibrational band of peptides (35–44).
In Nguyen and Stock (32), the technical aspects of the nonequilibrium MD method were considered, such as the sensitivity of the conformational dynamics to the photoisomerization model and the convergence of the ensemble average. In this work, we are concerned with 1) a comprehensive analysis of the global and local conformational arrangements of bcAMPB after photoisomerization and 2) to what extent this conformational dynamics is reflected in the transient infrared response of the peptide.
METHODS
Nonequilibrium simulation technique
It is useful to briefly summarize the main aspects of the nonequilibrium simulation method; see Nguyen and Stock (32) for details. The excitation of a molecular system by an optical laser pulse prepares the molecule in an excited electronic state. In the case of the azobenzene peptide bcAMPB, the system rapidly isomerizes within ≈0.2 ps from cis to trans. To model this photoisomerization process, we proposed a minimal model for the corresponding potential-energy surfaces that diabatically connects the excited-state S1 of the cis isomer with the ground-state S0 of the trans isomer. Although this strategy certainly represents a grossly oversimplified description of the ultrafast nonadiabatic photoreaction, it was shown to virtually not affect the subsequent peptide conformational dynamics (32).
Adopting this model, a nonequilibrium MD description of the photoinduced conformational dynamics in bcAMPB can be rationalized as follows. First, 400 statistically independent initial conformations were selected from a previously performed equilibrium MD simulation of cis-bcAMPB (24). Next, we mimic the photoexcitation of the system by an ultrashort laser pulse by instantly switching from the ground-state N = N torsional potential to the excited-state potential. After this nonequilibrium preparation at time t = 0, the system isomerizes along excited-state N = N potential within ≈0.2 ps. After isomerization (i.e., for times ≥ 500 fs), the N = N torsional potential is switched back to its ground-state form, and a standard MD simulation is performed up to 1 ns. After the nonequilibrium simulations, the time-dependent observables of interest are obtained via an ensemble average over the initial distribution. For example, to calculate the time evolution of some energy E, we obtain
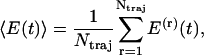 |
(1) |
where E(r) denotes the energy pertaining to an individual trajectory and Ntraj is the number of trajectories.
We used the GROMOS96 force-field 43a1 (45) to model the bcAMPB peptide and the united-atom model of Liu et al. (46) to describe the DMSO solvent. Additional force-field parameters for the azobenzene unit were derived from density functional theory as described in Nguyen et al. (24). All simulations were performed by employing the GROMACS program suite (47,48). The bcAMPB peptide was placed in an octahedral box containing ≈700 DMSO molecules. The equation of motion was integrated by using a leap-frog algorithm with a time step of 2 fs. Covalent bond lengths were constrained by the procedure SHAKE (49) with a relative geometric tolerance of 0.0001. We employed the particle-mesh Ewald method to treat the long-range electrostatic interactions (50). The nonbonded interaction pair-list was updated every 5 fs, using a cutoff of 1.4 nm.
To correctly describe the initial cooling of the hot photoproduct in the solvent, the following strategy was employed (32). For the first 50 ps, each trajectory was simulated at constant total energy (NVE ensemble) using a time step of 0.2 fs. Subsequently, the MD simulations were performed at a constant temperature of 300 K (NVT ensemble) with a time step of 2 fs, using the Berendsen coupling method (51) with a temperature coupling constant of 0.1 ps.
Time-windowed principal component analysis
Principal component analysis (PCA) is an efficient method to represent the conformational distribution of a 3N-dimensional system in terms of a few principal components (52–54). The basic idea is that the correlated internal motions are represented by the covariance matrix
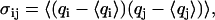 |
(2) |
where q1, …q3N are the mass-weighted Cartesian coordinates of the solute molecule and 〈…〉 denotes the average over all sampled conformations. By diagonalizing σ, we obtain 3N eigenvectors vn and eigenvalues λn, which are rank-ordered in descending order, i.e., λ1 represents the largest eigenvalue. The principal components Vn are then defined as the projections of the trajectory q(t) = {qi(t)} on the eigenvectors vn, i.e.,
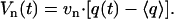 |
(3) |
It has been shown that a large part of the system's fluctuations can be described in terms of only a few principal components (52–54).
In this work, we performed a PCA based on the (φ, ψ) dihedral angles of the peptide (55). For flexible systems such as folding peptides, the use of internal coordinates may be advantageous, because problems associated with the mixing of internal and overall motions are circumvented. For example, in the case of a photoswitchable peptide the problem arises as to which reference structure (cis or trans state of bcAMPB) should be chosen for the rotational fit. To uniquely define the distance in the space of periodic dihedral angles, the dihedral PCA employs the variables (55)
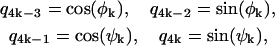 |
(4) |
where k = 1…Np, with Np being the number of peptide groups.
The concept of a PCA of equilibrium data can—to some extent—be generalized to a time-dependent PCA of nonequilibrium data (56). The basic idea is to perform at each time t (after the initial photoexcitation at t = 0) a PCA using an ensemble average over the initial distribution of nonequilibrium trajectories. In the limit of long times, when the system is again in equilibrium, the PCA is again independent of time and the ensemble average should give equivalent results as the time average. In the limit of short times, however, the PCAs obtained for different times have different eigenvalues and eigenvectors. As a consequence, it is not straightforward to compare the results of two PCAs obtained for different times. For example, the time-dependent probability distribution P(Vn, t) of the principal component Vn considered in Fig. 4 corresponds to different eigenvectors vn(t1) and vn(t2) at times t1 and t2. As a remedy, we choose to represent the data of PCAs for all times with respect to the eigenvectors of the PCA at time t = 0.
