

Abstract
The electrostatic potentials within the pore of the nicotinic acetylcholine receptor (nAChR) were determined using lanthanide-based diffusion-enhanced fluorescence energy transfer experiments. Freely diffusing Tb3+-chelates of varying charge constituted a set of energy transfer donors to the acceptor, crystal violet, a noncompetitive antagonist of the nAChR. Energy transfer from a neutral Tb3+-chelate to nAChR-bound crystal violet was reduced 95% relative to the energy transfer to free crystal violet. This result indicated that crystal violet was strongly shielded from solvent when bound to the nAChR. Comparison of energy transfer from positively and negatively charged chelates indicate negative electrostatic potentials of −25 mV in the channel, measured in low ionic strength, and −10 mV measured in physiological ionic strength. Debye-Hückel analyses of potentials determined at various ionic strengths were consistent with 1–2 negative charges within 8 Å of the crystal violet binding site. To complement the energy transfer experiments, the influence of pH and ionic strength on the binding of [3H]phencyclidine were determined. The ionic strength dependence of binding affinity was consistent with −3.3 charges within 8 Å of the binding site, according to Debye-Hückel analysis. The pH dependence of binding had an apparent pKa of 7.2, a value indicative of a potential near −170 mV if the titratable residues are constituted of aspartates and glutamates. It is concluded that long-range potentials are small and likely contribute little to selectivity or conductance whereas close interactions are more likely to contribute to electrostatic stabilization of ions and binding of noncompetitive antagonists within the channel.
INTRODUCTION
The nicotinic acetylcholine receptor (nAChR) is a pentameric ligand gated ion channel with a subunit stoichiometry of α2βγδ; the subunits are arranged with rotational pseudosymmetry about a central pore that constitutes the ion conductive pathway (1). Cryoelectron microscopic imaging has shown the channel pore to comprise a large extracellular vestibule with a diameter near 20 Å that narrows at the transmembrane domain (2–4). Within the transmembrane domain, the channel is lined by the second transmembrane helices (M2) of the five subunits and these helices have been shown to largely determine the conductivity of the channel ((5); see Fig. 1 for illustration).
FIGURE 1.

Cartoon of a cross section of the nAChR with two M2 α-helices shown. Charged residues are clustered at the extracellular and intracellular ends of the transmembrane pore. The putative location of CrV is shown within the extracellular half of the transmembrane pore.
Site-directed mutagenesis defined three rings of charges that particularly affected conduction: an inner ring at the intracellular end of the helix, an intermediate ring near the intracellular end, and an outer ring located near the extracellular end of the M2 helices (6). Changes in charge at these sites from negative to positive reduced net conductance, with the strongest effects at the intermediate ring of charges and smaller effects at the inner rings and outer rings. The role of the outer ring charges, and perhaps others within the large extracellular vestibule, have been proposed to concentrate ions within the vestibule through long-range electrostatic interactions, thereby providing a reservoir of conducting ions that contributes to net conductivity and ionic selectivity (7,8). However, detailed examination of charge changes in the outer ring showed effects of ionic strength changes on conductance that indicated that ionic effects at this locus were predominantly through close interactions or binding of conductive ions rather than long-range electrostatic interactions (9).
Computation using hypothetical charge distributions further argued, though, that a modest charge density within the vestibule could enhance selectivity and conductivity of the channel (10). Homology modeling of the nAChR based on the atomic resolution structure of the acetylcholine binding protein also showed substantial charge density in the upper parts of the vestibule as well (11). Individual, site-directed mutagenesis of these residues affected the overall function of the channel (12); they may also affect selectivity or conductivity through their vestibular charge distribution mechanisms, as proposed by computational modeling. The experimental observations clearly indicated that some charged residues have a strong impact on conduction, though the residues at the extracellular vestibule appeared to have a smaller effect. In contrast, modeling of vestibular potentials indicated a potentially significant role for the net charge that may have been missed by mutagenesis of individual residues.
To determine the impact of vestibular charges on ionic selectivity and conduction, we undertook a direct determination of the relative concentration of ions using lanthanide-based diffusion-enhanced luminescence resonance energy transfer (13,14). This technique compares the energy transfer of three homologous Tb3+-chelates to an energy transfer acceptor, crystal violet (CrV), bound to the nAChR ion channel (15–17). Comparison of changes in energy transfer with the change in chelate charge reveals the extent of the long-range electrostatic interactions within the vestibule. It was determined that the vestibule has a relatively small potential that is attenuated in physiological ionic strength. These results were juxtaposed to the more profound effects of pH and ionic strength on the binding of phencyclidine (PCP), a noncompetitive antagonist that is competitive with CrV, that suggest a larger impact of local potential.
The theoretical basis of lanthanide-based diffusion-enhanced fluorescence energy transfer (DEFET) is well established and permits direct computation of energy transfer if the optical properties of the fluorophores are known and an atomic resolution model of the acceptor and its environment is known (13–15). Computed DEFET rates include the effects of computed electrostatic potentials that, thereby, can be compared directly to experimentally measured potentials. This provides a mutual check on the predictions of computed potentials and experimentally determined potentials. Such a comparison seems warranted: computation of potentials at protein surfaces often relies on continuum electrostatic approximations using the Poisson-Boltzmann equation (18). However, this approach may not be adequate in cases where the protein surface is constricted (19) or can focus the potential, as is likely the case in the vestibule of the nAChR. A particular concern is that the vestibular volume is too small to include a significant number of ions and, therefore, the ionic screening included in the continuum electrostatics may not apply. The volume of the nAChR vestibule is expected to hold ∼3 ions; thus, the continuum approximation may not be valid in this instance. It was argued that perhaps Brownian dynamics simulations provide a better approximation to evaluate the effects of electrostatics on solution ions (19).
In the second article of this series (11), we directly compare computed and experimental potentials in the nAChR vestibule and find that computations tend to overestimate the experimental results severalfold. In the third article we utilize a combination of fluorescence stopped-flow binding kinetics, DEFET, and computation to determine the contribution of local electrostatic potentials to the binding energy at the nAChR active sites and at the AChE active site (20). The results indicate that, despite sizeable local potentials, especially at low ionic strength, the net contribution to binding energy at physiological conditions is smaller than −1 kcal/mol. The observations permit us to conclude that despite the strong charge density in many regions of the nAChR, the direct impact of long-range electrostatic attraction on function, such as binding rate, conductivity, and ionic selectivity is small; charged residues likely have more significant effects on function through direct interactions with substrates.
MATERIALS AND METHODS
Reagents
CrV was purchased from Aldrich Chemical (Milwaukee, WI) and crystallized from chloroform. Carbamylcholine (Carb), EDTA, N-(2-hydroxyethyl)ethylenediaminetriacetate (HED3A), and PCP were purchased from Sigma Chemical (St. Louis, MO). HEPES was from Boehringer Manheim (Roche, Indianapolis, IN). Sucrose and NaCl were purchased from Fisher. N,N′-bis(2-hydroxyethyl)ethylenediaminediacetate (BED2A) was synthesized as described (21). The Tb3+-chelates [Tb3+-EDTA]− (Tb−), [Tb3+-HED3A], (Tb0), and [Tb3+-BED2A]+ (Tb+) were prepared and purified by ion exchange chromatography as described previously (22) and stored as lyophilized powders.
