

Abstract
Cells contain many subcellular structures in which specialized proteins locally cluster. Binding interactions within such clusters may be analyzed in live cells using models for fluorescence recovery after photobleaching (FRAP). Here we analyze a three-dimensional FRAP model that accounts for a single spatially localized cluster of binding sites in the presence of both diffusion and impermeable boundaries. We demonstrate that models completely ignoring the spatial localization of binding yield poor estimates for the binding parameters within the binding site cluster. In contrast, we find that ignoring only the restricted axial height of the binding-site cluster is far less detrimental, thereby enabling the use of computationally less expensive models. We also identify simplified solutions to the FRAP model for limiting behaviors where either diffusion or binding dominate. We show how ignoring a role for diffusion can sometimes produce serious errors in binding parameter estimation. We illustrate application of the method by analyzing binding of a transcription factor, the glucocorticoid receptor, to a tandem array of mouse mammary tumor virus promoter sites in live cells, obtaining an estimate for an in vivo binding constant (10−7 M), and a first approximation of an upper bound on the transcription-factor residence time at the promoter (∼170 ms). These FRAP analysis tools will be important for measuring key cellular binding parameters necessary for a complete and accurate description of the networks that regulate cellular behavior.
INTRODUCTION
Knowledge of in vivo binding constants will be important for understanding molecular interactions within live cells. In vivo binding measurements can in principle be accomplished either by fluorescence auto- or cross-correlation spectroscopy (FCS), or by fluorescence recovery after photobleaching (FRAP). Compared to FCS, FRAP is currently more widely used and easier to implement. Unlike the limited number of FCS studies, the vast majority of FRAP studies do not attempt quantitative estimates of binding interactions, although they often make inferences about these interactions based on the shape of the FRAP curve. These interpretations, however, are suspect without a model that explains what underlying processes give rise to the FRAP curve. Thus realistic FRAP models are important for both qualitative and quantitative interpretation of FRAP experiments.
We are using FRAP to assay the in vivo binding interactions of a transcription factor with its promoter target site. We employ a cell line (1) containing a tandem array of mouse mammary tumor virus (MMTV) promoters and the GFP-tagged form of one of this promoter's cognate transcription factors, the glucocorticoid receptor (GFP-GR). This tandem array can be visualized as a single bright region of GFP-GR binding within the nucleus. We have shown that the FRAP recovery within this spatially localized cluster of specific binding sites is rapid (2), and that it contains information about the in vivo binding interactions of GFP-GR with the MMTV promoters (3). These transient binding interactions have challenged the notion that a stable transcriptional complex forms at a promoter. However, a detailed understanding of these transient interactions requires a model that first explains what the FRAP curve reflects, and then quantifies the underlying processes.
Quantitative analyses of FRAP have been performed for binding sites that are homogeneously and globally distributed throughout a cellular compartment (4–9), but fewer studies (10–14) have tackled the problem for heterogeneously distributed binding sites such as that which occurs at a spatially localized cluster of binding sites, despite its obvious biological importance. The primary reason is that the analysis is more complicated compared to homogeneously and globally distributed binding sites. A cluster of specific binding sites is always embedded in a larger domain throughout which molecules may freely diffuse or interact weakly with uniformly distributed nonspecific sites. Thus spatial variability must be considered in any reasonable model for localized binding. To simplify such an analysis, most previous FRAP models for spatially localized binding have presumed that diffusion plays a negligible role. In those cases where diffusion has been ignored, the consequences of this assumption have not been tested. In addition, previous analyses have been tailored to the specific problem under study, and general principles about expected FRAP behaviors at a spatially localized cluster of binding sites have not been elucidated.
Here we investigate a FRAP model that also incorporates diffusion within and around a spatially localized cluster of binding sites. We show that completely ignoring the localization of binding sites will introduce serious errors into the estimation of binding parameters. However, we find that a reasonable approximation can often be achieved by assuming a cylindrical column of binding sites that accounts for spatially localized binding in every xy cross section of the cell, but not along z. The resultant model is considerably easier to implement and faster to compute, and so should be more widely useful. We also identify limiting behaviors for FRAPs at spatially localized binding sites where either diffusion or binding dominate, and show how ignoring diffusion can also lead to serious errors in the estimation of binding parameters at the localized binding site cluster. Finally, we apply these methods to the analysis of GFP-GR binding at the MMTV promoter, and obtain a first approximation to an upper bound on the GR residence time of ∼170 ms. This figure underscores the transient nature of this binding interaction, and has broader implications for how assembly of the transcription complex may occur.
MATERIALS AND METHODS
Cell lines
Mouse adenocarcinoma cell line 3617 containing the MMTV array was used for all experiments. These cells stably express GFP-GR under the control of a tetracycline-off system (1). Cells were grown and prepared for fluorescence imaging as previously described (3). Thirty minutes prior to imaging, GFP-GR translocation to the nucleus was induced by the addition of 100 nM dexamethasone (synthetic hormone).
FRAP protocol
FRAP experiments were carried out on a Zeiss 510 confocal microscope (Carl Zeiss, Thornwood, NY) with a 100×/1.3 NA oil-immersion objective. Bleaching was performed using the 488- and 514-nm lines from a 40-mW argon laser operating at 75% laser power. Bleaching was done with a single scan that lasted 17 ms. Fluorescence recovery was monitored at low laser intensity (0.2% of the 40 mW laser) at 78 ms intervals. Normalized FRAP curves were generated from the raw data exactly as described in Stavreva and McNally (15).
MODELS
Essential features for modeling spatially localized binding at the MMTV array
The 2 Mb MMTV tandem array contains ∼200 copies of a basic repeat unit: the MMTV promoter (∼1 kb) followed by a ras reporter gene and additional sequence from the bovine papilloma virus (totaling ∼9 kb). With six binding sites for the glucocorticoid receptor (GR) at each of the 200 MMTV promoters, the MMTV array is a cluster of ∼1200 specific binding sites (Fig. 1, A and B). FRAP experiments at the MMTV array are typically performed by bleaching it with a circular spot equal to the diameter of the MMTV array (Fig. 1 C), with the axial extent of the bleach reasonably approximated as a cylinder (Fig. 1 D).
FIGURE 1.

The MMTV array in the 3617 mouse cell line. (A) The MMTV array appears as a bright region due to the local accumulation of GFP-GR. (B) An xz cross-sectional slice of the nucleus shows that the MMTV array does not extend throughout the full height of the nucleus. (C) The same (fixed) cell now bleached with a 2-μm-diameter circular spot (white dots). (D) An xz cross section of this cell reveals that the photobleach extends throughout the nucleus defining approximately a cylindrical profile. (E) The average nuclear and array dimensions from three-dimensional measurements of 10 cells are shown. The average radial coordinate of the array is also shown as determined by two-dimensional 2D measurements from 50 cells. The effective diffusion constant of GR in the nucleoplasm was measured to be D1eff = 1.2 μm2/s with 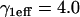 using the FRAP model for globally distributed binding sites described in Sprague et al. (8). Scale bars, 2 μm.
using the FRAP model for globally distributed binding sites described in Sprague et al. (8). Scale bars, 2 μm.
We measured typical nuclear dimensions in the MMTV array cell line, and found that the average diameter and axial height of the array were ∼2.0 μm, the average nuclear radius was ∼7.5 μm, and the average nuclear height was ∼4.5 μm (Fig. 1 E). We measured the average radial coordinate of the array (i.e., distance from the center of the nucleus), and found that most arrays occupied radial positions between 3.3 and 6.3 μm, with a mean radial coordinate of ∼4.8 μm. Finally, we measured the distance of the center of mass of arrays to the closest edge of nucleoli (dark regions where GFP-GR is largely absent), and found an average distance of 1.1 μm, with some arrays as far as 3.5 μm from a nucleolus.
Away from the MMTV array, GFP-GR is distributed rather uniformly throughout the rest of the nucleus, again with the exception of nucleoli (Fig. 1 A). To define GR interactions throughout the nucleoplasm, FRAP experiments can be performed at some distance from both the MMTV sites and nucleoli. These nucleoplasmic FRAP recoveries are consistently faster than FRAP recoveries at the MMTV array (3), suggesting that GR associates more strongly with the array sites than with the nucleoplasmic sites. Analysis of these nucleoplasmic FRAPs predicts that GFP-GR interacts predominantly with a single binding state in the nucleoplasm (8). Moreover, for bleach spots >1 μm, the FRAP recovery exhibits effective diffusion, which means that the free and bound GR in the nucleoplasm behave as if they were a single species diffusing at a rate much slower than free diffusion. The molecular identity of this nucleoplasmic binding state is unknown, but it may reflect nonspecific DNA binding, or some other generic association of this transcription factor with chromatin.
Mathematical model for FRAP at a single cluster of localized binding sites
Based on the above features, a minimal model for FRAP at the MMTV array must incorporate two types of binding: the nucleoplasmic sites, S1, and the MMTV array or spatially localized sites, S2 (for a listing of all model variables and parameters; see the Appendix, Tables 3 and 4). The S1 sites are found throughout the nucleus including within the array (since ∼90% of the DNA at the MMTV array is not promoter sequence). However, the array specific sites S2 are restricted to just the array (Fig. 2 A).
FIGURE 2.