FIGURE 4.

Time evolution of the probability distributions of the first two principal components V1 (A) and V2 (B), respectively. (C) Time-dependent width ΔVn of the two distributions.
Another, more practical problem concerns the convergence of the ensemble average in the time-dependent PCA. This is because the number of nonequilibrium trajectories is typically much smaller than the number of conformations of an equilibrium trajectory. Here, we combined ensemble and time average, by averaging (say, the coordinate q) over a time-window Δt according to
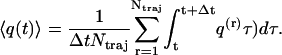 |
(5) |
By choosing Δt = 0.5 ps, we obtain a reasonable compromise between convergence of the averages and time resolution of the data.
Calculation of infrared response
To model the amide I infrared response of peptides in aqueous solution, we adopt an exciton model (22,23,33), which consists of local C=O vibrational modes with frequencies εn that are interacting via the vibrational couplings βnm. Employing harmonic-oscillator creation and annihilation operators 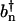 and bn that describe the local C=O mode at the nth peptide site, the amide I vibrational Hamiltonian in harmonic approximation reads
and bn that describe the local C=O mode at the nth peptide site, the amide I vibrational Hamiltonian in harmonic approximation reads
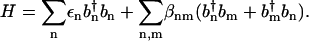 |
(6) |
To describe the dependence of the diagonal and off-diagonal matrix elements εn and βnm on the conformation of the peptide, various groups have performed ab initio calculations on small model peptides (38–41). In the calculations reported below we use a parameterization of εn and βn, n ± 1 as a function of the (φ, ψ) dihedral angles of the peptide backbone, which was obtained at the B3LYP/6-31+G(d) theoretical level for the model peptide Ac-Gly-NHCH3 (41).
To account for the effects of the aqueous solvent on the vibrational frequencies, several groups have performed electron structure calculations for various MD snapshots of the solute and a few surrounding water molecules (42–44). In a second step, the frequency change was empirically correlated with the external electrostatic potential or field at one or more solute sites, and this empirical relationship is then used to calculate the vibrational frequencies along a MD trajectory. In this work, we have employed a six-site parameterization developed by Skinner and co-workers (44), which is based on extensive ab initio calculations of n-methylacetamide in D2O. Although the empirical relationship was obtained for aqueous solvent, it was recently shown that the same parameterization can also be employed to estimate the frequency shift of other polar solvents such as DMSO (57).
COMPUTATIONAL RESULTS
After photoexcitation, the azobenzene unit of bcAMPB undergoes nonadiabatic photoisomerization from the excited S1 state of the cis isomer to the ground state S0 of the trans isomer (18,19). As shown in Nguyen and Stock (32), the classical model of diabatically connecting initial and final states reproduces the 0.2-ps timescale of this process. In particular, it was shown that the oversimplified modeling of the nonadiabatic photoisomerization process virtually does not affect the subsequent peptide conformational dynamics, which is the focus of this article. As explained in the Introduction, time-resolved experiments (18–20) have revealed a picosecond cooling of the hot photoproducts as well as a series of conformational rearrangements occurring on a picosecond-to-nanosecond timescale. In the following, these processes and their infrared spectral response are discussed.
Cooling
Due to the cis→trans photoisomerization of bcAMPB, the azobenzene chromophore receives ≈320 kJ/mole excess energy. This energy is rapidly redistributed to the vibrational modes of the peptide (by intramolecular vibrational relaxation) as well as to the surrounding solvent molecules (by vibrational cooling). Although the quantitative description of these processes in general requires a quantum-mechanical modeling (58–60), a simple classical approach to study vibrational energy transport and cooling is to consider the kinetic energy of various parts of the molecular system. The upper panels in Fig. 2 show the time evolution of the kinetic energy of the azobenzene photoswitch and the octapeptide, respectively. After photoexcitation, the kinetic energy of the azobenzene chromophore rises within only 100 fs. During the next 100 fs the vibrational energy is transferred to the peptide. As a consequence, the peptide kinetic energy increases from 240 kJ/mol to ∼290 kJ/mol, which is equivalent to the temperature jump of ≈100°C found in experiment (20). Subsequently, the vibrational energy of both azobenzene and peptide is dissipated into the solvent within 100 ps. A single exponential fit yields a decay time of 16 ps for the kinetic energy of the peptide. Considering the total (kinetic and potential) energy instead of only the kinetic part, the results look quite similar (data not shown), with a slightly shorter decay time of 13 ps.
FIGURE 2.

Time evolution of the mean kinetic energy of various parts of bcAMPB after photoexcitation. The upper panels show the kinetic energy of the azobenzene photoswitch (left) and the octapeptide (right). The decay of the latter can be well represented by a 16-ps exponential fit shown in shading. The lower panels show the time evolution of the kinetic energy pertaining to the individual peptide residues.
To study the energy transport along the peptide, the lower panels in Fig. 2 show the time evolution of the kinetic energy pertaining to the individual peptide residues. The residues Ala-1 and Phe-8, which are directly connected to the photoswitch, are seen to receive most of the transferred energy from the photoswitch. Although this process is ultrafast (0.2 ps) and carries substantial vibrational energy, the subsequent transfer to the remaining peptide residues is significantly slower (1–2 ps) and less prominent.