To eliminate binding of CrV, glassware was acid washed and then treated with one of the cationic silanizing reagents N,N-dimethyl-3-aminopropyl-trimethoxysilane or 3-aminopropyl-trimethoxysilane (United Chemical Technologies, Bristol, PA) by vapor diffusion in an 80°C vacuum oven at 25 mmHg vacuum from a 1:6 dilution of silanizing agent in toluene followed by a water rinse. nAChR-rich membranes were prepared from fresh or frozen Torpedo californica electric organ (Marinus, Long Beach, CA) by differential sucrose ultracentrifugation as described previously (23,24). Binding activity of membrane was assayed by [3H]acetylcholine binding and ranged in specific activity from 1.0 to 1.6 nmol/mg. nAChR concentrations are reported as one-half the measured ACh binding-site concentrations.
Spectral measurements
Tb3+-chelate emission spectra were measured on a SLM 8000C fluorescence spectrometer (SLM, Rochester, NY) with upgraded electronics (HORIBA Jobin Yvon; Edison, NJ) or an ISS (Urbana-Champaign, IL) PC1 fluorometer. Excitation and emission monochromators were set to 4 nm bandwidth. Excitation was at 370 nm and emission spectra were collected from 450 to 700 nm. When needed for quantitation, technical emission spectra were corrected for instrument response with manufacturer-provided correction factors. CrV absorbance spectra were measured as described previously (25) alone or in the presence of excess nAChR on a Cary 1/3 UV/VIS spectrophotometer (Varian; Sugarland, TX) using dual beam mode.
The Förster distance R0 is the distance at which energy transfer is 50% efficient for a pair of fluorophores (26) and is given by the following equation:
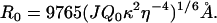 |
(1) |
The spectral overlap between the donor and acceptor fluorophores, J, was calculated from measured donor emission spectra F(λ) and the acceptor absorbance spectra, ɛ(λ), spectra over all wavelengths (λ):
 |
(2) |
Q0, the quantum yields for the donors, Tb+, Tb0, and Tb−, were determined from the lifetime of each chelate, τ0, and the intrinsic radiative lifetime of Tb3+ (τrad): Q0 = τ0/τrad. For τrad we used 4.4 ms, according to Stein and Wurzburg (27). These Q0 values (Table 1) agree with the value of 0.2 determined by Yeh and Meares (28) using standard spectroscopic methods. However, an approach using diffusion-enhanced energy transfer and lifetime measurements indicated a shorter τrad for Tb3+ near 3.3 ms (29); if this latter value is correct, our Q0 values would be systematically different. κ2 is the orientation factor between the donor and acceptor fluorophores and in this case is constrained by the isotropic emission fluorescence, which limits the value from 1/3 to 4/3; the average value of 2/3 was used (15). The refractive index η of water is 1.33.
TABLE 1.
Energy transfer characteristics for Tb3+-chelates and CrV
| nAChR-bound CrV
|
Free CrV
|
||||||
|---|---|---|---|---|---|---|---|
| Chelate | τ0(ms) | κ2 | Q | J* (M−1cm3) | R0 Å | J (M−1cm3) | R0 Å |
| Tb+ | 0.85 | 1/3–4/3 | 0.182 | 4.80 × 10−13 | 50.7 ± 8.2 | 4.79 × 10−13 | 50.7 ± 8.3 |
| Tb0 | 0.89 | 1/3–4/3 | 0.187 | 4.81 × 10−13 | 50.9 ± 8.3 | 4.83 × 10−13 | 50.9 ± 8.3 |
| Tb− | 1.16 | 1/3–4/3 | 0.244 | 4.83 × 10−13 | 53.2 ± 8.6 | 4.88 × 10−13 | 53.1 ± 8.6 |
J, Q0, η, and κ2 were determined as described in Materials and Methods. R0 was computed for κ2 = 2/3; the range reflects the span of κ2.
Luminescence lifetime measurements
Terbium fluorescent lifetimes were measured on a PhosphoCube fluorescence lifetime instrument from HORIBA Jobin Yvon IBH with a 370-nm wavelength SpectraLED light source passed through an ultraviolet-pass filter and TBX-04 photodetector with a 545-nm bandpass filter or a 595 cut-on filter (Oriel, Stratford, CT) for detection of emission. A homebuilt instrument was used in some experiments whereby the sample was excited by a mechanically chopped beam from an Ar-ion laser tuned to 363 nm. The instrument is described in detail by Lamture et al. (30). Luminescence decays were typically measured over 14–20 ms from 10,000 repeated flashes of ∼1 ms duration. Data were binned in 10 μs intervals. Tb3+-chelate fluorescence decays were fitted to an equation for a single exponential decay with a background.
The bimolecular rate of energy transfer from Tb3+-chelates to CrV, 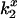 , where x refers to chelate charge, was calculated from the increase in observed fluorescence decay rate (kobs = 1/τobs, s−1) with increasing CrV concentration:
, where x refers to chelate charge, was calculated from the increase in observed fluorescence decay rate (kobs = 1/τobs, s−1) with increasing CrV concentration:
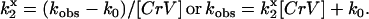 |
(3) |
Energy transfer was typically measured in 1 mM terbium chelate, with or without 3 μM nAChR rich Torpedo membranes desensitized with 1 mM Carb, and with increasing CrV concentrations titrated into samples up to 2 μM. This concentration of Carb was sufficient to fully desensitize the AChR and does not block noncompetitive antagonist binding to the channel (31). Low ionic strength buffer was 10 mM HEPES pH 7.0. High ionic strength buffer was either 10 mM HEPES, 300 mM NaCl, pH 7.0 or HTPS (250 mM NaCl, 5 mM KCl, 3 mM CaCl2, 2 mM MgCl2, and 20 mM HEPES, pH 7.0). Because of the low buffering capacity, the pH was titrated to the correct value immediately before each experiment.