Localized binding versus globally distributed binding. (A) The Axial and Radial Binding Model presumes a cylindrical (r,z) cluster of specific binding sites (MMTV promoters) 2 μm high and 2 μm in diameter, centered within a cylindrical nucleus 4.5-μm high and 15 μm in diameter filled with nonspecific binding sites and bleached with a cylindrical photobleach 2 μm in diameter (see Fig. 1 E). The FRAP recovery is calculated as the average fluorescent intensity within the specific binding zone, and corresponds to the measurement of intensity at the MMTV array from an optical section. (B) For the Global Binding Model specific binding sites are presumed to be distributed everywhere throughout the nucleus. In this model, variation in the axial dimension (z) is also ignored (dots indicate this symmetry in z). (C) Due to their different geometries, the two models generate different FRAP curves for the same binding parameters. (D) The Global Binding Model can be used to fit FRAP data generated by the Axial and Radial Binding Model, but the binding parameters predicted (red text) are quite different from the “true” values (black text). Thus the global binding model is a poor approximation to FRAPs at a cluster of binding sites. The FRAP curves in this and all subsequent figures have been normalized to their final recovery level. Units for k*2on and k2off in this and all subsequent figures are s−1.
The two binding sites S1 and S2 have different binding affinities, given by characteristic on and off rates (k1on, k1off and k2on, k2off). This leads to the following spatially restricted binding reactions:
 |
(1) |
where F represents freely diffusing (unbound) GFP-GR, S1 and S2 represent available binding sites of each type, C1 and C2 represent GFP-GR bound to the S1 or S2 sites, respectively,  is the radius of the zone containing the localized S2 binding sites (presumed to be circular), and
is the radius of the zone containing the localized S2 binding sites (presumed to be circular), and  is the half-height of this same zone. The nucleus is modeled as a cylinder with radial boundary at
is the half-height of this same zone. The nucleus is modeled as a cylinder with radial boundary at  and half-height of
and half-height of  and the array in this first idealization is presumed to be located at the center of the nucleus and surrounded by a homogeneous region without potential complications introduced by the presence of nucleoli.
and the array in this first idealization is presumed to be located at the center of the nucleus and surrounded by a homogeneous region without potential complications introduced by the presence of nucleoli.
To model a FRAP recovery, we write differential equations to describe the chemical kinetics for these binding reactions and the diffusion of the unbound molecules. As in other analyses of FRAP (4–6,8,9), we make two simplifying assumptions. First, we presume that the binding sites are immobile relative to the time- and lengthscale of the FRAP experiment. This is reasonable for the MMTV array because time-lapse movies have shown that this array moves very little during the 1-min FRAP recovery. This simplification eliminates a diffusion term for bound molecules (C1 or C2). Second, we assume that the system is at equilibrium before the bleach. This is satisfied if the total amount of the GFP-fusion protein does not change appreciably during the recovery period. This is also a reasonable assumption since the FRAP experiments were performed ∼18 h after induction of GFP-GR expression and ∼30 min after hormone induction has led to a constant nuclear concentration of GFP-GR. This simplification means that throughout the FRAP recovery the concentration of free binding sites for each binding state is constant and spatially uniform (namely S1eq and S2eq), since bleaching alters the fluorescence, but not the total concentration of either the free or bound GFP-GR.
With these two assumptions, we obtain two sets of differential equations that can be used to compute the FRAP recovery. Inside the array region, we use the equations for a homogeneous distribution of two binding states, while outside the array region we use the corresponding equations for a single binding state (see Sprague et al. (8)):
 |
(2) |
where  is the Laplacian operator in radial and axial coordinates, Df is the diffusion coefficient of the free molecules,
is the Laplacian operator in radial and axial coordinates, Df is the diffusion coefficient of the free molecules,  is an association rate constant (also known as a pseudo first order on rate or effective first order rate constant) for the nucleoplasmic binding sites,
is an association rate constant (also known as a pseudo first order on rate or effective first order rate constant) for the nucleoplasmic binding sites,  is an association rate constant for the MMTV specific binding sites, and
is an association rate constant for the MMTV specific binding sites, and 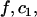 and
and 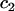 are the concentrations of the free and bound unbleached fluorescent molecules, F,
are the concentrations of the free and bound unbleached fluorescent molecules, F, 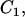 and C2, respectively.
and C2, respectively.
The effective diffusion behavior at the nucleoplasmic sites (described above and in Sprague et al. (8)) enables us to reduce the number of equations in Eq. 2 from five to three (see section A in the Appendix). This simplification arises by recognizing that with effective diffusion the free and bound fluorescence in the nucleoplasm behave as a single entity, an apparent “free” species 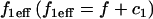 that appears to diffuse at a rate given by
that appears to diffuse at a rate given by 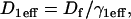 where
where 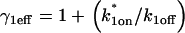 This yields the reduced system:
This yields the reduced system:
 |
(3) |
To simplify the analysis of these equations, we presume that the bleach spot radius is selected to coincide with the circular zone of MMTV specific binding sites, a constraint that is easily achieved experimentally. Equation 3 is then subject to the initial condition that the bleach depth (the initial fluorescence immediately after the bleach) is normalized to zero in the bleach zone, and therefore at 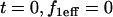 and
and 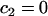 for all
for all 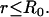 Outside the bleach zone, fluorescence is initially at its equilibrium concentration
Outside the bleach zone, fluorescence is initially at its equilibrium concentration 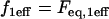 at
at  for all
for all 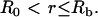 Note that for simplicity these initial conditions presume a cylindrical bleach (i.e., no z dependence), which is a reasonable first approximation to bleach patterns of the MMTV array cells (Fig. 1 D). Boundary conditions are no flux at
Note that for simplicity these initial conditions presume a cylindrical bleach (i.e., no z dependence), which is a reasonable first approximation to bleach patterns of the MMTV array cells (Fig. 1 D). Boundary conditions are no flux at 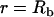 and
and 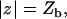 reflecting the fact that the nuclear membrane is impermeable to GR on the timescale of the FRAP recovery.
reflecting the fact that the nuclear membrane is impermeable to GR on the timescale of the FRAP recovery.
Note that solutions to Eq. 3 for spatially localized binding generalize in the following way. At the essence of the geometry described by these equations is a small domain of specific binding sites centered in a much larger domain where molecules diffuse freely. The solutions we obtain below should apply to all such scenarios. The diffusion constant may be that for either free or effective diffusion (as long as there are binding sites present throughout the larger region that exhibit effective diffusion behavior). Here we analyze the solutions to Eq. 3 using a diffusion constant corresponding to the effective diffusion constant for GFP-GR measured under our current conditions (D1eff ∼ 1.2 μm2/s) and a bleach-spot radius corresponding to the size of the MMTV array (R0 ∼ 1.0 μm). Fixing these parameters sets a characteristic time for diffusion (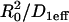 ) within the bleach spot, but does not alter the prototypical behaviors of the solution. Extending the applicability of the present findings to other binding site and ligand systems can be facilitated in future studies by nondimensionalizing the governing equations to remove the dependence upon the particular parameter values employed in the simulations for MMTV and GFP-GR.
) within the bleach spot, but does not alter the prototypical behaviors of the solution. Extending the applicability of the present findings to other binding site and ligand systems can be facilitated in future studies by nondimensionalizing the governing equations to remove the dependence upon the particular parameter values employed in the simulations for MMTV and GFP-GR.
Axial and radial binding model
We refer to Eq. 3 as the Axial and Radial Binding Model because it accounts for both the axial and radial localization of the binding site cluster (Fig. 2 A). We solved Eq. 3 numerically using a finite element method (see section B in the Appendix for details) implemented in the commercial software package FEMLAB (COMSOL, Burlington, MA). Running on a Pentium 3 processor, this approach required 5 min to generate a single FRAP curve for each set of model parameters.
To fit FRAP curves with this model, we first generated a series of possible solutions by independently varying 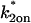 and
and 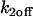 from 10−5 to 10+5 in increments of 100.5. This sampling produced a best (
from 10−5 to 10+5 in increments of 100.5. This sampling produced a best (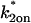 ,
, 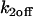 ) guess, defined as that yielding the smallest difference in the sum of residuals between the predicted FRAP and the experimental data. This best guess was then used as a starting point for finer sampling (increments of 100.1) within its vicinity. This then yielded the best fitting solution. The entire fitting procedure required ∼24 h.
) guess, defined as that yielding the smallest difference in the sum of residuals between the predicted FRAP and the experimental data. This best guess was then used as a starting point for finer sampling (increments of 100.1) within its vicinity. This then yielded the best fitting solution. The entire fitting procedure required ∼24 h.
Radial binding model
To obtain an analytical solution, Eq. 3 was simplified by assuming that the zone containing specific sites extended as a cylinder throughout the depth of the nucleus. This removes all axial (z) terms from the equations and so yields a model that accounts only for the radial localization of the specific binding sites. We refer to this model as the Radial Binding Model (Fig. 3 B), and solved the corresponding equations using a Laplace transform technique (see section C in the Appendix; final solutions summarized in Table 1). The Laplace transform was numerically inverted in Matlab (The MathWorks, Natick, MA). Running on a Pentium 4 processor, this approach required less than 1 s to generate a single FRAP curve for each set of model parameters.
FIGURE 3.

Restricted axial height of localized binding. (A) The Axial and Radial Binding Model accounts for a localized cluster of specific binding sites in three dimensions (see Fig. 2 A for details). (B) The Radial Binding Model accounts for a localized cluster of specific binding sites in each xy cross section of the cell, while ignoring localization in the z direction (dots indicate this presumed symmetry in z). (C) The presumption of uniformity in the axial direction has little effect on the FRAP curve when binding at the specific sites is relatively weak (small k*2on/k2off values), as the two models produce virtually identical FRAP curves for the same binding parameters. (D) However, with stronger binding at the localized sites (larger k*2on/k2off values), there is significant deviation between the recoveries predicted by the two models. (E) This discrepancy is largely due to the axial flux of fluorescence into the localized specific sites present in the Axial and Radial Binding Model. This flux makes up a significant portion of the total flux into the FRAP measurement zone when binding at the localized sites is strong. (F) Despite differences in the influx of axial fluorescence, the Radial Binding Model can yield a good fit to the Axial and Radial Binding Model with good estimates for the ratio k*2on/k2off (well within an order of magnitude, 48 vs. 100). (These particular FRAP data are in the local-equilibrium domain, so only the ratio k*2on/k2off can be reliably estimated).
TABLE 1.
Solutions for the radial binding model
| Radial binding model | Unique constants | ||||
|---|---|---|---|---|---|
| Full model |
|
|
|||
| Negligible specific binding |
|
|
|||
| Reaction dominant |
|
|
|||
| Local equilibrium |
|
|

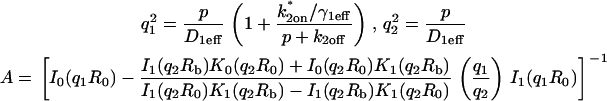


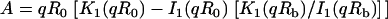
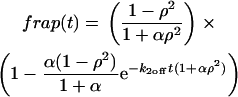
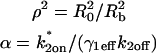


To fit FRAP curves with this model, we followed a similar procedure to that described above for the Axial and Radial Binding Model, except that possible solutions were sampled over a finer grid, namely increments of 100.1. This yielded an initial best guess that was then used as the starting guess for the Matlab nonlinear fit routine nlinfit. (The initial fine sampling was necessary because we found in a number of test cases that nlinfit did not converge correctly unless the starting guess was close to the correct solution). The entire fitting procedure was accomplished in ∼1 min.
Off-center radial binding model
To account for the fact that arrays are not located at the center of the nucleus, a numerical version of the Radial Binding Model was implemented. This Off-Center Radial Binding Model still presumed a cylindrical zone of array binding sites, but the numerical analysis was performed on a Cartesian (x,y) grid that enabled us to position the array at arbitrary locations throughout the nucleus (see Fig. 6 B, Section C in the Appendix and Müller (16)), and so determine the consequences of a nearby impermeable boundary such as the nuclear membrane.
FIGURE 6.