Global conformational rearrangement
To obtain from the MD simulations an overall picture of the photoinduced conformational changes of bcAMPB, we first consider several types of reaction coordinates describing its global conformational rearrangement. To this end, Fig. 3 shows the time evolution of the peptide's end-to-end distance and its radius of gyration as well as the root-mean squared deviation of the system.
FIGURE 3.

Time evolution (left) and probability distribution (right) of various reaction coordinates that describe the global conformational rearrangement of bcAMPB after photoexcitation. Shown are (from top to bottom) the peptide end-to-end distance dee, the radius of gyration Rg, and the RMSD. The time traces are shown including exponential fits (shaded lines) with the two main time constants of the fit indicated. The distributions are shown for the first (solid lines) and the last (dashed lines) picoseconds of the 1-ns simulation.
Let us first discuss the results for the end-to-end distance dee between the first and last residue of the peptide. Within 200 fs, the end-to-end distance rapidly increases from 0.6 to 0.82 nm, thus reflecting the initial isomerization of the photoswitch. The subsequent time evolution of dee up to 0.88 nm within ≈50 ps is governed by the competition between the driving force of the photoswitch and the restraining force of the peptide. A biexponential fit of dee yields rise times of 0.2 ps (75%) and 14 ps (25%) for this process.
The radius of gyration 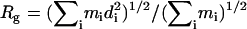 is defined as the average of the mass-weighted squared distances of all atoms to the center of mass and is therefore a measure of the overall size of the molecule. As a consequence of the stretching of the peptide backbone, the radius of gyration is found to increase from 0.58 to 0.65 nm within the first nanosecond and is obviously not yet completed. The exponential time constants 0.8 ps (18%), 44 ps (30%), and 470 ps (52%) reveal that the change of the overall size of the peptide monitored by Rg occurs on a slower timescale than the stretching of the peptide monitored by dee. This suggests that the initial backbone stretching is a prerequisite for the general rearrangement of the structure.
is defined as the average of the mass-weighted squared distances of all atoms to the center of mass and is therefore a measure of the overall size of the molecule. As a consequence of the stretching of the peptide backbone, the radius of gyration is found to increase from 0.58 to 0.65 nm within the first nanosecond and is obviously not yet completed. The exponential time constants 0.8 ps (18%), 44 ps (30%), and 470 ps (52%) reveal that the change of the overall size of the peptide monitored by Rg occurs on a slower timescale than the stretching of the peptide monitored by dee. This suggests that the initial backbone stretching is a prerequisite for the general rearrangement of the structure.
The root-mean square displacement (RMSD) of the octapeptide is another reaction coordinate that describes a global conformational rearrangement. The RMSD was evaluated for all atoms of the peptide and averaged over all trajectories, adopting the structure of the most prominent trans state of bcAMPB as reference geometry. (Note that we choose state I of the energy landscape in Fig. 4 a of Nguyen et al. (24) as a reference structure.) The decay time of the RMSD was found to virtually not depend on this specific choice. Fig. 3 shows that the RMSD decreases from 0.44 to 0.39 nm within 1 ns, thus reflecting the—as yet incomplete—conformational transition of the peptide from the cis to the trans form. The time constants of the RMSD decay are 1.5 ps (15%), 68 ps (30%), and 950 ps (55%)—that is, somewhat longer than for the radius of gyration.
For further illustration, Fig. 3 also shows the probability distributions of the considered reaction coordinates. Averaging over the first (solid lines) and last (dashed lines) picosecond of the 1-ns simulation, the distributions describe the initial and the final state of the photoswitchable peptide, respectively. The two states can be characterized by their RMSD distributions, which correspond to the conformational rearrangement of the photoprocess. Moreover, the distributions of the end-to-end distance dee and the Rg clearly reflect the transition from the disordered cis state to the well-defined trans state. Whereas the latter is dominated by a single conformational state, the many coexisting conformations of the former result in broad distribution of both dee and Rg, thus reflecting to the rugged free-energy surface of the system (24).
Each of the above discussed reaction coordinates provides only a one-dimensional view of the peptide motion. Moreover, these coordinates are not independent of each other. Principal-component analysis is a more systematic approach to describe the motion of a multidimensional system. It represents the motion in terms of an orthogonal basis, the “principal components,” which are ordered according to their content of RMSD fluctuations. To analyze the time evolution of the peptide structure, we performed a time-windowed principal component analysis, which is based on the dihedral angles of the peptide (see Methods). Only for the first three principal components were strongly non-Gaussian multimodal distributions found. The motions along these components contain a significant percentage of the fluctuations—that is, 20, 31, and 42% for the first, first two, and first three principal components, respectively.
Fig. 4, this article, shows the time evolution of the probability distributions P(V1) and P(V2) of the first two principal components, respectively. Initially, P(V1) exhibits a rather broad distribution that covers a range of 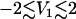 , thus reflecting the conformational heterogeneity of the initial cis state of the azopeptide. In the course of the time evolution, the overall width of P(V1) decreases and for times
, thus reflecting the conformational heterogeneity of the initial cis state of the azopeptide. In the course of the time evolution, the overall width of P(V1) decreases and for times 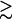 150 ps a pronounced feature at V1 ≈ −0.5 appears, which resembles the final trans state. Similarly, the time evolution of P(V2) shows a transition from a bimodal distribution, reflecting two conformational states of the initial cis state, to a unimodal distribution, corresponding to the final trans state. To further illustrate this narrowing of the conformational space visited by the peptide, Fig. 4 also displays the time-dependent width
150 ps a pronounced feature at V1 ≈ −0.5 appears, which resembles the final trans state. Similarly, the time evolution of P(V2) shows a transition from a bimodal distribution, reflecting two conformational states of the initial cis state, to a unimodal distribution, corresponding to the final trans state. To further illustrate this narrowing of the conformational space visited by the peptide, Fig. 4 also displays the time-dependent width 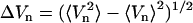 of the distribution, revealing decay times of 120 and 460 ps. A conformational analysis shows that the motion along the first principal component mainly contains transitions of the residues Ala-1 and Gly-7. The second principal component mainly describes another rearrangement of Gly-7.
of the distribution, revealing decay times of 120 and 460 ps. A conformational analysis shows that the motion along the first principal component mainly contains transitions of the residues Ala-1 and Gly-7. The second principal component mainly describes another rearrangement of Gly-7.