The electrostatic potential (ψ) was computed from energy transfer rate measurements using a Boltzmann relation:
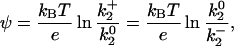 |
(4) |
where kB is Boltzmann's constant, e is the unit electron charge, and 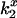 is the measured bimolecular rate of energy transfer for Tb+, Tb0, and Tb−. Because of the unit difference in charge distributed about the neutral chelate, the potential measured by the ratio of the Tb+ and Tb0 chelates is expected to equal that of the Tb0 and Tb−. The potentials from both pairs were averaged. Alternatively, the potential was computed from ψ = (kBT/e)*(slope of ln(
is the measured bimolecular rate of energy transfer for Tb+, Tb0, and Tb−. Because of the unit difference in charge distributed about the neutral chelate, the potential measured by the ratio of the Tb+ and Tb0 chelates is expected to equal that of the Tb0 and Tb−. The potentials from both pairs were averaged. Alternatively, the potential was computed from ψ = (kBT/e)*(slope of ln(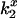 ) versus charge), which is equivalent to Eq. 4 but includes all three
) versus charge), which is equivalent to Eq. 4 but includes all three 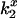 values simultaneously. The mean ± SE for the electrostatic potential σψ was determined from the mean ± SE of several independent determinations or by propagation of errors using the experimentally determined standard deviations for the
values simultaneously. The mean ± SE for the electrostatic potential σψ was determined from the mean ± SE of several independent determinations or by propagation of errors using the experimentally determined standard deviations for the  (σx) by Eq. 5 (32):
(σx) by Eq. 5 (32):
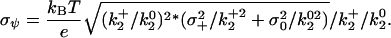 |
(5) |
The effect of ionic strength shielding on electrostatic potential was measured from changes in energy transfer rates from the Tb3+-chelates to bound CrV with increasing concentrations of NaCl, titrated into the sample from a concentrated, 5 M solution in HEPES, pH 7.0. The electrostatic potential as a function of ionic strength was fitted to a Debye-Hückel equation to estimate the effective interaction distance near the acceptor fluorophore (r) and the number of charges experienced in that region (z) using Eq. 6:
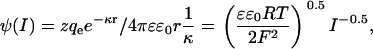 |
(6) |
where the potential at a given ionic strength 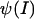 is a function of the elementary charge qe, the polarizability of free space, ɛ0, the dielectric constant, ɛ, and the Debye length 1/κ. The Debye length characterizes the screening effect of ionic strength on potential experienced about a point charge where R is the gas constant, F is Faraday's constant, and I is the ionic strength (33).
is a function of the elementary charge qe, the polarizability of free space, ɛ0, the dielectric constant, ɛ, and the Debye length 1/κ. The Debye length characterizes the screening effect of ionic strength on potential experienced about a point charge where R is the gas constant, F is Faraday's constant, and I is the ionic strength (33).
Binding data were fit to a distinct equation derived from Debye-Hückel theory that describes the change in dissociation constant with ionic strength (34,35):
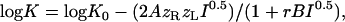 |
(7) |
where A and B are thermodynamic constants (A = 0.509 and B = 2.391 nm−1 at 298 K) and zR and zL are the charges on the receptor and ligand, respectively.
[3H]PCP binding
[3H]PCP binding was carried out by the centrifugation assay as described by Pedersen (36) and by Lurtz et al. (37). Typically, 50 or 100 μg of nAChR-rich membranes with activity of ∼0.1–0.2 nmol nAChR/mg in 400 μl (25–50 nM nAChR) were incubated with ∼1 nM [3H]PCP and 1 mM Carb, to desensitize the nAChR. At each condition, bound ligand was determined by counting the pellets; free ligand was determined by counting a 100-μl aliquot of the supernatant. Nonspecific binding was determined for each condition using parallel samples that also included 25 μM proadifen; this was subtracted from the total binding to yield specific binding. Because the concentration of nAChR was in large excess over [3H]PCP, free nAChR was essentially equivalent to the concentration added. From this value and from the measured concentrations of free and bound [3H]PCP the affinity constant was determined: K = [RL]/[L][R] where R is the nAChR, L is phencyclidine, and RL the bound complex. In experiments with varying salt and pH, the buffers were titrated to the desired pH at each salt concentration immediately before the experiment. The pKa of PCP is 10 as measured by titration and thus was predominantly protonated across the experimental pH range used for binding.
RESULTS
To determine the electrostatic potential within the nAChR pore, we measured DEFET rates from variously charged Tb3+-chelates to CrV bound to the nAChR. This technique was used previously to determine the electrostatic environment near myoglobin (15), DNA (17), and cytochrome c (38). It takes advantage of the long, millisecond luminescent lifetime of Tb3+. During the excited-state lifetime, the chelate effectively samples the nearby volume, including regions near acceptors where the probability of energy transfer is greater. Changes in lifetimes can be measured precisely using time-correlated photon counting to determine the extent of energy transfer, as given by the rate constant for energy transfer (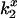 , Eq. 3). The efficiency of energy transfer depends on the acceptor accessibility and on the local electrostatic potential, due to changes in local concentration of charged Tb3+-chelates (13–15). The relative DEFET rates from the three homologous Tb3+-chelates, Tb−, Tb0, and Tb+, which vary in charge (see Fig. 2), determine an effective electrostatic potential in the region near the acceptor.
, Eq. 3). The efficiency of energy transfer depends on the acceptor accessibility and on the local electrostatic potential, due to changes in local concentration of charged Tb3+-chelates (13–15). The relative DEFET rates from the three homologous Tb3+-chelates, Tb−, Tb0, and Tb+, which vary in charge (see Fig. 2), determine an effective electrostatic potential in the region near the acceptor.
FIGURE 2.

Chemical structures of (A) Tb3+-chelates, (B) CrV, and (C) PCP.
CrV as an acceptor of Tb3+ energy transfer
DEFET depends directly on the acceptor concentration (Eq. 3) and the acceptor optical properties. To obtain sufficient energy transfer at nAChR concentrations in the low micromolar range, we required an acceptor with high extinction coefficient and with spectral overlap of Tb3+ emission, as characterized by the Förster distance for energy transfer, R0 (Eq. 1). CrV (Fig. 2) was previously identified for this purpose as a high affinity (KD ∼ 10 nM), fluorescent, noncompetitive antagonist of the nAChR that binds with a 1:1 stoichiometry (25). The overlap between the emission spectrum of the Tb3+-chelates and the absorbance spectra for CrV, free or bound to excess nAChR, are shown in Fig. 3. The spectral overlap integrals, J, for these compounds were calculated from corrected spectra according to Eq. 2 and are given in Table 1. The R0 values for the three chelates are ∼50 Å; variations between the three chelates are due to small differences in chelate emission spectra and in lifetimes. CrV exhibits a 10-nm shift in peak absorbance upon binding the nAChR, which yields a small change in R0 of <0.1 Å. The primary uncertainty in the R0 arises from the orientation factor κ2, though κ2 is limited by the isotropic Tb3+ emission, as described in Materials and Methods. The uncertainties in the R0 values, however, do not affect the interpretation of the DEFET rates and electrostatic potentials. These R0 values demonstrate that CrV is a competent acceptor and are primarily utilized for computational determination of DEFET rates, as described in the accompanying article (11). In those computations, κ2 was computed explicitly from the orientation of the acceptor, thereby reducing the uncertainty in R0.
FIGURE 3.

Spectral overlap of Tb− and CrV. The emission spectrum of 1 mM Tb− (solid line) was taken in H2O as described in Materials and Methods. Absorption spectra of 400 nM free CrV (dashed line) or bound to 512 nM nAChR (dotted line) were measured in HTPS (data from Lurtz and Pedersen (25)) and the data converted to the extinction coefficient.
