

Abstract
Human epidermal growth factor receptor 2 (HER2) overexpression has been associated with increased invasiveness in mammalian breast cancer cell lines, but the effects of overexpression on key underlying cell migration properties such as translational speed and directional persistence are not understood. Moreover, the differential effect of HER2 activation through heterodimerization with epidermal growth factor receptor versus human epidermal growth factor receptor 3 (HER3) on cell speed and persistence has not been studied. To investigate these issues, we developed a high-throughput wound closure assay in which individual cell locomotion and wound closure kinetics were quantified in human mammary epithelial cells with varying levels of HER2 under epidermal growth factor or heregulin (a HER3 ligand) stimulation. Increasing levels of HER2 elevated wound closure with closure kinetics dependent on ligand treatment. Cell speed increased with HER2 levels under epidermal growth factor treatment, but decreased under heregulin treatment. In contrast, directional persistence increased with HER2 levels under both ligand treatments. Increasing persistence quantitatively accounted for observed elevated wound closure, as measured by the effective diffusion of the cells. Taken together, the data show that the HER2 overexpression mediates cell migration through differential control of translational speed and directional persistence dependent on epidermal growth factor receptor-HER2 versus HER2-HER3 heterodimerization. Observed consistent increases in persistence associated with HER2 overexpression indicate a prospective mechanism for invasiveness previously documented in HER2-overexpressing human breast tumors.
Human epidermal growth factor receptor 2 (HER2) is overexpressed in 20–30% of breast cancers and correlates with poor prognosis and increased metastasis (1). HER2 belongs to the ErbB or HER family of receptors (comprised of HER1/EGFR, HER2, HER3, and HER4) and can be activated through concentration-dependent homodimerization or ligand-driven heterodimerization. Epidermal growth factor (EGF) and heregulin (HRG), two ErbB family ligands implicated in cancer progression, bind HER1 and HER3, respectively, to induce the activation of HER2 through heterodimerization (2). Because of HER2's role in breast cancer metastasis, a number of groups have investigated the effect of HER2 expression on aspects of cell motility, demonstrating that activation by EGF, HRG, or homodimerization leads to increased invasion and motility in breast cancer cell lines (3–5). In addition, these and other studies have implicated various downstream signaling molecules as effectors of HER2-increased motility. Primarily because many of these studies relied on invasion assays, however, there is little known about how HER2 overexpression affects cell migration parameters such as cell speed and persistence. Prior study of primary ductal breast carcinoma cells revealed that groups of cells tend to detach from primary tumor lesions and move away in a highly polarized and directionally persistent manner, indicating that the control of directional persistence may be distinct in highly motile breast cancer cells, such as those with HER2 overexpression (6). In addition, increased directional persistence has been identified as a hallmark of cell migration in highly invasive tumors, such as neuroepithelial tumors (7). Distinct signaling and biophysical mechanisms controlling directional persistence versus random motility have also been identified in recent studies (8–10). Thus, a more in-depth study of HER2's effect on cell migration, speed, and persistence could potentially serve two purposes: 1), to connect HER2 overexpression with persistent movement shown to be important in cancer systems; and 2), to provide a context within which to understand previously identified HER2-associated downstream signals by linking them to pathways that regulate directionally persistent migration.
We examined cell migration in a human mammary epithelial cell line. Two clones of the cell line, parental (with 200,000 EGFR, 20,000 HER2, and 20,000 HER3) and 24H (200,000 EGFR, 600,000 HER2, and 30,000 HER3), were studied in the presence of EGF (100 ng/ml), HRG (80 ng/ml), or serum-free media. Cell migration was tracked using a high-throughput 96-well migration assay that we developed for the rapid screening of cell motility. The movement of epithelial monolayers and the motility of hundreds of individual cells in monolayers were rapidly screened, generating time-resolved population-level statistics for all treatment conditions during one 15-h time course (Supplementary Fig. 1 and Supplementary Methods). This presages future assay application to drug-based screens designed to rapidly explore the biochemical basis of cell migration.
Increasing HER2 levels from 20,000 to 600,000 increased wound closure across all treatment conditions (Fig. 1, A–D). Absolute levels of wound closure differed depending on treatment condition, with EGF-treated cells being the most motile followed by HRG-treated cells under both low and high HER2 expression (Fig. 1 A). EGF-treated 24H cells exhibited the most rapid closure, completely sealing the wound in ∼6 h, and EGF treatment showed the most responsiveness to HER2 overexpression as measured by the difference in closure between parental and 24H cells at 15 h. Interestingly, HER2 overexpression caused elevated closure even in the absence of ligand, presumably due to low levels of autocrine production or concentration-dependent homodimerization.