Local conformational rearrangement
To investigate which residues are involved in the photoinduced conformational rearrangement of the system, we next study the structural changes of the individual peptide groups. To this end, Fig. 5 shows the time-dependent distributions of the backbone dihedral angles φn(t) and ψn(t) (n = 1…8) pertaining to the eight amino acids of bcAMPB. Apart from the glycine group, the peptide residues can be roughly characterized by three conformational states, that is, the right-handed helix conformation α located at (φ, ψ) ≈ (−70°, −40°) and two extended conformations, here referred to as β ((φ, ψ) ≈ (−120°, 130°)) and PII ((φ, ψ) ≈ (−70°, 130°)). For example, the residue Ala-3 is found in α-conformation, Cys-5 is mostly in β, and Thr-4 and Phe-8 are found in the mixtures α/β and β/PII, respectively. In the equilibrium simulations of Nguyen et al. (24), the dihedral angles for these residues were quite similar for the cis and trans isomers. As expected, these residues exhibit therefore only minor changes (such as reversible β→PII transitions of Phe-8) in the nonequilibrium simulation.
FIGURE 5.

Time evolution of the distributions of the (φ, ψ) backbone dihedral angles pertaining to the eight amino acids of bcAMPB.
The most significant conformational changes are observed for Ala-1, which is directly connected to the photoswitch and undergoes a β ↔ α transition, and for Gly-7, which is the most flexible residue found in the NMR (11) and MD (24) studies. Furthermore, we find a β→PII transition for Cys-2 and a weak α ↔ β transition for Asp-6. The short-time evolution of the mean dihedral angles monitoring these conformational transitions is shown in Fig. 6 together with exponential fits. The time constants are 57 ps for ψAla1, 63 ps for φCys2, 131 ps for ψAsp6, and 110 ps ψGly7, respectively. Considering the complex time evolution of the broad multimodal distributions in Fig. 5, however, the meaning of single time constants is obviously limited.
FIGURE 6.

Time evolution and exponential fits of various backbone dihedral angles of bcAMPB.
Photoinduced infrared response
Although the transient amide I spectrum reflects conformational transitions of the peptide, it is not straightforward to estimate this spectral change from the time evolution of the MD trajectories. This is because the infrared spectrum of a peptide depends, in a complex and nonlinear manner, on the individual conformations of the residues (22,23). Hence, a small conformational change might affect a large spectral difference, whereas a prominent conformational change might hardly be visible. To obtain a qualitative impression on the infrared response of bcAMPB, we study the photoinduced time evolution of the diagonal and off-diagonal matrix elements εn and βnm of the vibrational Hamiltonian in Eq. 6. Although it is clear that the calculation of transient infrared spectra requires more effort (which goes beyond the scope of this article), the time dependence of the vibrational Hamiltonian at least gives a rough idea of the spectral evolution of the system.
The vibrational frequencies of the system depend on the conformation of the peptide as well as on its solvation in the DMSO solvent. To account for the former effect, we consider the shift of the local mode frequency δεn = εn – 〈εn〉 of the nth residue and the vibrational coupling βnm between residues n and m, which was previously (41) calculated as a function of the (φn, ψn) dihedral angles of the peptide backbone (see Methods). Fig. 7 shows the short-time evolution of these quantities for the residues Ala-1, Cys-2, Asp-6, and Gly-7, which exhibit the most significant structural changes. Both local frequency shifts and vibrational couplings are seen to undergo changes of ≈0.5–2 cm−1 within the first 200 ps. Similar to the corresponding (φ, ψ) dihedral angles shown in Fig. 6, the conformational rearrangement of Ala-1 results in the largest changes of δε and β. It is interesting to note that the time constants found for the vibrational matrix elements and for the corresponding dihedral angles may differ considerably. While we find similar times for Ala-1 and Cys-2, the rise times of β for Asp-6 (15 ps) and Gly-7 (9 ps) are significantly shorter than the times constants for ψAsp6 (131 ps) and ψGly7 (110 ps). We have also studied the time-dependent distributions of δε and β (data not shown). As a consequence of the large conformational heterogeneity seen in the (φn, ψn) distributions (Fig. 5), the distributions of the vibrational matrix elements are quite broad, thus giving rise to a substantial broadening of the infrared spectrum.
FIGURE 7.

Time evolution and exponential fits of the intramolecular frequency shifts δε and vibrational couplings β, obtained for the residues Ala-1, Cys-2, Asp-6, and Gly-7.