Diffusion-enhanced fluorescence energy transfer measurements
Tb3+-chelates characteristically exhibit single-exponential luminescent decay with lifetimes near 1 ms. The data shown in Fig. 4 illustrate the decay of Tb+ (Fig. 4 A), Tb0 (Fig. 4 B), and Tb− (Fig. 4 C), each in the absence of added acceptor (black points), in the presence of free CrV (blue points), and in the presence of CrV bound to the nAChR (red points). Free CrV substantially decreases the lifetime of each chelate due to DEFET. The change was greatest for Tb− and smallest for Tb+ consistent with the presence of positive electrostatic potential about free CrV. Bound CrV alters the lifetimes less than free CrV, reflecting less DEFET, which is consistent with lower accessibility of the Tb3+-chelates to bound CrV; the  value for bound CrV is ∼5% of that of free CrV (see Fig. 4 B and Table 2). Table 2 shows decay-rate data and the calculated
value for bound CrV is ∼5% of that of free CrV (see Fig. 4 B and Table 2). Table 2 shows decay-rate data and the calculated  values for a typical DEFET experiment. The pattern of changes in DEFET indicated by the trend of the
values for a typical DEFET experiment. The pattern of changes in DEFET indicated by the trend of the  values with charge qualitatively indicates the electrostatic environment. The values decrease with charge for free CrV and increase for bound CrV, consistent with CrV bound in a negatively charged environment within the channel.
values with charge qualitatively indicates the electrostatic environment. The values decrease with charge for free CrV and increase for bound CrV, consistent with CrV bound in a negatively charged environment within the channel.
FIGURE 4.

Tb-chelate fluorescent lifetime decay. Fluorescent lifetimes for (A) Tb+, (B) Tb0, and (C) Tb− were measured with 1 mM Tb3+-chelate in 10 mM HEPES, pH 7.0. Decay rates for the chelate alone (black dots), with 2 μM CrV (blue dots), and with 2 μM CrV bound to 3 μM nAChR with 1 mM Carb (red dots). Solid lines indicate a single-exponential decay fitted to the data.
TABLE 2.
Tb3+-chelate DEFET to free and bound CrV
| Free CrV
|
|||
|---|---|---|---|
| [CrV] (μM) | Tb+ | Tb0kobs (s−1)* | Tb− |
| 0 | 1193 ± 1 | 1119 ± 3 | 865 ± 1 |
| 2 | 1463 ± 3 | 1689 ± 25 | 1925 ± 13 |
| k2 (μM−1s−1)† | 135 ± 2 | 285 ± 13 | 529 ± 7 |
| CrV, 3 μM nAChR
|
|||
| kobs (s−1) | |||
| 0 | 1107 ± 1 | 1082 ± 4 | 855 ± 3 |
| 2 | 1216 ± 8 | 1106 ± 5 | 865 ± 1 |
| k2 (μM−1s−1)† | 54 ± 4 | 12 ± 3 | 5 ± 2 |
Luminescence decays were recorded for 1 mM Tb3+-chelates in 10 mM HEPES, pH 7.0; 2 μM CrV was added without and with 3 μM nAChR present. Decay rates are presented as the average and standard deviation of triplicate measurements from the same sample.
DEFET rates, 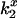 , were computed from kobs before and after addition of CrV (Eq. 3). Errors in
, were computed from kobs before and after addition of CrV (Eq. 3). Errors in 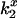 were derived by propagation of errors in kobs.
were derived by propagation of errors in kobs.
To gauge the effect of ionic strength on the electrostatic potential within the pore of the nAChR, DEFET measurements were made at both low and high ionic strength conditions. To improve precision, decay rates were determined by titrating samples with several CrV concentrations; DEFET rates,  , were determined from the slopes using the linear form of Eq. 3. The changes in decay rates are shown in Fig. 5. The DEFET rates, indicated by the slopes, for free CrV (Fig. 5, A and B) are much higher than those for CrV bound to the nAChR (Fig. 5, C and D). Free CrV shows the highest energy transfer to Tb− and the least to Tb+; the difference in slopes for the three chelates is reduced in high ionic strength, which indicates a smaller potential than in low ionic strength.
, were determined from the slopes using the linear form of Eq. 3. The changes in decay rates are shown in Fig. 5. The DEFET rates, indicated by the slopes, for free CrV (Fig. 5, A and B) are much higher than those for CrV bound to the nAChR (Fig. 5, C and D). Free CrV shows the highest energy transfer to Tb− and the least to Tb+; the difference in slopes for the three chelates is reduced in high ionic strength, which indicates a smaller potential than in low ionic strength.
FIGURE 5.

DEFET between Tb3+-chelates and CrV. Decay rates were measured from Tb+ (▵), Tb0 (○), and Tb− (▿) in the presence of CrV at the indicated concentrations in 10 mM HEPES, pH 7.0 (A and C) and in HTPS (B and D). Panels A and B are CrV free in solution. Panels C and D are CrV bound to 3 μM nAChR with 1 mM Carb. Duplicate determinations are shown at each condition. The slope of linear fits to CrV titration data yields the 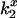 values according to Eq. 3.
values according to Eq. 3.
DEFET rates, 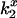 , for free and bound CrV in low and high ionic strength are summarized in Table 3 along with the corresponding electrostatic potentials, as calculated from a Boltzmann distribution (Eq. 4). This measure of potential assumes that changes in local concentrations of Tb3+-chelates are directly reflected by changes in dipolar energy transfer and therefore includes contributions from the surrounding volume as modulated by the distance dependence of energy transfer. Free CrV has an electrostatic potential of 14 mV in low ionic strength and 5 mV in high ionic strength. For CrV bound to the nAChR the potential was −27 mV in low ionic strength and −10 mV in high ionic strength. The electrostatic environment within the pore is negatively charged to an extent that the intrinsic charge of the CrV is overcome. The measured energy transfer rates and potentials are similar when measured in 300 mM NaCl and in HTPS, indicating that the electrostatic environment is modulated by ionic strength rather than by specific ion interactions.
, for free and bound CrV in low and high ionic strength are summarized in Table 3 along with the corresponding electrostatic potentials, as calculated from a Boltzmann distribution (Eq. 4). This measure of potential assumes that changes in local concentrations of Tb3+-chelates are directly reflected by changes in dipolar energy transfer and therefore includes contributions from the surrounding volume as modulated by the distance dependence of energy transfer. Free CrV has an electrostatic potential of 14 mV in low ionic strength and 5 mV in high ionic strength. For CrV bound to the nAChR the potential was −27 mV in low ionic strength and −10 mV in high ionic strength. The electrostatic environment within the pore is negatively charged to an extent that the intrinsic charge of the CrV is overcome. The measured energy transfer rates and potentials are similar when measured in 300 mM NaCl and in HTPS, indicating that the electrostatic environment is modulated by ionic strength rather than by specific ion interactions.
TABLE 3.
