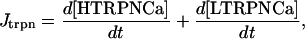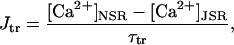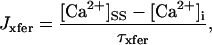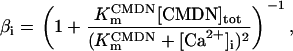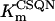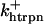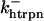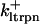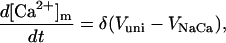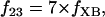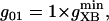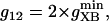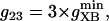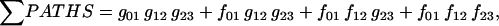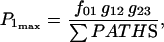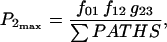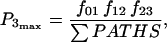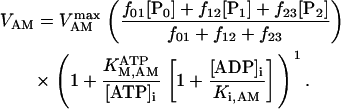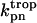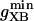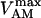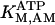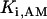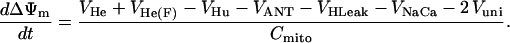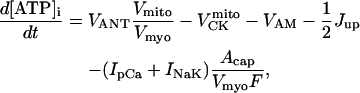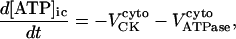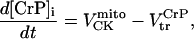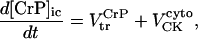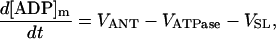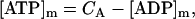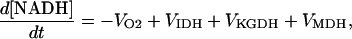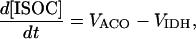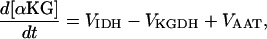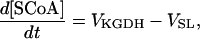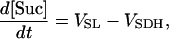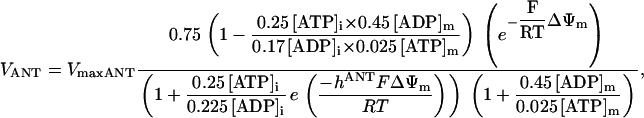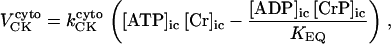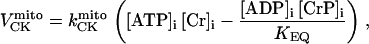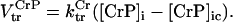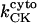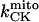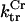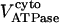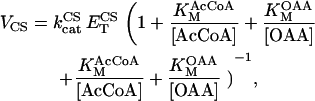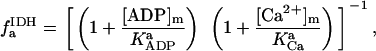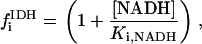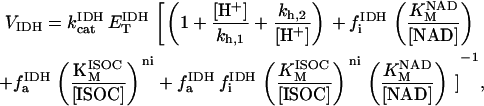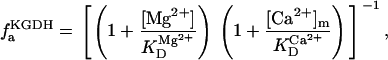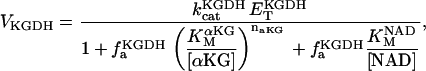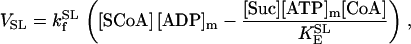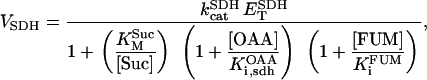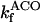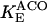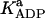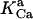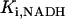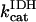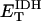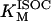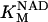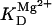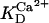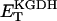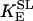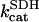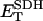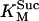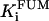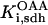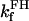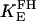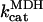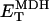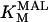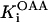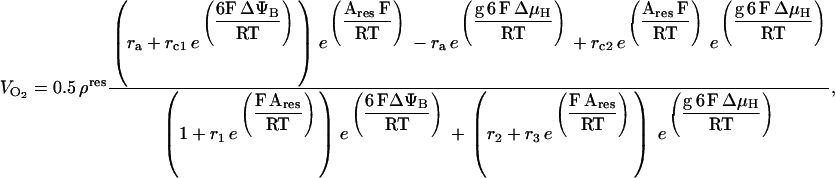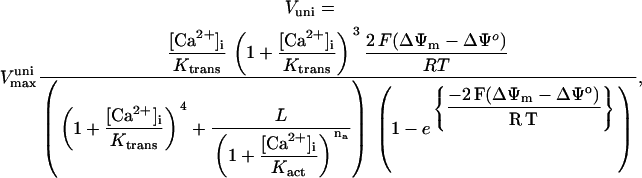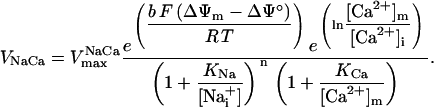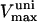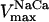An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
1The Johns Hopkins University, Institute for Computational Medicine, and Institute of Molecular Cardiobiology, Baltimore, Maryland
Address reprint requests to Raimond L. Winslow, The Johns Hopkins University, School of Medicine, Rm. 201B Clark Hall, 3400 N. Charles St., Baltimore MD 21218. Tel.: 410-516-5417; E-mail: rwinslow@jhu.edu.
Received 2005 Oct 14; Accepted 2006 Apr 24; Issue date 2006 Aug 15.