The conformational rearrangement of the peptide may also influence its solvation, which in turn may change the vibrational frequencies of the system. Adopting an empirical model (44) that estimates the solvent-induced amide I shifts δεn of each residue (see Methods), we have calculated the mean frequency shift 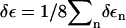 as a function of time. The time evolution of the solvent-induced amide I shift shown in Fig. 8 reveals a gradual increase of δε of ∼2 cm−1. An exponential fit of the data for the first 50 ps results in a time constant of 32 ps. Since the spatial distribution of solvent molecules around the peptide may vary significantly for different nonequilibrium trajectories, the convergence of δε with respect to the number of trajectories is rather slow and probably not fully achieved with 400 trajectories. Compared to aqueous solvent, the DMSO frequency shift is quite small, reflecting the fact that intermolecular hydrogen bonds play a comparatively minor role in this system.
as a function of time. The time evolution of the solvent-induced amide I shift shown in Fig. 8 reveals a gradual increase of δε of ∼2 cm−1. An exponential fit of the data for the first 50 ps results in a time constant of 32 ps. Since the spatial distribution of solvent molecules around the peptide may vary significantly for different nonequilibrium trajectories, the convergence of δε with respect to the number of trajectories is rather slow and probably not fully achieved with 400 trajectories. Compared to aqueous solvent, the DMSO frequency shift is quite small, reflecting the fact that intermolecular hydrogen bonds play a comparatively minor role in this system.
FIGURE 8.

Time evolution and exponential fit of the mean solvent-induced amide I frequency shift δε. The convergence with respect to the number of trajectories is indicated.
DISCUSSION AND CONCLUSIONS
The nonequilibrium MD simulation of bcAMPB draws a detailed picture of the photoinduced dynamics of the photoswitchable peptide. In particular, the time-dependent distributions of global and local reaction coordinates reveal that the conformational rearrangement of the peptide is rather complex and occurs on at least four timescales.
Femtosecond cis→trans photoisomerization. After photoexcitation, the azobenzene unit of bcAMPB undergoes nonadiabatic photoisomerization within 200 fs.
Picosecond-driven dynamics. On the picosecond timescale, two main processes take place. Cooling: The photoinduced excess energy of the azobenzene chromophore is rapidly redistributed to the vibrational modes of the peptide (1 ps) as well as to the surrounding solvent molecules (13 ps). Stretching: The isomerization causes an ultrafast (0.2 and 14 ps) stretching of the peptide, where the latter time constant reflects the competition between the driving force of the photoswitch and the restraining force of the peptide.
Fast conformational rearrangement. Most local and global reaction coordinates exhibit a 50–100 ps component (for example, ψAla1 (57 ps), ψGly7 (110 ps), Rg (44 ps), and RMSD (68 ps)), reflecting fast initial conformational transitions. The most significant conformational changes are observed for Ala-1, which is directly connected to the photoswitch and undergoes a β↔α transition, and for Gly-7, which is the most flexible residue. Since the photoinduced excess energy is dissipated on a 13-ps timescale, only the first part of this dynamics is directly driven by the photoisomerization.
Slow conformational equilibration. The slow 500–1000 ps component observed in the simulation accounts for the subsequent conformational equilibration of the system. This process is diffusion-controlled, as the peptide needs solvent-driven fluctuations to escape from traps on the way to the final state.
On a qualitative level, the MD description of the photoinduced molecular processes is in remarkable agreement with the experimental studies in Wachtveitl et al. (18) and Bredenbeck et al. (19,20). In particular, the experimental data also indicate the above-discussed timescales 1–4 of the photoreaction. From a more quantitative point of view, the MD simulation appears to reproduce the fastest (0.2 ps) as well as the slowest (500–1000 ps) timescales observed in experiment. For the two intermediate timescales reflecting the cooling and the fast conformational rearrangements, however, the situation is somewhat more complicated.
Let us compare the above results to the outcome of time-resolved experiments monitoring the cooling of the photoexcited bcAMPB. In transient infrared spectroscopy on bcAMPB (19), the cooling is reflected by a red-shifted hot band of the amide I spectrum, which occurs immediately after photoswitching and decays on a timescale of 4 ps. The red shift is caused by nonthermally excited low frequency modes of the peptide that are anharmonically coupled to the amide I vibrations. Similarly, optical pump probe spectroscopy yielded a 5.4 ps decay time, which is thought to reflect cooling (18). Assuming that these spectral features can be directly correlated to the decay of the peptide vibrational energy, the calculated time constant of 13 ps is somewhat too long. Most likely, this deviation is caused by the united-atom representation of the DMSO solvent model (46). Alternatively, one may employ a flexible all-atom model of the solvent (62), which adds more degrees of freedom, therefore increasing the heat capacity and the ability to cool the solute (albeit at the cost of other problems such as the classical description of high-frequency modes). In Nguyen and Stock (32), we have shown that the timescale of the cooling virtually does not affect the subsequent conformational rearrangements of the peptide. Taking this fact into account as well as the sum of all uncertainties in experiment (e.g., deconvolution and interpretation of the spectra) and computation (e.g., force field and the neglect of quantum effects), the agreement appears satisfactory.
Whereas the experimental assignment of the hot bands describing the cooling process is relatively straightforward, the interpretation of the spectral features accounting for the conformational rearrangement of the peptide is not that clear. From optical pump probe spectroscopy (18) of bcAMPB, two kinetic components with time constants of 100 and 1000 ps were obtained that presumably reflect conformational dynamics. The timescales are in general agreement with transient two-dimensional infrared spectroscopy (20), which revealed substantial changes of the spectra for times up to 1 ns. On the other hand, transient (one-dimensional) infrared spectroscopy (19) showed a blue-shifted signature, which is almost equivalent to the stationary FTIR difference spectrum and is formed on a timescale of only 6 ps.