Effects of a nearby impermeable boundary. (A) The Radial Binding Model accounts for a localized cluster of specific binding sites centered in the middle of the cell. (B) The Off-Center Radial Binding Model allows the binding site cluster to be positioned at an arbitrary radial distance Δ from the cell center. (C) Comparison of FRAP curves predicted by these two models shows little difference when the FRAP recovery is in the reaction-dominant domain (RD), but increasing disparities arise through the full model domain (FM) reaching a plateau in the local equilibrium domain. Boundaries between the domains are shown with dotted lines extracted from Fig. 4 B. Shown are the sum of the squared residuals between the Radial Binding Model FRAP curve and the Off-Center Radial Binding Model FRAP curve with Δ = 5 μm at a constant k*2on/k2off ratio of 100. (D) FRAP recoveries in the full model or local equilibrium domain (the latter case is shown here) slow down as the binding site cluster moves closer to the impermeable boundary. (E) The FRAP recovery in panel D for Δ = 2.5 μm fit with the Radial Binding Model resulting in a slight overestimate of the true ratio k*2on/k2off. (F) The FRAP recovery in panel D for Δ = 5.0 μm fit with the Radial Binding Model resulting in a 50% overestimate of the true ratio k*2on/k2off.
Global binding model
Finally, we derived a solution for the case in which the specific binding sites were neither radially or axially restricted, but rather distributed homogeneously and globally throughout the nucleus. We refer to this model as the Global Binding Model (Fig. 2 B). These equations were also solved by a Laplace transform technique (see section D in the Appendix), and calculation of a FRAP curve was also done in Matlab in <1 s. The fitting procedure was identical to that described above for the Radial Binding Model.
RESULTS
Effects of a spatially localized cluster of binding sites
The Axial and Radial Binding Model accounts in three dimensions for a spatially localized cluster of specific binding sites. We used this model to examine how FRAP recoveries and binding parameter estimation were influenced by spatially localized binding. First, we assessed the consequences of completely ignoring this spatial localization of specific binding sites. To this end, we compared FRAP recoveries generated by the Axial and Radial Binding Model (Fig. 2 A) with those from the Global Binding Model in which the same binding sites were distributed uniformly throughout a nucleus of the same size (Fig. 2 B). As expected, these FRAPs differed for all values of k*2on, k2off tested (Fig. 2 C), demonstrating that the spatial localization of binding sites leads to characteristically different FRAP behaviors.
Next we asked what the consequences of this difference were for binding parameter estimation. Here we used the Global Binding Model to fit the FRAP data generated by the Axial and Radial Binding Model. Interestingly, we found that we could obtain reasonable fits with the Global Binding Model, but the binding parameters estimated were incorrect by more than an order of magnitude (Fig. 2 D). These results demonstrate that when spatial localization of binding occurs, a localized-binding site model is essential for accurate FRAP analysis of the binding interactions.
The Axial and Radial Binding Model, however, required 5 min to generate a single FRAP curve and up to a day to generate the hundreds of curves required to fit experimental FRAP data (see “Axial and Radial Binding Model” in the Models section). Thus, we wondered whether the computationally less expensive Radial Binding Model (Fig. 3 B) could be substituted in some cases. This model requires ∼1 s to generate a single FRAP curve and ∼1 min to fit experimental FRAP data, but presumes that the localized sites form a cylindrical column spanning the height of the nucleus, thereby ignoring the spatial variation of binding sites only along the z axis. To evaluate the utility of this model, we first generated FRAPs with it and compared the curves to those generated with the same parameters for the Axial and Radial Binding Model. We found that for smaller values of k*2on/k2off (weaker binding at the specific sites), the two models yielded FRAP curves that were quite similar (Fig. 3 C), while for larger values of k*2on/k2off (tighter binding at the specific sites), the two models disagreed (Fig. 3 D).
A significant contributing factor to this difference was the additional flux of fluorescence that could enter the bleached zone in the Axial and Radial Binding Model from the regions above or below the localized binding sites (Fig. 3 E). This flux is completely absent in the Radial Binding Model because it lacks an axial component. The axial flux generated in the Axial and Radial Binding Model increased as the binding at the localized sites became tighter, as did the discrepancy between the FRAP curves predicted by this model and the simpler Radial Binding Model (data not shown). We conclude that a restricted axial height for the localized binding sites leads to detectable differences in the FRAP recovery, with greater differences arising when binding is tighter at the localized sites, and that much of this difference arises due to the axial flux of fluorescence into the zone of localized binding.
To determine the consequences of ignoring the restricted axial height of the localized binding sites on fitting FRAP data, we generated FRAP curves using the Axial and Radial Binding Model and then attempted to fit them with the Radial Binding Model. In some cases, like that in Fig. 3 C, the restricted axial height of the localized binding sites has little effect on the FRAP curve. As expected, we found for these cases that the Radial Binding Model yielded excellent fits to FRAP data generated by the Axial and Radial Binding Model with perfect estimates of the binding parameters (data not shown). Interestingly, for cases where the Axial and Radial Binding Model FRAP recovery was noticeably different from that produced by the simpler Radial Binding Model (such as Fig. 3 D), the simpler model could nevertheless produce reasonable fits to data generated by the Axial and Radial Binding Model (Fig. 3 F).
Of course in these cases, despite the good fit, the Radial Binding Model yielded incorrect estimates for the rate constants. However these estimates, either for k*2on, k2off, or their ratio k*2on/k2off (see Fig. 3 F for an example), were always within an order of magnitude of the “true” values used in the Axial and Radial Binding Model to generate the FRAP curve to be fit. We conclude that ignoring the axial flux of fluorescence into the localized sites affects parameter estimation when binding at the localized sites is strong, but good fits can still be obtained yielding errors smaller than an order of magnitude for these binding parameters. Thus the Radial Binding Model, though not completely accurate, may often be a useful tool for fitting FRAP data.
Different limiting FRAP behaviors at a cluster of localized binding sites
Several previous studies have demonstrated that FRAPs could sometimes be reduced to simpler equations, defining limiting behaviors in the general equations (4,5,8,9,14). Identifying when these limiting behaviors occur is important because the simpler equations can then be used to most efficiently extract binding information, and for certain behaviors there are limitations on exactly what binding information can be extracted. In addition, these domains introduce self-consistency checks (8,17).
To identify these domains for FRAPs at localized binding sites, we created simplified versions of the Axial and Radial Binding Model corresponding to each of the expected limiting behaviors (see section B in the Appendix). These behaviors are: 1), when binding at the cluster of localized sites is negligible; 2), when binding is not negligible, but the expected time to diffuse across the bleach spot is much shorter than the expected time to begin binding (reaction-dominant behavior); and 3), when binding is not negligible, but the expected time to diffuse across the bleach spot is much longer than the expected time to begin binding (local-equilibrium behavior).
To determine when these limiting behaviors of the Axial and Radial Binding Model were valid approximations to the true FRAP behavior, we compared the FRAPs they predicted to those produced using the full version of the model. Comparisons were performed over a wide range of biologically relevant rate constants at the localized sites ( ), while fixing the effective diffusion constant and the bleach-spot radius at values typical for our biological FRAPs. We identified three large domains of k*2on, k2off where at least one of the approximate solutions was reasonably accurate, and a fourth domain where only the full solution described the FRAP recovery (Fig. 4 A).
), while fixing the effective diffusion constant and the bleach-spot radius at values typical for our biological FRAPs. We identified three large domains of k*2on, k2off where at least one of the approximate solutions was reasonably accurate, and a fourth domain where only the full solution described the FRAP recovery (Fig. 4 A).
FIGURE 4.

Simplified domains for FRAP at localized binding sites. (A) We generated solutions for limiting behaviors of the Axial and Radial Binding Model (see section B in the Appendix). Then we asked when these simplified solutions were good approximations to the full solution of the Axial and Radial Binding Model. A good match between the two predicted FRAP curves indicates that for the binding parameters used, the FRAP data are well approximated by the limiting behavior, and so this simpler solution suffices. Binding parameters (k*2on, k2off) were varied from 10−5 to 105 while holding the effective diffusion constant and the bleach-spot radius constant at typical values (D1eff = 1.2 μm2/s, R0 = 1 μm). This subdivided (k*2on, k2off) parameter space into the four regions shown. (A fifth region defined by the black hatched zone was uncomputable due to memory limitations in the numerical analysis; see Fig. 8 and the associated subsection on model validation in section B of the Appendix). The lines separating the domains correspond to a difference of one for the summed residuals calculated at 200 equally spaced time points for the full-model FRAP curve compared to the limiting behavior FRAP curve. The resultant simplified domains define whether localized binding parameters can be measured (outside the negligible specific binding domain), whether diffusion contributes significantly to the FRAP (outside the reaction-dominant domain), and whether independent estimates of the binding parameters can be extracted (outside the local-equilibrium domain). (B) A comparable procedure was performed with the Radial Binding Model using the full solution for this model (first row in Table 1) compared to the solutions for limiting behaviors (second to fourth rows in Table 1). A comparable domain structure was detected, indicating that the simpler Radial Binding Model still captures the essential behaviors present in the more complex Axial and Radial Binding Model. (This model also yielded an uncomputable zone defined again by the black hatched region in which the inverse Laplace transform for the local equilibrium model could not be computed). (C) Times for FRAP recoveries at localized binding sites were calculated as a function of location in (k*2on, k2off) parameter space for the Radial Binding Model (time in seconds to reach 99% of complete recovery is shown on the contour lines). FRAP recoveries lasting several minutes were found in domains where diffusion contributes significantly, namely in the full-model and local-equilibrium domains. With the same bleach-spot size, a diffusing molecule such as GFP would fully recover in <1 s. Therefore, despite this significant difference in recovery time, diffusion may still contribute substantively to many slow FRAP recoveries.
Since we had found above that the Radial Binding Model for localized binding was a reasonable approximation to the Axial and Radial Binding Model, we also asked whether the Radial Binding Model would exhibit comparable simplified domains. To investigate this, we derived equations corresponding to the different limiting behaviors outlined above (see section C in the Appendix for the derivations, and the second to fourth rows in Table 1 for the solutions). We compared the solutions for these limiting behaviors to the solutions for the full-model version of the Radial Binding Model (first row, Table 1) over the same range of biologically relevant rate constants examined above. We found a comparable domain structure (Fig. 4 B) as determined above for the Axial and Radial Binding Model. These results demonstrate that although it is simplified, the Radial Binding Model possesses an analogous pattern of limiting behaviors as found for the Axial and Radial Binding Model, and so further support the utility of the Radial Binding Model for FRAP analysis at localized binding sites.
Interestingly, the resultant distribution of idealized domains for FRAPs at localized binding sites resembles that previously identified for FRAPs at homogeneously and globally distributed binding sites (compare Fig. 4, A and B, to Fig. 2 D in Sprague et al. (8)), even though the two models yield entirely different FRAP curves for the same k*2on, k2off (Fig. 2 C). This similarity in domain structure was not necessarily expected because the spatially localized binding model is based on an entirely different geometry, namely a cluster of one type of binding site overlaid on a much larger region throughout which diffusion (or effective diffusion) occurs. In contrast, the situation for globally distributed binding sites is much simpler. There is only one type of site, and it is distributed uniformly throughout the entire region. Nevertheless, despite the increase in complexity accompanying spatially localized binding, the distribution of simplified domains as a function of rate constants is preserved.
Given this similarity of both the domains and their distribution between the spatially localized and globally distributed binding models, a number of the general conclusions drawn about global binding (8,17) can now be extended to FRAPs at a spatially localized cluster of binding sites. These are: 1), the contribution of diffusion to a FRAP recovery can be safely ignored only in the reaction-dominant domain, where the expected time for diffusion across the bleach spot is much faster than the expected time for binding (and so diffusion is effectively instantaneous on the timescale of the FRAP recovery). This idealized behavior occupies ∼1/3 of the region of rate-constant parameter space where binding parameters can be estimated (Fig. 4, A and B). Thus diffusion is expected to contribute to the majority (∼2/3) of FRAP recoveries at a cluster of binding sites, given typical values for effective diffusion and bleach spot size. Although these proportions are rough estimates because they depend on the specific values for the diffusion constant and bleach-spot size, the results nevertheless point to the importance of carefully considering diffusion's role in a FRAP at a cluster of binding sites. 2), Long FRAP recoveries do not necessarily reflect reaction-dominant behavior, as is often presumed. By computing the time for complete recovery as a function of k*2on, k2off, we find that FRAP recoveries of up to 4 min. can occur at localized binding sites in domains where diffusion contributes substantively (Fig. 4 C), even though a FRAP of a freely diffusing molecule would be complete in ∼1 s. 3), Fits to FRAP data can be subjected to a consistency check based on the estimated rate constants. Essentially, the predicted rate constants should lie in the appropriate domain in k*2on, k2off parameter space consistent with the type of model used to fit the data (see Table 2 for details). These constraints may be used to rule out incorrect models for a cluster of binding sites that nevertheless yield a good fit. 4), FRAP recoveries lying in the local-equilibrium domain will not yield independent estimates for 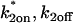 , but rather only the ratio
, but rather only the ratio  This is because the local-equilibrium solution for a cluster of binding sites depends only on the ratio
This is because the local-equilibrium solution for a cluster of binding sites depends only on the ratio 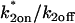 (see Table 1; fourth row and the definition for
(see Table 1; fourth row and the definition for 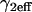 in Table 4).
in Table 4).
TABLE 2.
Consistency checks
| Model domain | Constraints on parameter estimates | |
|---|---|---|
| Reaction dominant |
|
|
| Full model |
|
|
| Local equilibrium |
|
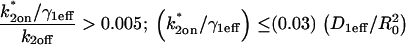
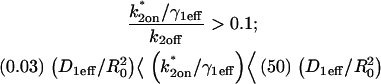