An intricate network of reactions is involved in matching energy supply with demand in the heart. This complexity arises because energy production both modulates and is modulated by the electrophysiological and contractile activity of the cardiac myocyte. Here, we present an integrated mathematical model of the cardiac cell that links excitation-contraction coupling with mitochondrial energy generation. The dynamics of the model are described by a system of 50 ordinary differential equations. The formulation explicitly incorporates cytoplasmic ATP-consuming processes associated with force generation and ion transport, as well as the creatine kinase reaction. Changes in the electrical and contractile activity of the myocyte are coupled to mitochondrial energetics through the ATP, Ca2+, and Na+ concentrations in the myoplasmic and mitochondrial matrix compartments. The pseudo steady-state relationship between force and oxygen consumption at various stimulus frequencies and external Ca2+ concentrations is reproduced in both model simulations and direct experiments in cardiac trabeculae under normoxic conditions, recapitulating the linearity between cardiac work and respiration in the heart. Importantly, the model can also reproduce the rapid time-dependent changes in mitochondrial NADH and Ca2+ in response to abrupt changes in workload. The steady-state and dynamic responses of the model were conferred by ADP-dependent stimulation of mitochondrial oxidative phosphorylation and Ca2+-dependent regulation of Krebs cycle dehydrogenases, illustrating how the model can be used as a tool for investigating mechanisms underlying metabolic control in the heart.
INTRODUCTION
Control mechanisms that modulate mitochondrial ATP production to match energy consumption are especially important in tissues with a high energetic demand like the heart (1). The proportionality between cardiac work and the rate of respiration was recognized almost 100 years ago (2), yet the mechanisms responsible for matching energy supply with demand remain controversial (3). The metabolic rate can change by 10- to 15-fold (4,5), but many investigators have observed that the levels of energy-rich adenine intermediates can be remarkably constant, indicating that oxidative phosphorylation is tightly controlled (for review, see (6,7)).
Several hypotheses have been put forward to explain the control of oxidative phosphorylation. The classical respiratory control hypothesis of Chance and co-workers (8), which has been subsequently confirmed in countless experiments on isolated mitochondria, implies that the rate of respiration is limited by the availability of ADP to the mitochondrial ATP synthase (as long as adequate substrates are available to maintain proton pumping). This demand-led control would be well suited to increase ATP production in response to cytoplasmic ATP consumption, but changes in bulk cytoplasmic ADP concentration have not been detected in intact hearts at high workloads by phosphorous NMR methods. Furthermore, a large increase in total ADP would have an unfavorable effect on cell processes that are sensitive to the free energy of ATP hydrolysis (i.e., the phosphorylation potential).
To explain how mitochondrial ADP delivery is accelerated without a large change in the cytoplasmic levels, models of intracellular dynamic compartmentation of adenine nucleotides have been proposed. This hypothesis is based on abundant evidence that different creatine kinase isozymes are localized near the sites of ATP hydrolysis (e.g., the myofibrils) and the mitochondrial inner membrane, contributing to intracellular-energetic units (9). This view implies that, whereas bulk ATP/ADP ratios remain constant, ADP signals generated by the myosin ATPase are rapidly transferred to the mitochondria via the creatine kinase shuttle. ATP can be similarly channeled back to the sites of utilization with alterations in the ATP/ADP ratio occurring only in microdomains close to the relevant sources and sinks of ATP (10). Evidence for compartmentalization of the adenine nucleotide pool has been addressed recently by combining biochemical models of phosphoryl transfer with estimations of CK fluxes by saturation transfer NMR methods (11).
Any mechanism that increases the rate of NADH oxidation would require concomitant activation of NADH production. Balaban and co-workers have proposed several mechanisms by which simultaneous stimulation of work and metabolism might be coordinated by cytoplasmic messengers. These include parallel activation of the respiratory chain and mitochondrial dehydrogenases by either Ca2+ (12) or Pi (1,13). As elaborated in these studies, very rapid responses in the NADH production and oxidation processes would virtually eliminate transients in metabolite levels during changes in work.
To gain a better understanding of the complex dynamics of the response of mitochondrial bioenergetics to alterations in the contractile and electrical activity of the heart cell, and to explore the feasibility of proposed control mechanisms, here we integrate models of the electrophysiological, Ca2+ handling, and force-generation subsystems of the cardiac myocyte (14,15) with a model of mitochondrial oxidative phosphorylation (16). The model is validated by performing parallel experiments measuring force and oxygen consumption in cardiac trabeculae and by comparison with earlier data describing transients in NADH and mitochondrial Ca2+ during abrupt perturbations of muscle workload (17).
MATHEMATICAL MODEL DEVELOPMENT
Excitation-contraction coupling/mitochondrial energetics (ECME) model
Fig. 1 shows the integrated model. The scheme emphasizes the links among mitochondrial metabolism, electrophysiology, and contraction.