Although the simulated 50–100 ps timescale found for the fast conformational rearrangements is in accordance with the results of optical and two-dimensional infrared spectroscopy, it is clearly in variance with the interpretation of the 6-ps time constant deduced from the transient infrared spectrum. Hence we arrive at the question to what extent 1) the calculated timescale is too slow and/or 2) the interpretation of the experimental timescale is not appropriate. Regarding the former point, it should be kept in mind that free energy barriers obtained from different MD force fields may vary significantly. For example, the frequency of α↔β conformational transitions of trialanine was found to differ by an order of magnitude when different force fields were employed (63). On the other hand, given the diffuse and structureless appearance of the amide I band of bcAMPB with an overall infrared shift of only ≈2 cm−1 between cis and trans conformations, it is clear that the interpretation of the experimental findings is rather difficult.
To investigate possible reasons for this apparent discrepancy, we have considered several aspects of the problem.
As a first step toward a more direct modeling of transient infrared spectra, we have calculated the time-dependent amide I frequency shift from the nonequilibrium simulations. Both intramolecular and solvent-induced contributions to the frequency shift were found to change by
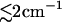 , in reasonable agreement with experiment. Interestingly, it was shown that the vibrational time constants found for the frequency shifts and the corresponding conformational motions may differ considerably. For the solvent-induced frequency shift, a rise time of 32 ps was found.
, in reasonable agreement with experiment. Interestingly, it was shown that the vibrational time constants found for the frequency shifts and the corresponding conformational motions may differ considerably. For the solvent-induced frequency shift, a rise time of 32 ps was found.We have performed various test calculations at higher temperature (350 K) and for a different force field (GROMOS 45A3 (64)). At least at short times, no significant changes of the overall conformation were observed.
To study the effect of the solvent, a further nonequilibrium simulation of bcAMPB in D2O was performed (unpublished). In direct agreement with optical pump-probe experiment on a similar bicyclic photoswitchable peptide in water (14), we obtain a twofold acceleration of both the cooling and the conformational dynamics.
Although the above investigations have confirmed the reliability of the nonequilibrium MD approach, it is clear that a quantitative comparison to experiment can only be obtained by a direct calculation of the measured transient spectra. In particular, the simulation of transient two-dimensional infrared spectra holds great promise to give a detailed description of the photoinduced conformational dynamics of biomolecules. Work along these lines is in progress.
Acknowledgments
We thank Peter Hamm and Josef Wachtveitl for numerous inspiring and helpful discussions.
This work has been supported by the Frankfurt Center for Scientific Computing, the Fonds der Chemischen Industrie, and the Deutsche Forschungsgemeinschaft.
References
- 1.Wand, A. J. 2001. Dynamics activation of protein function: a view emerging from NMR spectroscopy. Nat. Struct. Biol. 8:926–931. [DOI] [PubMed] [Google Scholar]
- 2.Leulliot, N., and G. Varani. 2001. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry. 40:7947–7956. [DOI] [PubMed] [Google Scholar]
- 3.Benkovic, S. J., and S. Hammes-Schiffer. 2003. A perspective on enzyme catalysis. Science. 301:1196–1202. [DOI] [PubMed] [Google Scholar]
- 4.Rief, M., and H. Grubmüller. 2002. Force spectroscopy of single biomolecules. ChemPhysChem. 3:255–261. [DOI] [PubMed] [Google Scholar]
- 5.Volk, M. 2001. Fast initiation of peptide and protein folding processes. Eur. J. Org. Chem. 14:2605–2621. [Google Scholar]
- 6.Chung, H., M. Khalil, A. W. Smith, Z. Ganim, and A. Tokmakoff. 2005. Conformational changes during the nanosecond-to-millisecond unfolding of ubiquitin. Proc. Natl. Acad. Sci. USA. 102:612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moerner, W. E., and M. Orrit. 1999. Illuminating single molecules in condensed phase. Science. 283:1670–1676. [DOI] [PubMed] [Google Scholar]
- 8.Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- 9.Feringa, B. L., editor. 2001. Molecular Switches. Wiley-VCH, Weinheim.