Consistency checks derived from the Radial Binding Model. The parameters estimated by each type of fit should satisfy the constraints shown. These constraints are derived from the structure of the simplified domains (Fig. 4, A and B). The first constraint in the second column ensures that the estimated binding parameters place the solution outside of the negligible-binding domain. The second constraint in this column ensures that 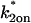 is of the proper magnitude to place the solution in the correct simplified domain. Note that for the local equilibrium constraint,
is of the proper magnitude to place the solution in the correct simplified domain. Note that for the local equilibrium constraint,  is annotated as “full model”. The local equilibrium fit produces only the ratio
is annotated as “full model”. The local equilibrium fit produces only the ratio 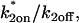 but a full-model fit should produce a series of nearly equivalent fits to the same data yielding a series of
but a full-model fit should produce a series of nearly equivalent fits to the same data yielding a series of 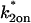 values that satisfy the inequality.
values that satisfy the inequality.
Effects of ignoring either diffusion or local equilibrium behavior on binding parameter estimation
In most current localized-site FRAP models, diffusion has been presumed to be fast and negligible. This simplification is invoked based on the fact that FRAP recoveries for GFP-tagged proteins typically last much longer than those for free diffusion, as assayed by a FRAP of unconjugated GFP. However, as shown in the domain analysis above, recoveries of up to 4 min often involve a role for diffusion (Fig. 4 C). Moreover, we found that for typical values of the effective-diffusion constant and the bleach-spot radius, many FRAP recoveries at a cluster of binding sites are expected to depend on diffusion (Fig. 4 A).
To calculate errors introduced by improperly ignoring diffusion, we generated FRAP curves using the Axial and Radial Binding Model in domains where the recovery was expected to depend on diffusion (the local-equilibrium domain and the full-model domain as defined in Fig. 4, A and B). We then attempted to fit these FRAP data with a reaction-dominant form of the Axial and Radial Binding Model produced simply by setting the free diffusion constant to a very large value (see section B in the Appendix). This reaction-dominant form is therefore equivalent to models that presume diffusion is so rapid that it can be ignored. When improperly applied to FRAP data that actually depended on diffusion, this reaction-dominant model nevertheless yielded fairly respectable fits to the diffusion-dependent data, but the binding parameters estimated were off by almost two orders of magnitude (Fig. 5 A). These poor estimates often produced k*2on estimates that were too large to be in the reaction-dominant domain (see Fig. 5 A legend and Table 2 for details), and so could be ruled out by this consistency check, if it were applied. We conclude that improperly ignoring diffusion's role in localized binding can lead to serious errors that might otherwise be identified by applying the appropriate consistency check.
FIGURE 5.

Consequences of ignoring either diffusion or local equilibrium behavior. (A) To investigate the impact of improperly neglecting diffusion, we used the Axial and Radial Binding Model and generated FRAP curves using binding parameters that would produce recoveries that depended on diffusion (either from the full model or the local equilibrium domains). These diffusion-dependent recoveries were then fit using the simplified solution of the Axial and Radial Binding Model that ignored diffusion (the reaction-dominant solution). We found we could obtain decent fits with this approach, but estimates of the binding parameters were often in error by almost two orders of magnitude (true parameters in black text, estimated parameters in red text). This improper fit could in fact be rejected by applying the reaction-dominant consistency check in Table 2, namely that  For the reaction-dominant fit here,
For the reaction-dominant fit here, 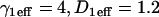 μm2/s and
μm2/s and  μm. Therefore, the estimated value for
μm. Therefore, the estimated value for 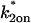 must satisfy
must satisfy 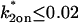 s−1, which is violated by the
s−1, which is violated by the 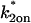 estimate of 6.3 s−1 (red text). (B) To investigate the consequences of ignoring the constraints imposed by local-equilibrium behavior, we generated FRAP data within this domain using the Axial and Radial Binding Model. We were able to achieve excellent fits to these data using the same model and a range of other binding parameters. Shown here is a case where the binding rates differ by an order of magnitude (purple versus orange text), but even larger differences were possible as long as the ratio k*2on/k2off was kept constant. (C) A constant ratio k*2on/k2off = 100 yields the equation log(k*2on) − log(k2off) = 2, which produces a line of slope +1 (dotted black line) in the log-log plot of rate constants. All points along this line yield virtually identical FRAP curves demonstrating that independent estimates of the rate constants cannot be obtained in the local equilibrium domain.
estimate of 6.3 s−1 (red text). (B) To investigate the consequences of ignoring the constraints imposed by local-equilibrium behavior, we generated FRAP data within this domain using the Axial and Radial Binding Model. We were able to achieve excellent fits to these data using the same model and a range of other binding parameters. Shown here is a case where the binding rates differ by an order of magnitude (purple versus orange text), but even larger differences were possible as long as the ratio k*2on/k2off was kept constant. (C) A constant ratio k*2on/k2off = 100 yields the equation log(k*2on) − log(k2off) = 2, which produces a line of slope +1 (dotted black line) in the log-log plot of rate constants. All points along this line yield virtually identical FRAP curves demonstrating that independent estimates of the rate constants cannot be obtained in the local equilibrium domain.
We also considered the consequences of improperly ignoring the constraints imposed by the local-equilibrium domain for a cluster of binding sites. For this, we generated a FRAP curve using the Axial and Radial Binding Model in a domain that we identified as local equilibrium from Fig. 4 A. Then we decreased both k*2on and k2off by an order of magnitude, and used the same Axial and Radial Binding Model to generate a second FRAP curve that nevertheless yielded an excellent fit to the first curve (Fig. 5 B). Indeed, we found that many pairs of (k*2on, k2off) values yielded excellent fits to the original data, with the set of successful (k*2on, k2off) values describing a line in the log-log plot of rate constants (dotted black line in Fig. 5 C). This line of good fits corresponds to the constant ratio k*2on/k2off that is the only parameter that can be estimated in the local equilibrium domain. We conclude that it is critical to demonstrate that FRAP data from a cluster of localized binding sites are not in the local-equilibrium domain before assigning unique values to k*2on and k2off, otherwise meaningless, independent estimates of rate constants can result.
Effects of ignoring proximity to an impermeable boundary
Both the Axial and Radial Binding Model and the simpler Radial Binding Model presume that the binding site cluster is at the center of the nucleus. This is, however, rarely the case either for the MMTV array (Fig. 1 E) or for other typical binding site clusters. To evaluate the effects of an off-center location, we used the Off-Center Radial Binding Model to position the cluster of binding sites at an arbitrary position within the nucleus (Fig. 6, A and B).
We varied k*2on, k2off over a wide range of values, and then compared FRAP curves for a 2-μm-diameter binding site cluster located either at the nuclear center or 5 μm distant from the center (i.e., 2.5 μm from the nuclear membrane). The two FRAP curves were essentially identical for values of k*2on, k2off lying within the reaction-dominant domain, but became increasingly different as k*2on increased and the FRAP curves entered first the full model domain and eventually the local equilibrium domain (Fig. 6 C). Within these domains, clusters closer to the impermeable nuclear membrane always showed slower recoveries (Fig. 6 D), consistent with a previous analysis of the effects of an impermeable boundary on a purely diffusive FRAP recovery (6). These slower recoveries reflect a decrease in the diffusive flux into the bleach spot due to the nearby impermeable boundary (6). However, for a reaction-dominant scenario, this difference in diffusive flux should be restricted to only the earliest time points, and so as we observed for these recoveries, the effect of proximity to the impermeable boundary should be negligible. In sum, our results indicate that only FRAP recoveries in the full model or local equilibrium regimes will be significantly retarded by their proximity to an impermeable boundary.
We evaluated the consequences of ignoring this boundary effect by using the analytical solution of the Radial Binding Model to fit the FRAP recoveries generated by the Off-Center Radial Binding Model for the worst-case scenario, namely FRAP data in the local equilibrium domain. As expected, when the displacement from the nuclear center was zero, the Off-Center Radial Binding Model yielded an identical FRAP recovery as the Radial Binding Model, and the fit of the Radial Binding Model yielded the same rate constants as the “true” values used to generate the FRAP curve by the Off-Center Radial Binding Model (data not shown). However, as displacement from the nuclear center increased and the binding site cluster came closer to the nuclear membrane, the fits became less accurate as did the estimates of the binding parameters (Fig. 6, E and F). This error in estimating k*2on/k2off was ∼50% for a case corresponding to the average MMTV array location at ∼5 μm from the nuclear center (Fig. 6 F). Despite the fact that errors arise due to ignoring an impermeable boundary, the errors are considerably smaller than those produced by other simplifications, such as ignoring either the role of diffusion, the constraints of the local equilibrium domain, or the localization of specific binding sites.
Application to GFP-GR FRAP recoveries at the MMTV promoter sites
To fit experimental FRAP data from the MMTV array, we used both the Axial and Radial Binding Model (Fig. 3 A and section B in the Appendix) and the Radial Binding Model (Fig. 3 B and section C in the Appendix). Calculation of a single FRAP curve required ∼5 min for the Axial and Radial Binding Model and ∼1 s for the Radial Binding Model. Unfortunately, finding a good fit took longer because we generated a large number of FRAP curves (at least 500) to find the one producing the closest match to the experimental data (see Models section). For this reason, the fitting procedure required ∼1 min with the Radial Binding Model, and ∼1 day with the Axial and Radial Binding Model. These times could be dramatically shortened if a more efficient fitting procedure were identified.
Using GFP-GR FRAP data from the MMTV array, we achieved a good fit with the local-equilibrium version of the Axial and Radial Binding Model, using a single free parameter, namely the ratio k*2on/k2off = 2.3 (Fig. 7 A). We also achieved a good fit with the local equilibrium form of the Radial Binding Model, again just using a single free parameter, in this case yielding the estimate k*2on/k2off = 2.5 (Fig. 7 B). Note that as expected from our analysis above, the difference in this estimate of k*2on/k2off (2.3 vs. 2.5) is well within an order of magnitude.
FIGURE 7.