Diffusion-enhanced energy transfer to CrV
| DEFET rate*
|
|||||
|---|---|---|---|---|---|
| Buffer | 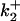 |
 μM−1 s−1 μM−1 s−1
|
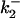 |
Potential†(mV) | |
| Free CrV | 10 mM HEPES | 168 ± 10 (10) | 299 ± 12 (10) | 499 ± 18 (10) | 14 ± 1 |
| 300 mM NaCl | 285 ± 42 (3) | 343 ± 43 (3) | 392 ± 45 (3) | 4 ± 3 | |
| HTPS | 237 ± 36 (3) | 281 ± 13 (3) | 348 ± 13 (3) | 5 ± 2 | |
| Bound CrV | 10 mM HEPES | 51 ± 6 (10) | 13 ± 1 (8) | 6 ± 1 (7) | −27 ± 3 |
| 300 mM NaCl | 24 ± 2 (8) | 15 ± 3 (7) | 11 ± 2 (6) | −10 ± 4 | |
| HTPS | 28 ± 4 (5) | 17 ± 2 (5) | 12 ± 1 (5) | −10 ± 3 | |
| CrV + PCP | 10 mM HEPES | 147 ± 24 (3) | 78 ± 24 (3) | 51 ± 15 (3) | −13 ± 7 |
| HTPS | 77 ± 11 (3) | 76 ± 8 (3) | 57 ± 1 (3) | −4 ± 3 | |
DEFET rates ( ) were determined for Tb3+-chelates in the presence of free CrV, CrV, and 3 μM nAChR, and of CrV, nAChR, and 25 μM PCP, and measured in low and high ionic strength buffers. The average and standard deviation of (n) experiments are presented where
) were determined for Tb3+-chelates in the presence of free CrV, CrV, and 3 μM nAChR, and of CrV, nAChR, and 25 μM PCP, and measured in low and high ionic strength buffers. The average and standard deviation of (n) experiments are presented where  values were determined either by single CrV concentrations or by fits to kobs from several concentrations (0–2 μM).
values were determined either by single CrV concentrations or by fits to kobs from several concentrations (0–2 μM).
The presence of free, unbound CrV could potentially skew the measured DEFET rates in the samples of nAChR-bound CrV because the DEFET rate to free CrV is 20-fold higher than to bound CrV. From the known 10 nM KD for CrV (25), experimental conditions were designed for <1% free CrV in the high ionic strength buffer; the affinity of CrV is substantially higher in low ionic strength (KD < 1 nM; data not shown). Measurements of free CrV by HPLC showed free concentrations of <3 nM, however, CrV partitions strongly into the lipid bilayer (25). Therefore, we measured parallel samples containing excess PCP to determine the effective DEFET rate of unbound CrV in the presence of nAChR-rich membranes (Table 3). Examination of 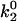 values for this case shows that DEFET was reduced fourfold relative to free CrV in the absence of membranes but is nonetheless substantially higher (approximately fivefold) than for bound CrV. The trend of
values for this case shows that DEFET was reduced fourfold relative to free CrV in the absence of membranes but is nonetheless substantially higher (approximately fivefold) than for bound CrV. The trend of 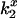 values, and corresponding estimated potential, indicate a negative electrostatic field (Table 3) near unbound CrV, which is consistent with its partitioning into the anionic lipid bilayer. If
values, and corresponding estimated potential, indicate a negative electrostatic field (Table 3) near unbound CrV, which is consistent with its partitioning into the anionic lipid bilayer. If 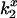 values for bound CrV are compensated for the contribution of free and membrane-associated CrV, the largest correction occurs for
values for bound CrV are compensated for the contribution of free and membrane-associated CrV, the largest correction occurs for 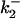 in low ionic strength and yields twofold lower values; these corrections have less than a 2 mV effect on the potentials for all cases. Because the effect is small, the data shown were not corrected.
in low ionic strength and yields twofold lower values; these corrections have less than a 2 mV effect on the potentials for all cases. Because the effect is small, the data shown were not corrected.
Charge distribution at the pore entrance
To estimate the number and distribution of charges near the CrV binding site the change in potential with ionic strength was determined by DEFET measurements and then fit to the Debye-Hückel formulation given in Eq. 6. Fig. 6 A shows the effect of ionic strength titration on DEFET rates to free CrV (open symbols) and to nAChR-bound CrV (solid symbols). In both cases, the separation between 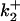 and
and  is greatest in low ionic strength and decreases with increasing ionic strength, presumably due to ionic screening. DEFET rates from Tb0,
is greatest in low ionic strength and decreases with increasing ionic strength, presumably due to ionic screening. DEFET rates from Tb0,  , were unchanged across the ionic strength titration for CrV free in solution, but rose somewhat for CrV bound to the nAChR. Fig. 6 B shows the electrostatic potentials calculated from the data series in Fig. 6 A as described in Materials and Methods. For free CrV, the positive potential drops from 15 mV at low ionic strength to nearly undetectable levels at high ionic strength (2 ± 4 mV). The potential for nAChR-bound CrV was −26 mV at low ionic strength and approaches to −5 mV in high ionic strength.
, were unchanged across the ionic strength titration for CrV free in solution, but rose somewhat for CrV bound to the nAChR. Fig. 6 B shows the electrostatic potentials calculated from the data series in Fig. 6 A as described in Materials and Methods. For free CrV, the positive potential drops from 15 mV at low ionic strength to nearly undetectable levels at high ionic strength (2 ± 4 mV). The potential for nAChR-bound CrV was −26 mV at low ionic strength and approaches to −5 mV in high ionic strength.
FIGURE 6.

Increasing ionic strength attenuates local potential near CrV. (A) DEFET rates were measured for 2 μM CrV, free (▵, ○, ▿), or bound to 3 μM nAChR (▴, •, ▾), in 10 mM HEPES, pH 7.0 with 1 mM Carb. The DEFET rates 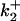 (▵, ▴),
(▵, ▴), 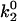 (○, •), and
(○, •), and 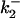 (▿, ▾) are plotted versus NaCl concentration. Errors in
(▿, ▾) are plotted versus NaCl concentration. Errors in 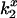 were determined by propagation of errors from individual lifetime measurements. (B) Electrostatic potentials computed from energy transfer rates for free CrV (○) and bound CrV (•). The free CrV potential subtracted from the bound CrV potential (▿) is shown to estimate the potential of the unoccupied nAChR. Curves are the best fit of potentials to Eq. 6. The charge (z) and mean distance of closest approach (r) were: free CrV (○), z = 0.81 ± 0.03, r = 8.3 ± 0.2 Å; bound CrV (•), z = −1.16 ± 0.12, r = 7.1 ± 0.5 Å; bound minus free CrV (▿), z = −1.97 ± 0.1, r = 7.5 ± 0.3 Å.
were determined by propagation of errors from individual lifetime measurements. (B) Electrostatic potentials computed from energy transfer rates for free CrV (○) and bound CrV (•). The free CrV potential subtracted from the bound CrV potential (▿) is shown to estimate the potential of the unoccupied nAChR. Curves are the best fit of potentials to Eq. 6. The charge (z) and mean distance of closest approach (r) were: free CrV (○), z = 0.81 ± 0.03, r = 8.3 ± 0.2 Å; bound CrV (•), z = −1.16 ± 0.12, r = 7.1 ± 0.5 Å; bound minus free CrV (▿), z = −1.97 ± 0.1, r = 7.5 ± 0.3 Å.
The solid lines are the best fits of the potential to Eq. 8 that optimize the number of charges (z) and a mean distance of closest approach (r), which represent the effective charge environment in the vicinity of CrV (35). By this analysis, CrV in solution has a charge of z = 0.81 and a r = 8.3 Å; these values are consistent with the known charge and size of CrV. For nAChR-bound CrV, z = −1.16 and r = 7.1 Å. Because electrostatic fields are additive, the potential of the nAChR pore in the absence of CrV may be estimated by simply subtracting the contribution of free CrV (Fig. 6 B, open triangles). In this case, the CrV binding locus is characterized by z = −1.97 and r = 7.5 Å.