- 10.Kumita, J. R., O. S. Smart, and G. A. Woolley. 2000. Photo-control of helix content in a short peptide. Proc. Natl. Acad. Sci. USA. 97:3803–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renner, C., J. Cramer, R. Behrendt, and L. Moroder. 2000. Photomodulation of conformational states. II. Mono- and bicyclic peptides with (4-aminomethyl)-phenylazobenzoic acid as backbone constituent. Biopolymers. 54:501–514. [DOI] [PubMed] [Google Scholar]
- 12.Cattani-Scholz, A., C. Renner, C. Cabrele, R. Behrendt, D. Oesterheld, and L. Moroder. 2002. Photoresponsive cyclic bis(cysteinyl)peptides as catalysts of oxidative protein folding. Angew. Chem. Int. Ed. Engl. 41:289–292. [DOI] [PubMed] [Google Scholar]
- 13.Renner, C., U. Kusebauch, M. Löweneck, A. G. Milbradt, and L. Moroder. 2004. Photomodulation of conformational states. II. Mono- and bicyclic peptides with (4-aminomethyl)-phenylazobenzoic acid as backbone constituent. J. Pept. Res. 65:4–14. [Google Scholar]
- 14.Satzger, H., C. Root, C. Renner, R. Behrendt, L. Moroder, J. Wachtveitl, and W. Zinth. 2004. Picosecond dynamics in water-soluble azobenzene-peptides. Chem. Phys. Lett. 396:191–197. [Google Scholar]
- 15.Zhao, J., D. Wildemann, M. Jakob, C. Vargas, and C. Schiene-Fischer. 2003. Direct photomodulation of peptide backbone conformations. Chem. Commun. 22:2810–2811. [DOI] [PubMed] [Google Scholar]
- 16.Helbing, J., H. Bregy, J. Bredenbeck, R. Pfister, P. Hamm, R. Huber, J. Wachtveitl, L. De Vico, and M. Olivucci. 2004. A fast photoswitch for minimally perturbed peptides: investigation of the cis-trans photoisomerization of n-methylthioacetamide. J. Am. Chem. Soc. 126:8823–8834. [DOI] [PubMed] [Google Scholar]
- 17.Spörlein, S., H. Carstens, C. Renner, R. Behrendt, L. Moroder, P. Tavan, W. Zinth, and J. Wachtveitl. 2002. Ultrafast spectroscopy reveals subnanosecond peptide conformational dynamics and validates molecular dynamics simulation. Proc. Natl. Acad. Sci. USA. 99:7998–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wachtveitl, J., S. Spörlein, H. Saltger, B. Fonrobert, C. Renner, R. Behrendt, D. Oesterhelt, L. Moroder, and W. Zinth. 2004. Ultrafast conformational dynamics in cyclic azobenzene peptides of increased flexibility. Biophys. J. 86:2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bredenbeck, J., J. Helbing, A. Sieg, T. Schrader, W. Zinth, C. Renner, R. Behrendt, L. Moroder, J. Wachtveitl, and P. Hamm. 2003. Picosecond conformational transition and equilibration of a cyclic peptide. Proc. Natl. Acad. Sci. USA. 100:6452–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bredenbeck, J., J. Helbing, C. Renner, L. Moroder, J. Wachtveitl, and P. Hamm. 2003. Transient 2D-IR spectroscopy: snapshots of the nonequilibrium ensemble during the picosecond conformational transition of a small peptide. J. Phys. Chem. B. 107:8654–8660. [Google Scholar]
- 21.Mukamel, S., and R. M. Hochstrasser, editors. 2001. Special Issue on Multidimensional Spectroscopies. Chem. Phys. 266:2–3. [Google Scholar]
- 22.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformations of peptides, polypeptides, and proteins. Adv. Protein Chem. 38:181–364. [DOI] [PubMed] [Google Scholar]
- 23.Torii, H., and M. Tatsumi. 1996. Infrared Spectroscopy of Biomolecules. Wiley-Liss, New York.
- 24.Nguyen, P. H., Y. Mu, and G. Stock. 2005. Structure and dynamics of a photoswitchable peptide: a replica exchange molecular dynamics study. Proteins. 60:485–494. [DOI] [PubMed] [Google Scholar]
- 25.Carstens, H., C. Renner, A. G. Milbradt, L. Moroder, and P. Tavan. 2005. Multiple loop conformations of peptides predicted by molecular dynamics simulations are compatible with nuclear magnetic resonance. Biochemistry. 44:4829–4840. [DOI] [PubMed] [Google Scholar]
- 26.Berweger, C. D., F. Müller-Plathe, and W. F. van Gunsteren. 1998. Molecular dynamics simulation with an ab initio potential energy function and finite element interpolation: the photoisomerization of cis-stilbene in solution. J. Chem. Phys. 108:8773–8781. [Google Scholar]
- 27.Scheurer, C., A. Piryatinski, and S. Mukamel. 2001. Signatures of β-peptide unfolding in two-dimensional vibrational echo spectroscopy: a simulation study. J. Am. Chem. Soc. 123:3114–3124. [DOI] [PubMed] [Google Scholar]
- 28.Röhrig, U. F., L. Guidoni, and U. Röthlisberger. 2002. Early steps of the intramolecular signal transduction in rhodopsin explored by molecular dynamics simulations. Biochemistry. 41:10799–10809. [DOI] [PubMed] [Google Scholar]
- 29.Ben-Nun, M., F. Molnar, H. Lu, J. C. Philips, T. J. Martinez, and K. Schulten. 1998. Quantum dynamics of the femtosecond photoisomerization of retinal in bacteriorhodopsin. Faraday Discuss. 110:447–462. [DOI] [PubMed] [Google Scholar]
- 30.Saam, J., E. Tajkhorshid, S. Hayashi, and K. Schulten. 2002. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys. J. 83:3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groenhof, G., M. Bouxin-Cademartory, B. Hess, S. P. de Visser, H. J. C. Berendsen, M. Olivucci, A. E. Mark, and M. A. Robb. 2004. Photoactivation of the photoactive yellow protein: why photon absorption triggers a trans-to-cis isomerization of the chromophore in the protein. J. Am. Chem. Soc. 126:4228–4283. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen, P. H., and G. Stock. 2006. Nonequilibrium molecular dynamics simulation of a photoswitchable peptide. Chem. Phys. 323:36–44. [Google Scholar]
- 33.Mukamel, S. 1995. Principles of Nonlinear Optical Spectroscopy. University Press, Oxford.