Fitting FRAP of GFP-GR at the MMTV array. (A) The FRAP recovery curve for GFP-GR at the MMTV array (average of 10 experiments) was well fit with the local equilibrium form of the Axial and Radial Binding Model. This fit was obtained with a single free parameter, namely the ratio 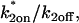 which was estimated by the fit to be 2.3. All other parameters in the model were fixed at measured values (see Fig. 1 E). (B) The same experimental GFP-GR recovery curve was also well fit with the local-equilibrium version of the Radial Binding Model (fourth row, Table 1), again with a single free parameter yielding a second estimate of the binding constant ratio of
which was estimated by the fit to be 2.3. All other parameters in the model were fixed at measured values (see Fig. 1 E). (B) The same experimental GFP-GR recovery curve was also well fit with the local-equilibrium version of the Radial Binding Model (fourth row, Table 1), again with a single free parameter yielding a second estimate of the binding constant ratio of 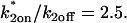 (C) Experimental FRAPs on a set of arrays whose centers were either 0.5–1.0 μm away from nucleoli or 1.7–3.0 μm away from nucleoli. No significant difference was detected. Error bars have been omitted for clarity, but are comparable to those in panels A and B. (D) Although only the ratio k*2on/k2off can be estimated from these local equilibrium data, a lower bound on the off rate is available based on the structure of the domain space. The estimate k*2on/k2off = 1.7 defines a series of points along the thick black line, each of which is consistent with the experimental FRAP data. At the star, this line intersects the contour line specifying the boundary separating the local-equilibrium domain from the full-model domain (see also Fig. 4, A and B). Therefore, the thick black line cannot be extrapolated beyond this boundary, otherwise the experimental data would not have been fit by the local-equilibrium form of the model. Thus the smallest value for k2off is given by the location of the star, yielding k2off > 10.8 = 6 s−1.
(C) Experimental FRAPs on a set of arrays whose centers were either 0.5–1.0 μm away from nucleoli or 1.7–3.0 μm away from nucleoli. No significant difference was detected. Error bars have been omitted for clarity, but are comparable to those in panels A and B. (D) Although only the ratio k*2on/k2off can be estimated from these local equilibrium data, a lower bound on the off rate is available based on the structure of the domain space. The estimate k*2on/k2off = 1.7 defines a series of points along the thick black line, each of which is consistent with the experimental FRAP data. At the star, this line intersects the contour line specifying the boundary separating the local-equilibrium domain from the full-model domain (see also Fig. 4, A and B). Therefore, the thick black line cannot be extrapolated beyond this boundary, otherwise the experimental data would not have been fit by the local-equilibrium form of the model. Thus the smallest value for k2off is given by the location of the star, yielding k2off > 10.8 = 6 s−1.
We next subjected these fits to a consistency check. We used the full-model form of the Axial and Radial Binding Model to fit the FRAP data. This model should be able to fit all FRAP data from a localized binding site cluster regardless of whether it lies in a simplified domain or not, but as we show here this fit can be used to confirm the assignment to a simplified domain. We found that the full model version of the Axial and Radial Binding Model actually yielded a family of good-fitting curves for which the sum of residuals between the fit and the experimental data was negligibly different. All of these good fits shared the property that k*2on/k2off = 2.3 (data not shown), consistent with the local equilibrium constraint that only this ratio is determined by the data. Further, nearly all of the successful fits yielded k*2on values that were in the local equilibrium domain as defined by the constraints in Table 2. We conclude that the current MMTV FRAP data are in the local equilibrium domain and that only the ratio k*2on/k2off can be reliably estimated.
These self-consistent results from the modeling analysis were satisfying; however, we wondered how the estimates of k*2on/k2off might be affected by “real-life” complications, specifically the off-center position of the array and the presence of nucleoli near the array. The Axial and Radial Binding Model and the Radial Binding Model both presume a central location of the array surrounded by a homogeneous distribution of “nonspecific” GR binding sites. In reality, MMTV arrays are distributed throughout the nucleus with an average radial location of ∼5 μm from the nuclear center, and are often very near nucleoli.
To account for the off-center location of the array, we used the analysis of FRAP recoveries produced by the Off-Center Radial Binding Model. Comparing this model to the Radial Binding Model, we showed above (Fig. 6 F) that the Radial Binding Model will overestimate k*2on/k2off by ∼50% for a binding site cluster that is ∼5 μm from the nuclear center. This example (Fig. 6 F) corresponds precisely to the array data, as both the Radial Binding Model in Fig. 6 F and the array data yield k*2on/k2off = 2.5 instead of the “true” k*2on/k2off = 1.7. Thus to account for the array's off-center location, the ratio k*2on/k2off = 2.5 should be corrected to 1.7.
All of the models analyzed in this study ignore the presence of nucleoli in the vicinity of the array, and instead presume that the concentration of fluorescence surrounding the array is uniform before the bleach and that the diffusion and binding properties of the nucleoplasm surrounding the array are homogeneous. However, compared to the rest of the nucleoplasm, nucleoli contain little or no GFP-GR. This could arise because there may be very few binding sites for GR within nucleoli, or because nucleoli might be fairly impermeable to GR, or some combination of these two factors. Fewer GR binding sites within nucleoli should lead to faster FRAP recoveries near nucleoli because GR diffusion through the neighboring nucleolus would be less retarded by binding interactions. Conversely, a less permeable nucleolus should lead to slower FRAP recoveries near nucleoli due to slower diffusion of GR through the nucleolus.
To test the impact of nucleoli on GR recoveries at the MMTV array, we performed FRAPs first on one set of arrays whose centers were 0.5–1.0 μm away from nucleoli, and second on another set of arrays whose centers were 1.7–3.0 μm away from nucleoli. Since arrays are ∼1 μm in radius, all of the arrays from the first data set directly contacted nucleoli while none of the arrays in the second data set did. We expected to detect some difference between these two sets of FRAP data, at least at early time points, if the nucleolus either slows down or speeds up the FRAP recovery. However, the measured recoveries from the two data sets were within experimental error at all time points (Fig. 7 C). This suggests that the nucleolus has at best a modest effect on the FRAP recovery, perhaps because the properties of the nucleolus counteract each other, or because cell-to-cell variability overshadows small differences arising from nucleoli. We conclude that the estimate of k*2on/k2off = 1.7 for the MMTV array is not significantly altered by the presence of nucleoli.
This estimate of k*2on/k2off for the array enables an estimate of an in vivo binding constant. Using GFP-tagged viral particles, we have previously determined that on average ∼900 of the possible 1200 GFP-GR binding sites at the MMTV array are occupied (18). Therefore, there are ∼300 free binding sites at the array, which permits an estimate of S2eq, the equilibrium concentration of free binding sites at the array. Using our measurements of the array volume (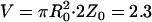 μm3), we obtain S2eq ∼ 220 nM. Since k*2on/k2off = (k2on/k2off)S2eq = (1/Kd)S2eq we arrive at an order of magnitude estimate for the binding dissociation constant, Kd ∼ 10−7 M for GFP-GR binding to MMTV.
μm3), we obtain S2eq ∼ 220 nM. Since k*2on/k2off = (k2on/k2off)S2eq = (1/Kd)S2eq we arrive at an order of magnitude estimate for the binding dissociation constant, Kd ∼ 10−7 M for GFP-GR binding to MMTV.
Although we cannot extract independent estimates of k*2on or k2off from these FRAP data, we can use the structure of the domain space (Fig. 4, A and B) to obtain a rough upper-bound estimate of the GFP-GR residence time at the MMTV promoter. This is because local-equilibrium behavior imposes a lower bound of ∼6 s−1 for k2off (see Fig. 7 D). Since the residence time is given by τR = 1/k2off, a lower bound of 6 s−1 for k2off yields an upper bound for the GFP-GR residence time at the MMTV promoter of ∼170 ms.
DISCUSSION
Models for FRAP analysis at localized binding sites
We have analyzed models for FRAP recoveries at a single cluster of binding sites that extend current techniques for the evaluation of such data. The Radial Binding Model incorporates the effects of diffusion and binding and also accounts for finite, impermeable cell boundaries. Using this model implemented on a PC, a FRAP curve could be computed in <1 s. The Axial and Radial Binding Model further extends this analysis by incorporating the effects of a finite axial height for the region of localized binding sites. Although more realistic, this model is considerably slower, requiring ∼5 min to generate a FRAP curve. A second disadvantage of this approach is that it also requires specialized numerical-analysis software (FEMLAB).
Even though the Radial Binding Model presumed an axial zone of localized binding extending throughout the entire nucleus, we could still use this model to achieve “good” fits of simulated FRAP data generated from the Axial and Radial Binding Model, which accounted for not only the radial but also the axial restriction in localized binding. By “good” fit, we mean that we could find a FRAP curve using the Radial Binding Model that nearly overlapped the one produced by the Axial and Radial Binding Model. The parameters estimated from this fit were in all cases less than an order of magnitude different from the true parameters used to produce the FRAP curve in the Axial and Radial Binding Model. This suggests that the faster and more easily implemented Radial Binding Model might still be used in many cases to achieve order of magnitude estimates of binding parameters, even though it ignores the restricted height of the localized binding zone. This result is consistent with the observation that a two-dimensional FRAP analysis also yields reasonable estimates for binding parameters obtained for the heterogeneous three-dimensional distribution of certain chromatin associated proteins (14). Our results, however, demonstrate that there are limits to this simplification, for example as the binding strength increases so too do the errors in parameter estimation. An additional cautionary note is that our results are only valid for a single binding site cluster like the MMTV array that occupies ∼50% or more of the nuclear height. Further numerical analysis would be required to quantify the errors introduced for a smaller cluster, such as a centrosome, which may occupy <10% of the nuclear height, or for multiple clusters such as nuclear pores, which may be close enough together to influence each other.
Insights provided by a knowledge of limiting FRAP behaviors at localized sites
We also investigated several additional assumptions that are often made in fitting FRAP data at localized binding sites. A role for diffusion is frequently ignored, first because FRAP recoveries of many GFP fusion proteins are much slower than those for unconjugated GFP, and second because the spatially localized cluster of specific binding sites occupies a small domain in which diffusion should occur rapidly. We found however that these presumptions are often incorrect: FRAP recovery times within a cluster of localized binding sites may last several minutes and still involve diffusion. Based on typical parameters for cellular binding, bleach-spot diameter and diffusion, we estimate that a sizeable proportion (perhaps as much as 2/3) of FRAP recoveries at a cluster of binding sites are likely to involve diffusion. Moreover, we demonstrate here that improperly ignoring this role for diffusion at the binding site cluster will have serious consequences leading to very poor estimates of the binding parameters.
A second common assumption in all FRAP models at localized sites is that independent estimates of the on and off rates of binding can be recovered. Using the models for a binding site cluster, we showed that in a simplified domain that we called local equilibrium, the FRAP recovery depended only on the ratio of these rates. If we improperly ignored this constraint, we found that the models could produce vastly different estimates for these rates. These radically different estimates still yielded a constant ratio, which is the only parameter that can be accurately estimated in the local-equilibrium domain. This domain can be recognized using the inequalities in Table 2, or simply by checking to see if a series of equally good fits can be achieved for different on and off rates that are constrained to yield a constant ratio. Our results underscore the importance of determining when FRAP data are in the local-equilibrium domain before assigning unique values to the on and off rates.
Importantly, these preceding features echo those that we found for FRAPs at homogeneously and globally distributed binding sites (8), and they are also similar to observations of Beaudouin et al. (14) for a heterogeneous distribution of binding sites. This suggests that these features are generic to all FRAP analyses.
Comparison of analytical and numerical approaches
In this study, we employed parallel numerical and analytical approaches. For ultimate precision, numerical models are preferable since they can incorporate all essential features of the real experiment. We used our numerical analysis to account both for the effects of a restricted axial height of localized binding sites and for the effects of proximity to an impermeable boundary. In a more general numerical approach, Beaudoin et al. (14) account for FRAPs arising from an arbitrary distribution of binding sites that can be measured directly from the image data.
These numerical methods can also be used for additional future tests of the many assumptions still present in most FRAP models. These assumptions often overlook the complicated geometries of real bleaching patterns (19), the finite time involved in bleaching (6,20,21), and the methods for correcting the unintentional bleaching that occurs during the measurement of the FRAP recovery (6,22). Given all of these approximations, we believe that our current measured values for binding parameters are likely to be order-of-magnitude estimates at best. As further refinements in these numerical methods are implemented, additional improper assumptions can be identified and remedied, ultimately enabling more precise estimates of binding parameters.
While a numerical approach offers many advantages, the analytical approach also has merit. From a practical standpoint, analytical solutions are much faster to compute, in our case, by more than two orders of magnitude. Typically, an analytical approach also provides more insights into the structure and general behavior of a model. For FRAP this has led to the identification of limiting behaviors, and these in turn have generated the consistency checks on estimated parameters. Finally, analytical solutions provide test cases to validate numerical models, which often suffer from implementation defects. In this way, we identified a defect in the initial implementation of our numerical method (see section B and C in the Appendix).
Application of the models to transcription factor binding at a promoter
We have applied these models to the analysis of FRAP recoveries at a tandem array of MMTV promoters. We found that FRAPs at this site were well fit by the local equilibrium solutions of both the Axial and Radial Binding Model and the Radial Binding Model. These fits passed the consistency checks enforced by the domain structure of the models. Namely, the full-model solution yielded a set of nearly equivalent fits in the local-equilibrium domain each with the same ratio k*2on/k2off, and the local-equilibrium solution itself yielded a good fit demonstrating that only the ratio k*2on/k2off could be determined.
We could obtain good fits for the MMTV FRAP data with the Axial and Radial Binding Model or the Radial Binding Model using only a single free parameter, namely the ratio k*2on/k2off. The predicted ratio differed only by ∼10% between these two models, consistent with our detailed comparison of these alternate approaches. This ratio was found to be an overestimate by ∼50% based on the proximity of the average array to the impermeable nuclear membrane, leading to a final estimate of k*2on/k2off = 1.7. With this ratio combined with an earlier estimate of the number of free binding sites at the MMTV array (18), we could calculate an in vivo binding dissociation constant for GR at MMTV of Kd ∼ 10−7 M. This first in vivo estimate indicates weaker binding than previous in vitro estimates, which have ranged from Kd ∼ 10−8–10−10 (23,24). This may reflect the fact that the in vitro estimates were obtained using naked DNA and a truncated GR molecule that contained primarily the DNA binding domain, while our in vivo estimates are based on an intact GFP-GR molecule interacting with chromatin in the ionic environment found within a living nucleus.
Since the FRAP data for GFP-GR were in the local-equilibrium domain, we could not obtain independent estimates of k*2on and k2off. However, we could deduce an approximate lower bound estimate for k2off of 6 s−1, which led to an upper bound estimate of ∼170 ms for the GFP-GR residence time at the MMTV promoter. This estimate of an in vivo residence was derived indirectly from the domain structure of the model, rather than from a direct estimate of k2off based exclusively on the FRAP curve. As a result, this value should be treated with caution until more direct estimates of k2off can be achieved. Nevertheless, it seems safe to conclude that the in vivo GR residence time will be significantly shorter than in vitro estimates, which have been as long as 108 min (23). Shorter in vitro residence times (∼20 s) however have been measured as the concentration of nonspecific DNA present in the in vitro reaction is increased to 22 μg/ml (24), at which point the temporal resolution of the in vitro gel-shift assay is reached. The concentration of DNA within the nucleus (∼10 mg/ml) greatly exceeds these in vitro concentrations, and so this might contribute to the faster in vivo residence times that we measured. However, other cellular factors may also be at play, as the environment of the nucleus is far more complicated than the simple conditions used in the in vitro assays. The tools developed here will be useful for identifying these various cellular factors and their influence on GR binding to MMTV in live cells.
Regardless of the factors influencing transient binding, our current in vivo estimate for the GR residence time suggests that the binding interaction is quite rapid. In the simplest model for transcription, the full transcription complex including coactivators, remodelers, and the polymerase might be assembled during the GR residence time at the promoter. Alternatively, partial and progressive assembly of the complex may occur with repeated visits of GR to this site (25). The procedures developed here can now be used to help distinguish between such models by determining if most other factors at the promoter exhibit comparable or vastly different residence times.
Acknowledgments
We thank Dr. Tatiana Karpova for assistance with the imaging, and Dr. Carolyn Smith for comments on the manuscript.
This research was supported in part by the intramural program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
APPENDIX OVERVIEW
The Appendix is divided into four sections. In section A, we show how the FRAP equations for binding at the MMTV promoter sites can be reduced to a simpler set when the nucleoplasmic sites exhibit effective diffusion. In section B, we explain how these equations are solved numerically for the Axial and Radial Binding Model and its three simplified domains, and we provide some validation of the numerical analysis. In section C, we derive analytical solutions for the Radial Binding Model and its corresponding three simplified domains. We also discuss our numerical implementation of the Off-Center Radial Binding Model using a Cartesian coordinate system. In section D, we derive the analytical solution for the Global Binding Model. For convenience, we list in Table 3 all variables used in the models, and in Table 4 all parameters derived from these variables.
TABLE 3.
Variables and basic nomenclature
| Name | Meaning |
|---|---|
| t | Time |
| p | Laplace transform variable |
| S1 | Nonlocalized binding sites |
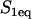 |
Equilibrium concentration of unoccupied nonlocalized binding sites |
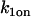 |
On rate at the nonlocalized binding sites |
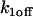 |
Off rate at the nonlocalized binding sites |
| C1 | Fluorescent molecules bound to 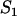 binding sites binding sites |
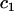 |
Concentration of fluorescent molecules bound to 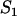 binding sites binding sites |
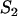 |
Localized binding sites |
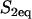 |
Equilibrium concentration of unoccupied localized binding sites |
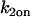 |
On rate at the localized binding sites |
 |
Off rate at the localized binding sites |
| C2 | Fluorescent molecules bound to 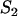 binding sites binding sites |
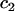 |
Concentration of fluorescent molecules bound to 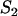 binding sites binding sites |
| F | Freely diffusing fluorescent molecules |
| f | Concentration of freely diffusing fluorescent molecules |
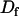 |
Free diffusion constant |
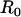 |
Radius of both the localized binding region and of the bleach spot |
 |
Radius of the cell nucleus |
 |
Half-height of the localized binding zone |
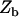 |
Half-height of the cell nucleus |
The names and definitions of all variables and directly related parameters used in the models.
TABLE 4.
Model parameters
| Name | Meaning | Definition | |
|---|---|---|---|
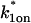 |
Association rate constant at the nonlocalized binding sites |  |
|
 |
Association rate constant at the localized binding sites |  |
|
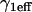 |
Effective diffusion retardation factor accounting for local equilibrium at 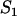 binding sites binding sites |
|
|
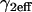 |
Effective diffusion retardation factor accounting for local equilibrium at 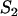 binding sites binding sites |
|
|
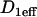 |
Effective diffusion constant incorporating local equilibrium behavior at 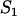 binding sites binding sites |
|
|
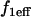 |
Apparent free species incorporating free fluorescence and local equilibrium behavior at 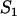 binding sites binding sites |
|
|
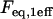 |
Equilibrium concentration of fluorescence in the apparent free species |
|
|
 |
Equilibrium concentration of fluorescence bound to the localized  sites sites |
|
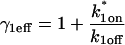
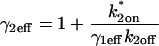
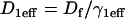
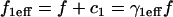