Electrostatic potentials in the nAChR pore affect [3H]PCP binding
An alternative method for determining the electrostatic environment within the nAChR ion conductive pore is to measure the effect of ionic strength on the binding affinity of a cationic noncompetitive antagonist. [3H]PCP binding was measured as described in Materials and Methods and is shown in Fig. 7 A over a range of ionic strengths. Increased ionic strength decreased the apparent affinity of [3H]PCP, but the block of PCP binding at high ionic strength was incomplete, indicating that inhibition was likely due to ionic screening effects rather than direct competition of cations for the PCP binding site. Fitting the binding affinities to a Debye-Hückel equation (34,35) from several experiments (n = 5) yielded averages of zR = −3.33 ± 1.4 charges and r = 4.7 ± 2.1 Å.
FIGURE 7.

Ionic strength and pH effects on [3H]PCP binding. [3H]PCP affinity was determined with the centrifugation assay described in Materials and Methods. (A) Binding of ∼1 nM [3H]PCP to 54 nM nAChR was determined in varying concentrations of NaCl in 1 mM carbamylcholine, 10 mM HEPES, pH 7.0 (▿) or in HTPS (▾). The affinity constant at each concentration is plotted against the square root of the total ionic strength (I). The curve represents the best fit to a Debye-Hückel analysis of binding (Eq. 7). This fit yields a net charge of z = −5.4 and size of r = 8. (B) The affinity of [3H]phencyclidine to 40 nM nAChR with 1 mM carbamylcholine was measured at various pHs in 10 mM concentrations of the following buffers: acetate (▪, pH 4–5.5), MES (▵, pH 5.5 and 6.0), HEPES (▴, pH 6.0–7.5), BICINE (○, pH 7.5–8.5), and glycine (•, pH 8.5–9.5). The curve shows the best fit to an equation for competitive inhibition at a single titratable site: Kobs = Kmax/(1 + [H+]/Ka) where Kmax = 5.2 × 107 M−1 and Ka = 6.39 × 10−8 M or pKa = 7.19.
The effect of pH on PCP binding affinity was also determined from binding assays in buffers that varied from pH 4 to 9.5. PCP binding affinity was maximized and stable above pH 8.0, and decreases at more acidic pH. The data were well fit to an equation for single-site competition of [H+] against PCP binding. The fit to this curve yields a pKa of 7.19. The data can also be interpreted in terms of neutralization of glutamate or aspartate residues in the vicinity of the PCP binding site. Compared to the intrinsic pKa of these residues (pKa = 4.25), the measured pKa differs by nearly 3 pH units. This corresponds to a negative potential of ∼−170 mV, assuming that the pKa shift is due to electrostatic attraction of protons (ΔV = 2.303 RT ΔpH/F).
DISCUSSION
To understand the role of long-range electrostatic attraction on the function of the nAChR, several distinct experimental approaches were applied to determine the electrostatic environment within the extracellular vestibule. Tb3+-chelate DEFET methods show the potential near the CrV binding site to be from −10 to −14 mV at physiological ionic strength. Evaluation of the change in potential with ionic strength using this technique indicated the presence of 1–2 negative charges with a 7-Å radius. Debye-Hückel analysis of changes in the binding of PCP with ionic strength indicated that the binding site had three negative charges with a 5-Å radius. These size estimates are consistent with the expected dimensions of the entrance to the narrow aspects of the channel. In contrast, estimation of the potential by measuring the effective pK for binding indicated a much large local potential of −170 mV, at low ionic strength. The stark discrepancy between this result and those of the DEFET and Debye-Hückel type analyses suggests that other mechanisms than long-range electrostatic attraction of protons account for the pH-induced changes in PCP affinity. In the following article we evaluate these results by comparison to potentials computed from an atomic resolution model of the nAChR.
Determination of surface electrostatic potentials by DEFET
Long-range surface electrostatic attraction of substrates can enhance association rates (39,40) and increase (or decrease) local concentrations of ions. Anionic residues at the entrance to the narrow part of the nAChR pore (7) or on the interior surface of the extracellular vestibule (10) have been proposed to enhance conductivity and selectivity by local concentration of ions, where the vestibule may serve to focus the electrostatic potential. To provide a direct measure of changes in local ion concentrations we applied the DEFET technique of Wensel and Meares (15,21). This technique has been applied to several model systems (15–17,21,38) and has a firm theoretical basis (13,14) that makes it possible to compute DEFET rates from a structural model. Because the three, homologous Tb3+-chelates are similar in structure and optical properties, changes in DEFET rates can be inferred to reflect changes in local concentration due to electrostatic attraction or repulsion. The Boltzmann distribution can be applied to extract an effective voltage for the acceptor locus. Because dipolar energy transfer can occur over substantial distances, the voltage estimate reflects a value integrated over the nearby volume as modulated by the distance-dependent likelihood of energy transfer. An advantage of this measure is that it can be directly compared to potentials computed from structural models and the optical properties of CrV and the Tb-chelates.
The DEFET rates at the CrV binding site in the nAChR differ about twofold per charge change, in low ionic strength buffer, which corresponds to a potential of −27 mV. In physiological ionic strength buffer, this potential was reduced to −10 mV. If we assume that the potential of CrV is additive with that of the pore, we can calculate a corrected potential for the pore in the absence of CrV of −14 mV. This potential reflects ∼50% changes in ion concentration at the pore relative to the bulk concentration. Debye-Hückel analysis of the change in potential with ionic strength indicate the presence of −1 (or −2 if we compensate for the presence of CrV), charges near the pore with a distribution of 7 Å. Long-range electrostatic attraction, therefore, constitutes a small contribution to localizing cations at the pore and is a primary determinant of neither conductivity nor selectivity. The saturating effect of increasing electrolyte concentrations on the local potential was predicted for ion channels at pore entrances (41,42). However, it seems unlikely that changes in potential are sufficient to account for the saturating response of conductivity to increased concentration of conductive ions. A model that requires binding of the conductive ion within the pore may be more likely to account for this phenomenon (9).
The DEFET experiments determined the potential of a ligand-bound nAChR that is in the desensitized conformation and may not reflect the potential of the open channel. The various conformations of the nAChR could have distinct charge distributions near the pore entrance. However, these would seem unlikely to affect long-range electrostatic attraction unless there is substantial shielding or neutralization of the charge. CrV itself can influence the local potential by contributing one positive charge, and, potentially, by influencing the protonation state of adjacent side chains. The extent of DEFET is highly sensitive to the distance of closest approach of the Tb3+-chelates to the acceptor and, therefore, the electrostatic field of bound CrV could dominate the DEFET signal. However, CrV has a delocalized charge and its potential in solution is just 4 mV at physiological ionic strength, as measured by this approach. Direct subtraction, therefore, seems a valid approximation to the potential of the pore without CrV bound. The Tb3+-chelates are substantially larger than even a hydrated sodium or potassium ion and, therefore, it is possible that the restricted volume sampled biases the potential and leads to an underestimate of the effects on smaller ions. These issues will be addressed by direct computation of DEFET rates on the basis of a structural model in the following article. Although these are caveats for interpretation of the data, they are unlikely to alter the main conclusions.