- 34.Corcelli, S. A., C. P. Lawrence, and J. L. Skinner. 2004. Combined electronic structure/molecular dynamics approach for ultrafast infrared spectroscopy of dilute HOD in liquid H2O and D2O. J. Chem. Phys. 120:8107–8117. [DOI] [PubMed] [Google Scholar]
- 35.la Cour Jansen, T., W. Zhuang, and S. Mukamel. 2004. Stochastic Liouville equation simulation of multidimensional vibrational line shapes of trialanine. J. Chem. Phys. 121:10577–10598. [DOI] [PubMed] [Google Scholar]
- 36.Gnanakaran, S., R. M. Hochstrasser, and A. E. Garcia. 2004. Nature of structural inhomogeneities on folding a helix and their influence on spectral measurements. Proc. Natl. Acad. Sci. USA. 101:9229–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hahn, S., S. Ham, and M. Cho. 2005. Simulation studies of amide I IR absorption and two-dimensional IR spectra of β-hairpins in liquid water. J. Phys. Chem. B. 109:11789–11901. [DOI] [PubMed] [Google Scholar]
- 38.Torii, H., and M. Tasumi. 1998. Ab initio molecular orbital study of the amide I vibrational interactions between the peptide groups in di- and tripeptides and considerations on the conformation of the extended helix. J. Raman Spectrosc. 29:81–86. [Google Scholar]
- 39.Cha, S., S. Ham, and M. Cho. 2002. Amide I vibrational modes in glycine dipeptide analog: ab initio calculation studies. J. Chem. Phys. 117:740–750. [Google Scholar]
- 40.Moran, A. M., and S. Mukamel. 2004. The origin of vibrational mode couplings in various secondary structural motifs of polypeptides. Proc. Natl. Acad. Sci. USA. 101:506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gorbunov, R. D., D. S. Kosov, and G. Stock. 2005. Ab initio-based exciton model of amide I vibrations in peptides: definition, conformational dependence, and transferability. J. Chem. Phys. 122:224904–224915. [DOI] [PubMed] [Google Scholar]
- 42.Bour, P., and T. Keiderling. 2003. Empirical modeling of the peptide amide I band IR intensity in water solution. J. Chem. Phys. 119:11253–11262. [Google Scholar]
- 43.Ham, S., J.-H. Kim, H. Lee, and M. Cho. 2003. Correlation between electronic and molecular structure distortions and vibrational properties. II. Amide I modes of NMA–ND2O complexes. J. Chem. Phys. 118:3491–3498. [Google Scholar]
- 44.Schmidt, J. R., S. A. Corcelli, and J. L. Skinner. 2004. Ultrafast vibrational spectroscopy of water and aqueous n-methylacetamide: comparison of different electronic structure/molecular dynamics approaches. J. Chem. Phys. 121:8887–8896. [DOI] [PubMed] [Google Scholar]
- 45.van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Vdf Hochschulverlag AG an der ETH Zürich, Zürich.
- 46.Liu, H., F. Müller-Plathe, and W. F. van Gunsteren. 1995. A force field for liquid dimethyl sulfoxide and physical properties of liquid dimethyl sulfoxide calculated using molecular dynamics simulation. J. Am. Chem. Soc. 117:4363–4366. [Google Scholar]
- 47.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 91:43–56. [Google Scholar]
- 48.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Modeling. 7:306–317. [Google Scholar]
- 49.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical-integration of Cartesian equations of motions of a system with constraints-molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 50.Darden, T., D. York, and L. Petersen. 1993. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 51.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. Dinola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 52.Ichiye, T., and M. Karplus. 1991. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 11:205–217. [DOI] [PubMed] [Google Scholar]
- 53.Garcia, A. E. 1992. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 68:2696–2699. [DOI] [PubMed] [Google Scholar]
- 54.Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 55.Mu, Y., P. H. Nguyen, and G. Stock. 2005. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins. 58:45–52. [DOI] [PubMed] [Google Scholar]
- 56.Moritsugu, K., and A. Kidera. 2004. Protein motions represented in moving normal coordinates. J. Phys. Chem. B. 108:3890–3898. [Google Scholar]
- 57.DeCamp, M. F., D. DeFlores, J. M. McCracken, A. Tokmakoff, K. Kwac, and M. Cho. 2005. Amide I vibrational dynamics of n-methylacetamide in polar solvents: the role of electrostatic interactions. J. Phys. Chem. B. 109:11016–11026. [DOI] [PubMed] [Google Scholar]
- 58.Egorov, S., K. F. Everitt, and J. L. Skinner. 1999. Quantum dynamics and vibrational relaxation. J. Phys. Chem. A. 103:9494–9499. [Google Scholar]
- 59.Leitner, D. M. 2005. Heat transport in molecules and reaction kinetics: the role of quantum energy flow and localization. Adv. Chem. Phys. 130B:205–256. [Google Scholar]
- 60.Fujisaki, H., and J. E. Straub. 2005. Vibrational energy relaxation in proteins. Proc. Natl. Acad. Sci. USA. 102:6726–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reference deleted in proof.
- 62.Strader, M. L., and S. E. Feller. 2002. A flexible all-atom model of dimethyl sulfoxide for molecular dynamics simulations. J. Phys. Chem. A. 106:1074–1080. [Google Scholar]
- 63.Mu, Y., D. S. Kosov, and G. Stock. 2003. Conformational dynamics of trialanine in water. II. Comparison of AMBER, CHARMM, GROMOS, and OPLS force fields to NMR and infrared experiments. J. Phys. Chem. B. 107:5064–5073. [Google Scholar]
- 64.Schuler, L. D., X. Daura, and W. F. van Gunsteren. 2001. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 22:1205–1218. [Google Scholar]