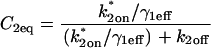
The names of all derived parameters used in the models, and their definition in terms of either other parameters or the model variables listed in Table 3.
APPENDIX A: REDUCTION OF THE MODEL EQUATIONS WHEN THE NUCLEOPLASMIC SITES EXHIBIT EFFECTIVE DIFFUSION
Equation 2 in the text is:
 |
(4) |
These equations can be simplified when the nucleoplasmic sites (S1) exhibit effective diffusion. We have shown that this occurs for bleach spot sizes of ∼1 μm radius (8). Since the MMTV array is also of ∼1 μm radius, effective diffusion can be used to model the nucleoplasmic sites both within and outside this zone. Adding the last two equations from Eq. 4 for the nucleoplasmic sites yields:
 |
(5) |
The effective diffusion simplification arises when the expected time for binding at the nucleoplasmic sites (S1) is rapid compared to the expected time for diffusion across the bleach spot. Under these conditions, the nucleoplasmic binding interaction is locally at equilibrium, yielding  This implies by the last equation in Eq. 4 that
This implies by the last equation in Eq. 4 that  , where:
, where:
 |
(6) |
Thus substituting into Eq. 5, we obtain:
 |
(7) |
The term  is the sum of the free and bound fluorescence in the nucleoplasm, and therefore equals the total fluorescence measured in a FRAP recovery due to the nucleoplasm. By defining an apparent free species
is the sum of the free and bound fluorescence in the nucleoplasm, and therefore equals the total fluorescence measured in a FRAP recovery due to the nucleoplasm. By defining an apparent free species  and an effective diffusion constant
and an effective diffusion constant  :
:
 |
(8) |
we can rewrite Eq. 7 as:
 |
(9) |
This is the diffusion equation, and therefore demonstrates how the nucleoplasmic fluorescence (free plus bound) behaves as a diffusing species with diffusion constant D1eff.
A similar strategy can be applied to the first three equations of Eq. 4 yielding a reduction in the number of these equations from three to two, dependent now on f1eff and D1eff.
 |
(10) |
Thus the effective diffusion behavior of the nucleoplasm reduces the five equations of Eq. 4 to the three equations of Eqs. 9 and 10. Two equations disappear because the variables f and 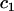 are absorbed into a single variable, the apparent free species
are absorbed into a single variable, the apparent free species  This yields Eq. 3 in the text.
This yields Eq. 3 in the text.
The initial conditions within the bleach zone are that  since the FRAP data are normalized to zero within this zone. The initial conditions outside the bleach zone are that
since the FRAP data are normalized to zero within this zone. The initial conditions outside the bleach zone are that  the equilibrium concentration for
the equilibrium concentration for  since this outer zone is not initially affected by the bleach. This equilibrium concentration for
since this outer zone is not initially affected by the bleach. This equilibrium concentration for  is obtained by setting
is obtained by setting  in Eq. 10, yielding:
in Eq. 10, yielding:
 |
(11) |
Since the FRAP data are normalized to one,  Combining this with Eq. 11 yields:
Combining this with Eq. 11 yields:
 |
(12) |
The boundary conditions are no flux at  and
and 
Summarizing, the FRAP problem at a central cluster of binding sites is defined by:
- Governing equations:

(13) - Initial conditions:

(14) - Boundary conditions:

(15)
APPENDIX B: AXIAL AND RADIAL BINDING MODEL AND ITS SIMPLIFIED DOMAINS
Full model
Equations 13–15 were solved numerically with FEMLAB version 3.1, using the mass balance application within the chemical engineering module. An axi-symmetric, two-dimensional cylindrical geometry was created, in which two fluorescent species existed: an “effective free” species, f1eff, and a bound species, c2. The parameters were those defined in Fig. 1 E, namely  and
and  The time-dependent solver was then used to compute the total fluorescence, f1eff + c2, as a function of time. This quantity was integrated over the blue cylindrical zone of localized binding illustrated in Fig. 2 A, and then normalized by dividing it by the final total fluorescence within the blue cylindrical zone of localized binding.
The time-dependent solver was then used to compute the total fluorescence, f1eff + c2, as a function of time. This quantity was integrated over the blue cylindrical zone of localized binding illustrated in Fig. 2 A, and then normalized by dividing it by the final total fluorescence within the blue cylindrical zone of localized binding.
Negligible specific binding solution
To investigate the simplified case when binding at the localized sites was nonexistent, we set the association rate to be very small and the off rate to be very high, namely  and
and  These rate constants ensure that fluorescent molecules will never be found at these specific binding sites, and so there will be no binding at these sites.
These rate constants ensure that fluorescent molecules will never be found at these specific binding sites, and so there will be no binding at these sites.
Reaction-dominant solution
To create a reaction-dominant form of the Axial and Radial Binding Model, the effective diffusion constant D1eff was set very large (108 μm2/s), such that the effectively diffusing species would equilibrate “instantaneously” in the 1-μm diameter bleach spot relative to the specific binding rates tested (maximum association rate constant at the localized sites  was 105 s−1).
was 105 s−1).
Local-equilibrium solution
The local-equilibrium version of the Axial and Radial Binding Model was created by first deriving the appropriate governing differential equations based on the local-equilibrium assumption for specific binding. Starting with Eq. 13, a strategy was employed similar to that used for the effective diffusion simplification of nucleoplasmic binding in Eqs. 5–10. Local equilibrium at the specific binding sites arises when  and so by the middle equation in Eq. 13:
and so by the middle equation in Eq. 13:
 |
(16) |
where
 |
(17) |
Note then that:
 |
(18) |
By adding the first two equations of Eq. 13 and employing Eq. 18, we obtain:
 |
(19) |
Outside of the localized sites, the last equation of Eq. 13 still holds, namely:
 |
(20) |
Thus the local equilibrium case reduces to Eqs. 19 and 20. To implement these equations in FEMLAB, the factor 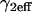 in Eq. 19 is accounted for by using a timescale coefficient of
in Eq. 19 is accounted for by using a timescale coefficient of  in the inner zone (
in the inner zone ( ).
).
The initial conditions for the local equilibrium equations are obtained by reduction of the initial conditions for the full equations (Eq. 14). This yields:
 |
(21) |
where the last equality arises from Eq. 18 and the fact that the FRAP is normalized to one within the localized binding zone. The boundary conditions for the local equilibrium case are identical to those in Eq. 15 for the full equations.
Validation of the axial and radial binding model
We tested whether the Axial and Radial Binding Model was correctly implemented by simplifying it to mimic the Radial Binding Model, which can be solved analytically (see section C below). We generated a numerical version of the Radial Binding Model by extending the axial column of localized sites throughout the height of the nucleus to create a numerical model with no axial dependence. The FRAP recoveries generated by this model were then compared to those obtained analytically using the Radial Binding Model.
Initially we detected some discrepancies between the FRAPs predicted by each model (Fig. 8 A), despite the adaptations to induce a match between them. To determine how these discrepancies arose, we compared the fluorescent intensity distributions at different time points predicted by each model. These distributions always differed, whether at late (Fig. 8 B) or early times (Fig. 8 C) after the bleach, with the difference consistently arising around the boundary defining the transition between the spatially localized binding sites and the surrounding diffusive zone (also defined as the edge of the bleach spot in each of the models). By increasing the number of finite elements (the mesh size) at this boundary in the numerical model, we could always improve and sometimes even eliminate these differences to produce FRAP curves that matched those from the analytical model (Fig. 8 D).
FIGURE 8.