PCP as a probe of the electrostatic environment
Ligand binding of ionic substrates is expected to be sensitive to changes in ionic strength and the effects of ionic screening can be modeled by a Debye-Hückel equation. This approach successfully predicted the number of charges associated with binding of acetylcholine to acetylcholinesterase (35). When applied to [3H]PCP binding to the nAChR, the analysis revealed −3 charges over a 5-Å radius. This distance is similar to that extracted from Debye-Hückel analysis of the change in potential at the CrV binding site and similar to the dimension of the channel. Structural models of the extracellular end of the M2 α-helix indicate a net sum of −3, assuming all ionic side chains have unitary charge (4,43). This sum assumes no self-neutralization of the anionic residues due to attraction of protons. In the following article (11), we will show that this assumption is essentially correct near neutral pH.
PCP binding was profoundly affected by pH and the data were well fit assuming titration of a single residue with a pK of 7.2. If this pKa reflects electrostatically increased local proton concentration it indicates a potential of at least −170 mV, a value that differs substantially from the −27 mV measured under similar conditions by DEFET. It is unlikely that other residues are involved; Lys, Arg, and Tyr all titrate at higher pH and Cys and His are not present in the vicinity of the binding site. The difference in potentials may represent a fundamental difference between PCP-binding and DEFET measurements. PCP binding reflects direct interaction with the unoccupied pore and is itself the probe of the charge environment whereas CrV binds, occludes, and reduces the net charge within the channel. In addition, the binding sites for PCP and CrV may be overlapping but with distinct electrostatic environments resulting from highly localized charged residues.
The role of surface potentials in nAChR function
Discerning the impact of electrostatic effects on ion channel conduction and selectivity has been hindered by the lack of direct methods for determining potentials themselves. In the constrained environment such as the nAChR vestibule, electrostatic potentials may also exhibit focusing and leading to unusual ionic strength dependencies (19). The advantage of the DEFET approach, therefore, lies in its direct detection of concentration changes of variously charged Tb3+-chelates. The ionic strength, PCP binding data, and DEFET data agree on the charge number and distribution. However, these results differ from computational estimates that have argued for a strong influence of pore charges and vestibular charges (7,10). Other experimental techniques for assessing the impact of electrostatics on nAChR function appear to be consistent with our data. The substituted cysteine accessibility methods evaluated the reaction rates of variously charged methanethiosulfonate compounds in the M2 α-helix and found potentials at the extracellular end of ∼−25–0 mV for the open channel and near +50 mV for the closed channel. The work of Kienker et al. (9) carefully evaluated the rate of conduction at various ionic strengths upon mutation of the outer ring residues. They concluded that these charges did not strongly influence the ionic strength dependence of conduction and thus did not contribute to long-range attraction of ions to the pore, but that they likely influenced conduction through binding of conductive ions. That work alone, however, did not rule out the possible influence of other vestibular charges on local cation concentrations.
The DEFET data are fundamentally consistent with previous work, though our data are restricted to the desensitized conformation of the nAChR. The results are likely to apply to the resting and open conformations as well because conformational changes are unlikely to profoundly affect the overall number and distribution of charges that determine long-range ionic attraction. Such conformational changes, however, likely will affect close interactions, such as PCP binding, ion chelation, or ion dehydration.
The data presented here show that the net vestibular negative charge does not contribute substantially to long-range electrostatic attraction. Because it considers both negative and positive chelates, it provides an internal check on DEFET rates. However, the basic inconsistency with the predictions from computational work that predicted more profound consequences of the presence of vestibular residues remains. In the following article, computed potentials and their influence on DEFET are compared directly with these experimental results.
Acknowledgments
The authors thank Arlene Samano for excellent technical assistance in the [3H]PCP binding assays. We also thank Iraida Andreeva and Nasrin Latif, for assistance with Torpedo californica preparations, as well as helpful discussions.
This work was supported by Public Health Service grants RO1-NS35212 (S.E.P.) and RO1-EY07981 (T.G.W.), and by grants Q-1406 (S.E.P.) and Q-0035 (T.G.W.) from the Robert A. Welch Foundation. R.H.M. was supported by T32 HL07676 to the Department of Molecular Physiology and Biophysics and T32 GM088280 to the Houston Area Molecular Biophysics Program.
Robert H. Meltzer's present address is Dept. of Physiology & Biophysics, University of Alabama, Birmingham, AL.
Monica M. Lurtz's present address is Dept. of Neuroscience, University of California, Riverside, CA.
References
- 1.Corringer, P. J., N. Le Novere, and J. P. Changeux. 2000. Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40:431–458. [DOI] [PubMed] [Google Scholar]
- 2.Unwin, N. 1993. Nicotinic acetylcholine receptor at 9 Å resolution. J. Mol. Biol. 229:1101–1124. [DOI] [PubMed] [Google Scholar]
- 3.Miyazawa, A., Y. Fujiyoshi, M. Stowell, and N. Unwin. 1999. Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J. Mol. Biol. 288:765–786. [DOI] [PubMed] [Google Scholar]
- 4.Unwin, N. 2005. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346:967–989. [DOI] [PubMed] [Google Scholar]
- 5.Imoto, K., C. Methfessel, B. Sakmann, M. Mishina, Y. Mori, T. Konno, K. Fukuda, M. Kurasaki, H. Bujo, Y. Fujita, et al. 1986. Location of a delta-subunit region determining ion transport through the acetylcholine receptor channel. Nature. 324:670–674. [DOI] [PubMed] [Google Scholar]
- 6.Imoto, K., C. Busch, B. Sakmann, M. Mishina, T. Konno, J. Nakai, H. Bujo, Y. Mori, K. Fukuda, and S. Numa. 1988. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 335:645–648. [DOI] [PubMed] [Google Scholar]