FIGURE 1 .

HER2 overexpression increases cell migration. Normalized wound area as a function of time for (A) parental and 24H cells under EGF (100 ng/ml), HRG (80 ng/ml), or serum-free treatment; (B) parental and 24H cells in serum-free conditions; (C) EGF-treated parental and 24H cells; and (D) HRG-treated parental or 24H cells. All time points shown ± SE after time averaging (see Supplementary Methods). For raw data time courses see Supplementary Fig. 3.
Detailed inspection of wound-closure kinetics demonstrates time-dependent control of migration differing across treatment conditions. For instance, in the presence of HRG, HER2 overexpression increased wound closure significantly only after the first 3.5 h, with both parental and 24H wounds measuring ∼68% of the original area before separation (p > 0.9; means are the same). In contrast, serum-free and EGF-treated 24H cells close more wound than similarly treated parentals at 3.5 h (p < 0.01), demonstrating that HRG exerts temporally distinct control of migration. Movement after 3.5 h differentiates the HRG-treated cells, with 24H cells closing ∼25% more normalized wound area than parentals at 15 h. Another interesting kinetic trend is the cessation of wound closure that occurs at early times under certain conditions. For instance, HRG-treated 24H cells close the wound throughout the duration of the experiment, but parental cells stop their movement after ∼5 h (Fig. 1 D). Interestingly, even after wound closure has stopped, HRG-treated cells continue to move in the monolayer, suggesting that cell movement perpendicular to the wound front is controlled separately from cell movement within the monolayer and parallel to the wound front (data not shown). EGF-treated parental cells exhibit similar behavior as do both parental and 24H cells in serum-free conditions. The time at which wound closure stops varies from ∼2 to 6 h depending on treatment conditions, but cell movement in the monolayer and parallel to the wound continues in all cases. These data are consistent with wound closure observations from other groups, and although the mechanistic basis of this “stopping” behavior is not fully understood, it is clear that HER2 overexpression in the context of HRG or EGF treatment provides necessary information to overcome the signals governing the termination of wound closure before the wound is sealed (11). Fig. 1 A also reveals that cells under all treatment conditions close similar wound areas at early times (21–29% wound area closed under all conditions at 1.5 h). This suggests that early wound closure is primarily governed by mechanical induction and associated start signals that operate independently of ligand/receptor conditions (Supplementary Fig. 2).
We considered whether the demonstrated differences in migration might arise from changes in cell speed, directional persistence, or both. We wondered further whether cells use these two migration “levers” in a similar or different manner to increase migration under different ligand treatments. To explore these questions, we tracked the wound closure response of fluorescent cells diluted in unlabeled cells. Dilution allowed for more accurate cell tracking within the monolayer (see Supplementary Methods). We then calculated the mean-squared displacement for each cell trajectory and fit it to the following equation:
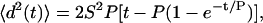 |
(1) |
where S is cell speed and P is cell directional persistence (12). Data demonstrated that although HER2 overexpression increased wound closure for all ligand conditions studied, it did not necessarily increase cell speed. Indeed, when HER2 was overexpressed, serum-free cell speed did not differ and HRG-treated cells exhibited slightly decreased speed (Fig. 2 A). EGF-treated cells, however, increased cell speed with HER2 overexpression (Fig. 2 A). The magnitude of cell speed was similar between HRG treatment and serum-free conditions, but significantly higher for EGF-treated cells. In contrast to cell speed data, HER2 overexpression increased directional persistence across all treatments. EGF treatment stimulated the highest degree of persistence, whereas HRG treatment induced the largest change between low and high HER2 conditions. Cell speed and persistence data together demonstrate that HER2 overexpression causes more rapid wound closure under EGF treatment due to increases in speed and persistence. However, in the absence of ligand or under HRG treatment, changes in closure rates are due primarily to increased cell persistence alone. HER2's influence on the effective diffusion of cells is found by equating Eq. 1 to 4 Dt when t ≫ P, where D is the diffusion constant or random motility coefficient (as it is more often referred to in migration literature). Expressing D as
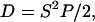 |
(2) |
FIGURE 2 .