Validating the numerical analysis used in the Axial and Radial Binding Model. (A) To confirm that the numerical models were solved correctly, a numerical solution for the Radial Binding Model was obtained and compared to the analytical solution (Table 1, first row). With our initial numerical solver protocol, we found a discrepancy between the two models that increased as a function of time during the recovery. This discrepancy only arose in cases where binding at the specific sites was very tight (i.e., for large values of k*2on/k2off; curves shown here are for k*2on = 100, k2off = 0.01). (B) To investigate the source of this discrepancy, we examined the spatial distribution of fluorescence as a function of time. At the endpoint of the FRAP, when the fluorescence has equilibrated, the analytical solution reveals a constant fluorescence within the zone of specific binding sites, and a much lower constant fluorescence outside of this zone. The largest discrepancy between the numerical and analytical solutions is found at the boundary of the spatially localized binding sites (at r = 1 μm). (C) A discrepancy between the analytical and numerical solutions also arises at early time points in the FRAP recovery. Once again the largest difference is found at the boundary between the localized and nonlocalized sites. (D) By refining the finite element mesh around the interface (r = 1 μm), we could obtain good agreement between both the radial distributions of fluorescence (data not shown) and the FRAP recoveries (comparison shown in panel D for k*2on = 100, k2off = 0.01; note that the dashed curve is essentially superimposed on the solid curve). Here, the grid was refined once at  again at
again at  and twice again at
and twice again at  over all z in each instance. This procedure eliminated the discrepancy between the models except when the binding at the localized sites was extremely tight (k*2on/k2off > ∼30,000), and further mesh refinements became prohibitive due to memory limitations.
over all z in each instance. This procedure eliminated the discrepancy between the models except when the binding at the localized sites was extremely tight (k*2on/k2off > ∼30,000), and further mesh refinements became prohibitive due to memory limitations.
However, available memory on the computer ultimately set a limit on the number of finite elements in the model, and so for a cluster of binding sites of very high affinity (and therefore sharp discontinuities in fluorescence), the numerical method failed to produce the sharp discontinuity at the boundary. Sharp discontinuities in fluorescence are not uncommon for localized binding sites within cells, or for the bleach-spot patterns that can be produced by laser scanning confocal microscopes, so an awareness of these limitations in a finite element implementation is important. However, as shown in the text, the binding at the MMTV array is not so tight, and so we do not expect errors in the numerical model to impact our analysis of this system.
Importantly, for all analyses involving the Axial and Radial Binding Model, we have used the finest mesh possible at the localized-site boundaries both radially and axially, and have avoided comparisons at large values of k*2on/k2off where we know the numerical solution becomes inaccurate.
APPENDIX C: RADIAL BINDING MODEL AND ITS SIMPLIFIED DOMAINS
Full model
Equations 13–15 can be solved analytically if we presume that the circular cluster of binding sites extends throughout the height of the nucleus ( ). This removes all z dependence from the equations, and permits a Laplace transform solution.
). This removes all z dependence from the equations, and permits a Laplace transform solution.
To obtain this solution, we first make the change of variables  and
and  thereby transforming Eqs. 13 and 14 to Eq. 22.
thereby transforming Eqs. 13 and 14 to Eq. 22.
 |
(22) |
Taking the Laplace transform of Eq. 22 yields Eq. 23:
 |
(23) |
Using the second equation, we can solve for  in terms of
in terms of  :
:
 |
(24) |
Substituting this into the first equation of Eq. 23, and then rearranging the first and third equations yields:
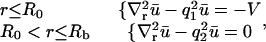 |
(25) |
where
 |
(26) |
Equations 25 and 26 are similar to those we previously derived to describe a FRAP recovery for globally and homogeneously distributed binding sites (see Eq. 15 in Sprague et al. (8)) bleached by a circular spot. Following the strategy outlined in Sprague et al. (8), we can write solutions comprised of Bessel functions for the two different zones of the problem. In the current scenario, the inner zone corresponds to both the bleach spot and the localized and nonlocalized binding sites, whereas the outer zone corresponds to the nonbleached region and the nonlocalized binding sites:
 |
(27) |
where I0 and K0 are modified Bessel functions of the second kind.
There are two key differences between this current solution and our previous solution (Eq. 16 in Sprague et al. (8)). First, in our earlier analysis the expression for  held both inside and outside the bleach spot, reflecting the fact that binding sites were uniformly distributed. Now, however, the localized binding sites are present only in the bleach spot, and so a different value,
held both inside and outside the bleach spot, reflecting the fact that binding sites were uniformly distributed. Now, however, the localized binding sites are present only in the bleach spot, and so a different value,  is obtained outside the bleach spot. Second, our current analysis requires a boundary condition, namely no-flux at
is obtained outside the bleach spot. Second, our current analysis requires a boundary condition, namely no-flux at  (Eq. 15). This we allow for by adding a second term (
(Eq. 15). This we allow for by adding a second term ( ) to the solution for
) to the solution for  in the zone surrounding the localized binding sites.
in the zone surrounding the localized binding sites.
The three constants  in Eq. 27 are determined by the continuity of
in Eq. 27 are determined by the continuity of  and its first derivative
and its first derivative  at the interface
at the interface  and by the requirement of an impermeable boundary at
and by the requirement of an impermeable boundary at  namely
namely  The solution for
The solution for  is:
is:
 |
(28) |
With this expression for  we can derive a solution for the FRAP recovery according to the procedure outlined in Eqs. 18–21 in Sprague et al. (8) to yield:
we can derive a solution for the FRAP recovery according to the procedure outlined in Eqs. 18–21 in Sprague et al. (8) to yield:
 |
(29) |
With the aid of Eq. 11 relating  and
and  it can be shown that:
it can be shown that:
 |
(30) |
Thus Eq. 29 simplifies to:
 |
(31) |
Negligible-binding solution
When specific binding at the localized sites is negligible, then the entire domain  contains only homogeneously distributed nucleoplasmic binding sites that exhibit effective diffusion behavior. For this simplified scenario, Eq. 22 reduces to:
contains only homogeneously distributed nucleoplasmic binding sites that exhibit effective diffusion behavior. For this simplified scenario, Eq. 22 reduces to:
 |
(32) |
Laplace transformation and rearrangement yields:
 |
(33) |
where:
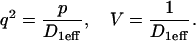 |
(34) |
This system can be solved analogously to Eq. 27 above leading to the reduced FRAP solution:
 |
(35) |
where A is still given by Eq. 28. However, with  and with the Bessel function identity
and with the Bessel function identity  the equation for A now simplifies to Eq. 36.
the equation for A now simplifies to Eq. 36.
 |
(36) |
Reaction-dominant solution
In the reaction-dominant scenario, diffusion is instantaneous on the timescale of the FRAP recovery, and so there is no longer spatial dependence in the problem. Rather,  and
and  depend only on time, and so an ordinary differential equation results describing the uptake of fluorescence within the localized binding region:
depend only on time, and so an ordinary differential equation results describing the uptake of fluorescence within the localized binding region:
 |
(37) |
This equation cannot be directly solved because  also depends on t. However,
also depends on t. However,  can be expressed in terms of
can be expressed in terms of  by imposing the impermeable boundary condition. This requires that the total fluorescence after the bleach is conserved, and given by:
by imposing the impermeable boundary condition. This requires that the total fluorescence after the bleach is conserved, and given by:
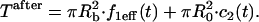 |
(38) |
The FRAP data are normalized such that the photobleach initially reduces the fluorescence to zero in the bleach zone. Thus  can be calculated by multiplying the initial concentration of fluorescence outside of the bleach zone (
can be calculated by multiplying the initial concentration of fluorescence outside of the bleach zone ( ) by the area of this outer zone:
) by the area of this outer zone:
 |
(39) |
with the last term obtained by using Eq. 12 for  Substitution of Eq. 39 into Eq. 38 yields:
Substitution of Eq. 39 into Eq. 38 yields:
 |
(40) |
where  and
and  Next with substitution of Eq. 40 into Eq. 37 we obtain the following first-order linear differential equation:
Next with substitution of Eq. 40 into Eq. 37 we obtain the following first-order linear differential equation:
 |
(41) |
With the initial condition  the solution to Eq. 41 is:
the solution to Eq. 41 is:
 |
(42) |
Since the FRAP measures the total fluorescence, it can be calculated by adding Eqs. 40 and 42, which after some simplification yields:
 |
(43) |
Local-equilibrium solution
We begin with the defining equations from the Axial and Radial Binding Model for local equilibrium (Eqs. 19 and 20), except now without z dependence:
 |
(44) |
We make the change of variables  with the latter equality arising from Eq. 21. Laplace transformation of Eq. 44 then yields:
with the latter equality arising from Eq. 21. Laplace transformation of Eq. 44 then yields:
 |
(45) |
This can be written as:
 |
(46) |
where
 |
(47) |
The solution can be obtained according to the procedure outlined above for Eq. 25:
 |
(48) |
where A is still given by Eq. 28, except that  and
and  are now specified by Eq. 47.
are now specified by Eq. 47.
Off-center radial binding model
The most general equations for a binding site cluster (Eq. 2 in the text) were converted to Cartesian coordinates in x,y but without z dependence. Allowing for a binding site cluster centered at an arbitrary location (a,b) yields two zones. Zone 1 contains the cluster of specific binding sites and Zone 2 contains the rest of the nucleus:
 |
(49) |
The differential equations within each zone are:
 |
(50) |
The boundary conditions are no flux across the circular boundary of Zone 2. The initial conditions are  in Zone 1, and
in Zone 1, and  and
and 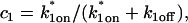 the equilibrium concentrations of f and
the equilibrium concentrations of f and  in Zone 2.
in Zone 2.
These equations were solved numerically using the partial differential equation (PDE) toolbox in Matlab based on the method described in Müller (16). Analogous to our observations for the numerical analysis of the Axial and Radial Binding Model, we found that for  the numerical solutions of the Off-Center Radial Binding Model failed to match the analytical solutions of the Radial Binding Model unless the finite element mesh at the boundary of the binding site cluster was successively refined. We found that 200 mesh points along this boundary provided sufficient accuracy to yield an excellent match between the numerical and analytical models. For a nucleus of 15 μm in diameter containing a 2-μm-diameter binding site cluster, this resulted in ∼900 finite elements within the cluster and ∼2600 elements in the rest of the nucleus.
the numerical solutions of the Off-Center Radial Binding Model failed to match the analytical solutions of the Radial Binding Model unless the finite element mesh at the boundary of the binding site cluster was successively refined. We found that 200 mesh points along this boundary provided sufficient accuracy to yield an excellent match between the numerical and analytical models. For a nucleus of 15 μm in diameter containing a 2-μm-diameter binding site cluster, this resulted in ∼900 finite elements within the cluster and ∼2600 elements in the rest of the nucleus.
APPENDIX D: GLOBAL BINDING MODEL
In the case of global binding, the specific binding sites are distributed throughout the nucleus ( ), and not just confined to the bleach zone (
), and not just confined to the bleach zone ( ). Thus the same partial differential equations apply both inside and outside of the bleach zone, with the only difference now arising from the different initial conditions inside and outside of the bleach zone. The correlate of Eq. 22 for this case is therefore:
). Thus the same partial differential equations apply both inside and outside of the bleach zone, with the only difference now arising from the different initial conditions inside and outside of the bleach zone. The correlate of Eq. 22 for this case is therefore:
 |
(51) |
Taking the Laplace transform and proceeding as described above once again yields equations of the form:
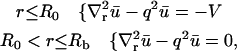 |
(52) |
where now  and
and  The FRAP recovery is:
The FRAP recovery is:
 |
(53) |
where A is given by:
 |
(54) |
Note that this newly derived solution for global binding in a bounded domain reduces to the solution that we derived previously for global binding in an unbounded domain (8). As  and so
and so  This is the solution found in Eq. 22 in Sprague et al. (8), as can be shown by application of Eq. 30 above to this earlier solution and by noting that this previous solution considered the case of pure diffusion, not effective diffusion, and so
This is the solution found in Eq. 22 in Sprague et al. (8), as can be shown by application of Eq. 30 above to this earlier solution and by noting that this previous solution considered the case of pure diffusion, not effective diffusion, and so  and
and 
References
- 1.Walker, D., H. Htun, and G. L. Hager. 1999. Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods. 19:386–393. [DOI] [PubMed] [Google Scholar]
- 2.McNally, J. G., W. G. Müller, D. Walker, R. Wolford, and G. L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 287:1262–1265. [DOI] [PubMed] [Google Scholar]
- 3.Stavreva, D. A., W. G. Müller, G. L. Hager, C. L. Smith, and J. G. McNally. 2004. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell. Biol. 24:2682–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufman, E. N., and R. K. Jain. 1990. Quantification of transport and binding parameters using fluorescence recovery after photobleaching. Potential for in vivo applications. Biophys. J. 58:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman, E. N., and R. K. Jain. 1991. Measurement of mass transport and reaction parameters in bulk solution using photobleaching. Reaction limited binding regime. Biophys. J. 60:596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrero, G., D. McDonald, E. Crawford, G. de Vries, and M. J. Hendzel. 2003. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods. 29:14–28. [DOI] [PubMed] [Google Scholar]
- 7.Carrero, G., E. Crawford, J. P. H. Th'ng, G. de Vries, and M. J. Hendzel. 2004. Quantification of protein-protein and protein-DNA interactions in vivo, using fluorescence recovery after photobleaching. Methods Enzymol. 375:415–442. [DOI] [PubMed] [Google Scholar]
- 8.Sprague, B. L., R. L. Pego, D. A. Stavreva, and J. G. McNally. 2004. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 86:3473–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrero, G., E. Crawford, M. J. Hendzel, and G. de Vries. 2004. Characterizing fluorescence recovery curves for nuclear proteins undergoing binding events. Bull. Math. Biol. 66:1515–1545. [DOI] [PubMed] [Google Scholar]
- 10.Dundr, M., U. Hoffmann-Rohrer, Q. Hu, I. Grummt, L. I. Rothblum, R. D. Phair, and T. Misteli. 2002. A kinetic framework for a mammalian RNA polymerase in vivo. Science. 298:1623–1626. [DOI] [PubMed] [Google Scholar]
- 11.Rabut, G., V. Doye, and J. Ellenberg. 2004. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol. 6:1114–1121. [DOI] [PubMed] [Google Scholar]
- 12.Handwerger, K. E., C. Murphy, and J. G. Gall. 2003. Steady-state dynamics of Cajal body components in the Xenopus germinal vesicle. J. Cell Biol. 160:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ficz, G., R. Heintzmann, and D. J. Arndt-Jovin. 2005. Polycomb group protein complexes exchange rapidly in living Drosophila. Development. 132:3963–3976. [DOI] [PubMed] [Google Scholar]
- 14.Beaudouin, J., F. Mora-Bermudez, T. Klee, N. Daigle, and J. Ellenberg. 2006. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 90:1878–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stavreva, D. A., and J. G. McNally. 2004. Fluorescence recovery after photobleaching (FRAP) methods for visualizing protein dynamics in living mammalian cell nuclei. Methods Enzymol. 375:443–455. [DOI] [PubMed] [Google Scholar]
- 16.Müller, F. Numerical simulations of fluorescence recovery after photobleaching experiments. Masters thesis. 2005. Karl Franzens University of Graz, Graz, Austria.
- 17.Sprague, B. L., and J. G. McNally. 2005. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 15:84–91. [DOI] [PubMed] [Google Scholar]
- 18.Dundr, M., J. G. McNally, J. Cohen, and T. Misteli. 2002. Quantitation of GFP-fusion proteins in single living cells. J. Struct. Biol. 140:92–99. [DOI] [PubMed] [Google Scholar]
- 19.Braga, J., J. M. Desterro, and M. Carmo-Fonseca. 2004. Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Mol. Biol. Cell. 15:4749–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiss, M., and T. Nilsson. 2004. In a mirror dimly: tracing the movements of molecules in living cells. Trends Cell Biol. 14:267–272. [DOI] [PubMed] [Google Scholar]
- 21.Waharte, F., C. M. Brown, S. Coscoy, E. Coudrier, and F. Amblard. 2005. A two-photon FRAP analysis of the cytoskeleton dynamics in the microvilli of intestinal cells. Biophys. J. 88:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phair, R. D., S. A. Gorski, and T. Misteli. 2004. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 375:393–414. [DOI] [PubMed] [Google Scholar]
- 23.Perlmann, T., P. Eriksson, and O. Wrange. 1990. Quantitative analysis of the glucocorticoid receptor-DNA interaction at the mouse mammary tumor virus glucocorticoid response element. J. Biol. Chem. 265:17222–17229. [PubMed] [Google Scholar]
- 24.Lieberman, B. A., and S. K. Nordeen. 1997. DNA intersegment transfer: how steroid receptors search for a target site. J. Biol. Chem. 272:1061–1068. [DOI] [PubMed] [Google Scholar]
- 25.Nagaich, A. K., D. A. Walker, R. Wolford, and G. L. Hager. 2004. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol. Cell. 14:163–174. [DOI] [PubMed] [Google Scholar]