- 7.Dani, J. A. 1986. Ion-channel entrances influence permeation. Net charge, size, shape, and binding considerations. Biophys. J. 49:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dani, J. A., and G. Eisenman. 1987. Monovalent and divalent cation permeation in acetylcholine receptor channels. Ion transport related to structure. J. Gen. Physiol. 89:959–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kienker, P., G. Tomaselli, M. Jurman, and G. Yellen. 1994. Conductance mutations of the nicotinic acetylcholine receptor do not act by a simple electrostatic mechanism. Biophys. J. 66:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adcock, C., G. R. Smith, and M. S. Sansom. 1998. Electrostatics and the ion selectivity of ligand-gated channels. Biophys. J. 75:1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meltzer, R. H., W. Vila-Carriles, J. O. Ebalunode, J. M. Briggs, and S. E. Pedersen. 2006. Computed pore potentials of the nicotinic acetylcholine receptor. Biophys. J. 91:1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrapani, S., T. D. Bailey, and A. Auerbach. 2003. The role of loop 5 in acetylcholine receptor channel gating. J. Gen. Physiol. 122:521–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stryer, L., D. D. Thomas, and C. F. Meares. 1982. Diffusion-enhanced fluorescence energy transfer. Annu. Rev. Biophys. Bioeng. 11:203–222. [DOI] [PubMed] [Google Scholar]
- 14.Thomas, D. D., W. F. Carlsen, and L. Stryer. 1978. Fluorescence energy transfer in the rapid-diffusion limit. Proc. Natl. Acad. Sci. USA. 75:5746–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wensel, T. G., and C. F. Meares. 1983. Electrostatic properties of myoglobin probed by diffusion-enhanced energy transfer. Biochemistry. 22:6247–6254. [Google Scholar]
- 16.Wensel, T. G., C. H. Chang, and C. F. Meares. 1985. Diffusion-enhanced lanthanide energy-transfer study of DNA-bound cobalt(III) bleomycins: comparisons of accessibility and electrostatic potential with DNA complexes of ethidium and acridine orange. Biochemistry. 24:3060–3069. [DOI] [PubMed] [Google Scholar]
- 17.Wensel, T. G., C. F. Meares, V. Vlachy, and J. B. Matthew. 1986. Distribution of ions around DNA, probed by energy transfer. Proc. Natl. Acad. Sci. USA. 83:3267–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madura, J. D., J. M. Briggs, R. C. Wade, M. E. Davis, B. A. Luty, A. Ilin, J. Antosiewicz, M. K. Gilson, B. Bagheri, L. R. Scott, and J. A. McCammon. 1995. Electrostatics and diffusion of molecules in solution - simulations with the University of Houston Brownian dynamics program. Comput. Phys. Commun. 91:57–95. [Google Scholar]
- 19.Moy, G., B. Corry, S. Kuyucak, and S. H. Chung. 2000. Tests of continuum theories as models of ion channels. I. Poisson-Boltzmann theory versus Brownian dynamics. Biophys. J. 78:2349–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meltzer, R. H., E. Thompson, K. V. Soman, X.-Z. Song, J. O. Ebalunode, T. G. Wensel, J. M. Briggs, and S. E. Pedersen. 2006. Electrostatic Steering at Acetylcholine Binding Sites. Biophys. J. 91:1302–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wensel, T. G. 1984. Energy transfer studies of proteins and nucleic acids (electrostatics, intercalators, DNA). PhD thesis. University of California at Davis, Davis, CA.
- 22.Wensel, T. G., and C. F. Meares. 1983. Electrostatic properties of myoglobin probed by diffusion-enhanced energy transfer. Biochemistry. 22:6247–6254. [Google Scholar]
- 23.Sobel, A., M. Weber, and J. P. Changeux. 1977. Large-scale purification of the acetylcholine-receptor protein in its membrane-bound and detergent-extracted forms from Torpedo marmorata electric organ. Eur. J. Biochem. 80:215–224. [DOI] [PubMed] [Google Scholar]
- 24.Pedersen, S. E., E. B. Dreyer, and J. B. Cohen. 1986. Location of ligand-binding sites on the nicotinic acetylcholine receptor alpha-subunit. J. Biol. Chem. 261:13735–13743. [PubMed] [Google Scholar]
- 25.Lurtz, M. M., and S. E. Pedersen. 1999. Aminotriarylmethane dyes are high-affinity noncompetitive antagonists of the nicotinic acetylcholine receptor. Mol. Pharmacol. 55:159–167. [DOI] [PubMed] [Google Scholar]
- 26.Förster, T. 1948. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 437:55–75. [Google Scholar]
- 27.Stein, G., and E. Wurzberg. 1975. Energy gap law in the solvent isotope effect on radiationless transitions or rare earth ions. J. Chem. Phys. 62:208–213. [Google Scholar]
- 28.Yeh, S. M., and C. F. Meares. 1980. Characterization of transferin metal-binding sites by diffusion enhanced energy transfer. Biochemistry. 19:5057–5062. [DOI] [PubMed] [Google Scholar]
- 29.Xiao, M., and P. R. Selvin. 2001. Quantum yields of luminescent lanthanide chelates and far-red dyes measured by resonance energy transfer. J. Am. Chem. Soc. 123:7067–7073. [DOI] [PubMed] [Google Scholar]
- 30.Lamture, J. B., Z. H. Zhou, A. S. Kumar, and T. G. Wensel. 1995. Luminescence properties of terbium(iii) complexes with 4-substituted dipicolinic acid analogs. Inorg. Chem. 34:864–869. [Google Scholar]
- 31.Papineni, R. V., J. U. Sanchez, K. Baksi, I. U. Willcockson, and S. E. Pedersen. 2001. Site-specific charge interactions of alpha-conotoxin MI with the nicotinic acetylcholine receptor. J. Biol. Chem. 276:23589–23598. [DOI] [PubMed] [Google Scholar]
- 32.Bevington, P. R. 1969. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill, New York, NY.
- 33.Hille, B. 2001. Ion Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- 34.Song, X. Z., and S. E. Pedersen. 2000. Electrostatic interactions regulate desensitization of the nicotinic acetylcholine receptor. Biophys. J. 78:1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nolte, H. J., T. L. Rosenberry, and E. Neumann. 1980. Effective charge on acetylcholinesterase active sites determined from the ionic strength dependence of association rate constants with cationic ligands. Biochemistry. 19:3705–3711. [DOI] [PubMed] [Google Scholar]
- 36.Pedersen, S. E. 1995. Site-selective photoaffinity labeling of the Torpedo californica nicotinic acetylcholine receptor by azide derivatives of ethidium bromide. Mol. Pharmacol. 47:1–9. [PubMed] [Google Scholar]
- 37.Lurtz, M. M., M. L. Hareland, and S. E. Pedersen. 1997. Quinacrine and ethidium bromide bind the same locus on the nicotinic acetylcholine receptor from Torpedo californica. Biochemistry. 36:2068–2075. [DOI] [PubMed] [Google Scholar]
- 38.Northrup, S. H., T. G. Wensel, C. F. Meares, J. J. Wendoloski, and J. B. Matthew. 1990. Electrostatic field around cytochrome c: theory and energy transfer experiment. Proc. Natl. Acad. Sci. USA. 87:9503–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wade, R. C., R. R. Gabdoulline, S. K. Ludemann, and V. Lounnas. 1998. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proc. Natl. Acad. Sci. USA. 95:5942–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan, R. C., T. N. Truong, J. A. McCammon, and J. L. Sussman. 1993. Acetylcholinesterase: electrostatic steering increases the rate of ligand binding. Biochemistry. 32:401–403. [DOI] [PubMed] [Google Scholar]
- 41.Jordan, P. C., R. J. Bacquet, J. A. McCammon, and P. Tran. 1989. How electrolyte shielding influences the electrical potential in transmembrane ion channels. Biophys. J. 55:1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jordan, P. C. 1987. How pore mouth charge distributions alter the permeability of transmembrane ionic channels. Biophys. J. 51:297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyazawa, A., Y. Fujiyoshi, and N. Unwin. 2003. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 423:949–955. [DOI] [PubMed] [Google Scholar]