HER2 effects on cell speed and persistence. Cell migration of parental and 24H cells treated with EGF (100 ng/ml), HRG (80 ng/ml), or in serum-free conditions was recorded and quantified for (A) cell speed in μm/h, (B) directional persistence in minutes, and (C) diffusion constant in μm2/h. D shows inequalities at 95% confidence as evaluated using Kolmogorov-Smirnov test for nonnormal distributions. All data reported as mean ± SE. Number of cells per condition equals 153–196. (See Supplementary Methods and Supplementary Fig. 4 for raw data histograms.)
we can evaluate the net effect of changes in speed and persistence on cell migration. Fig. 2 C shows that the diffusion constant describes differences in the magnitude of migration between conditions that are consistent with those observed using the wound closure assay (Figs. 1 A and 2 C). Thus, depending on the ligand treatment, the cell regulates speed and persistence in qualitatively different ways to achieve HER2-mediated increases in migration as measured by wound closure or diffusion.
The biological regulators of directional movement are not fully understood, but recent work has implicated Rac1, microtubules, and myosin IIb as possible candidates (7–9). Whatever the downstream regulators, our data indicate that HER2 overexpression affects cellular components involved with directional movement independently from those responsible for increased cell speed. We speculate that differential phosphorylation under the various ligand treatment and receptor levels considered initiates downstream signaling differences that affect cell speed and persistence differently. Our data show, however, that under a variety of activating conditions, HER2 overexpression increases cell persistence, perhaps indicating the existence of a redundant phospho-site or signaling mechanism responsible for increasing persistence. This strong connection between HER2 and directionally persistent migration may, in turn, be an important facet to HER2's documented ability to increase invasion and metastasis in human breast cancer cells.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors thank Dr. James Evans and Winston Timp for technical assistance. We acknowledge support from the National Institutes of Health-Biotechnology Training Program (N.K.), the Sokol Foundation (M.H.Z.), National Cancer Institute Integrative Cancer Biology Program P50-CA112967 (D.A.L., H.D.K.), National Cancer Institute Bioengineering Research Partnership R01-CA96504 (D.A.L.), and a grant from AstraZeneca (D.A.L.).
Muhammad H. Zaman and Hyung-Do Kim contributed equally to this work.
References
- 1.Hynes, N., and D. Stern. 1994. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta. 1198:165–184. [DOI] [PubMed] [Google Scholar]
- 2.Harari, D., and Y. Yarden. 2000. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 19:6102–6114. [DOI] [PubMed] [Google Scholar]
- 3.Brandt, B. H., A. Roetger, T. Dittmar, G. Nikolai, M. Seeling, A. Merschjann, J. Nofer, G. Dehmer-Moller, R. Junker, G. Assmann, and others. 1999. c-erbB-2/EGFR as dominant heterodimerization partners determine a motogenic phenotype in human breast cancer cells. FASEB J. 13:1939–1949. [DOI] [PubMed] [Google Scholar]
- 4.Dittmar, T., A. Husemann, Y. Schewe, J. Nofer, B. Niggemann, K. Zanker, and B. Brandt. 2002. Induction of cancer cell migration by epidermal growth factor is initiated by specific phosphorylation of tyrosine 1248 of c-erbB-2 receptor via EGFR. FASEB J. 16:1823–1825. [DOI] [PubMed] [Google Scholar]
- 5.Spencer, K. S., D. Graus-Porta, J. Leng, N. Hynes, and R. Klemke. 2000. ErbB2 is necessary for induction of carcinoma cell invasion by ErbB family receptor tyrosine kinases. J. Cell Biol. 148:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedl, P., P. Noble, P. Walton, D. Laird, P. Chauvin, R. Tabah, M. Black, and K. Zanker. 1995. Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer Res. 55:4557–4560. [PubMed] [Google Scholar]
- 7.Deisboeck, T. S., T. Demuth, and Y. Mansury. 2005. Correlating velocity patterns with spatial dynamics in glioma cell migration. Acta Biotheor. 53:181–190. [DOI] [PubMed] [Google Scholar]
- 8.Lo, C. M., D. Buxton, G. Chua, M. Dembo, R. Adelstein, and Y. Wang. 2004. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Mol. Biol. Cell. 15:982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pankov, R., Y. Endo, S. Even-Ram, M. Araki, K. Clark, E. Cukierman, K. Matsumoto, and K. Yamada. 2005. A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 170:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verkhovsky, A. B., T. Svitkina, and G. Borisy. 1999. Self-polarization and directional motility of cytoplasm. Curr. Biol. 9:11–20. [DOI] [PubMed] [Google Scholar]
- 11.Jacinto, A., A. Martinez-Arias, and P. Martin. 2001. Mechanisms of epithelial fusion and repair. Nat. Cell Biol. 3:E117–E123. [DOI] [PubMed] [Google Scholar]
- 12.Dickinson, R., and R. Tranquillo. 1993. Optimal estimation of cell-movement indexes from the statistical-analysis of cell tracking data. AIChE J. 39:1995–2010. [Google Scholar]