General scheme of the EC coupling/mitochondrial energetics (ECME) model. The electrophysiological module includes the main ion transport processes involved in EC-coupling, accounting for the transport of Ca2+, Na+, and K+ across the sarcolemma, Ca2+ transport inside and across the SR membrane, and Ca2+ handling by mitochondria. Five different Ca2+ compartments are defined including the mitochondrial matrix, the dyadic subspace, the junctional and network SR, and the myoplasmic compartments. Extracellular Ca2+ is considered an adjustable parameter. The mitochondrial module describes the production (F1, F0 ATPase) and transport (ANT) of ATP, Ca2+ transport, and Ca2+ activation of the TCA cycle dehydrogenases. The CK reaction occurs near the mitochondria (but does not include potential limitations imposed by the outer membrane) and the CrP diffuses to the cytoplasmic compartment where there is another pool of CK that catalyzes the regeneration of ATPic to fuel constitutive cytoplasmic ATPases (labeled “cyto”). Also the total pool of adenine (CA) and creatine (CC) metabolites are indicated by dark shading. The main ATP-consuming processes related to EC coupling are the myofibrillar ATPase (VAM) and SERCA (Jup), and in the sarcolemma, the Na+, K+ ATPase (INaK), and the Ca2+ ATPase (IpCa).
Energy intermediates as links between mitochondrial energetics and excitation-contraction coupling
The excitation-contraction (EC) coupling (guinea-pig ventricular myocyte) and mitochondrial energetic models (14,15) are linked by the concentrations of ATP, ADP, creatine and creatine phosphate, and mitochondrial and cytoplasmic Ca2+ and Na+.
As discussed above, the currently accepted view that CK functions as both a cellular energy buffer and a high-energy phosphate shuttle (11,18,19) required us to introduce into the model a mechanism to simulate both the rapid transmission of an ADP signal to the mitochondria from the myofilaments and the buffering of the bulk cytoplasmic ATP/ADP ratio by the creatine kinase (CK) system. To avoid the computational challenge of incorporating a spatial dimension and partial differential equations into the model, we approximated metabolic compartmentation by adding two pools of CK, creatine, and adenine nucleotides in the cytoplasm (Fig. 1). One pool is associated with mitochondrial transport through the adenine nucleotide translocator (ANT) and is linked to the EC coupling processes. The other pool accounts for energy provision to constitutive cytoplasmic processes, e.g. transport processes and macromolecule synthesis. In our model, adenine nucleotide levels provide a strong link between mitochondrial energetics and the electrophysiological and contractile processes (see Discussion).
Our model also defines two different pools of ATP in the cytoplasm: ATPi (EC coupling-linked) and ATPic (associated with constitutive cytoplasmic ATPases) (see Eqs. 1–5). In each one of these pools the total concentration of adenine nucleotides is constant, and the total concentration of adenine nucleotide, CA, is the same (8 mM). Thus, the following equations apply:
(1)
(2)
This approach implies that ADP levels are determined by ATP in each cytoplasmic pool, linked through the creatine metabolites that are exchanged with the adenine metabolites by the CK catalyzed reaction:
The rate of CK was modeled using mass action kinetics assuming that it operates close to thermodynamic equilibrium:
(3)
Creatine phosphate (CrP) is synthesized from ATP by the mitochondrial CK () (Eq. A141) and transported to the cytoplasm for its utilization by constitutive energy-consuming processes through (Eq. A142). The CK at the cytoplasmic site, (Eq. A140), catalyzes the phosphorylation of ADP from CrP to generate ATP (CK reaction as written above in the reverse direction) that will be used by the constitutive cytosolic ATPases ruled by (fixed parameter, Table 12). Thus, the direction of the CK reaction depends on the enzyme localization, i.e., toward CrP synthesis in the mitochondria and toward the synthesis of ATP in the cytoplasm. The creatine metabolite levels in each pool, like the adenine nucleotide concentrations, are linked through conservation relationships as follows:
(4)
(5)
Equations 4 and 5 indicate that the Cr levels in both cytoplasmic pools are determined by the level of CrP. The parameter Cc (the total concentration of creatine metabolites) is the same in both pools, 25 mM (Table 12).
Ionic linkages between mitochondrial energetics and EC coupling
In addition to the ATP and ADP links between mitochondrial and cytoplasmic compartments, the mitochondrial concentration of Ca2+ ([Ca2+]m) is related to the myoplasmic Ca2+ concentration through the activity of the Ca2+ uniporter and the mitochondrial Na+/Ca2+ exchanger (NCE). The NCE senses the cytoplasmic Na+ level that is exchanged with . Thus, mitochondria act as an additional Ca2+ and Na+ sink in our model.
The rate expressions accounting for the dynamics of the ATP-consuming processes in the cytoplasm have been modified to account for their dependence on ATP and ADP concentrations. Among those energy-dependent processes are: the sarcoplasmic reticulum Ca2+ pump (SERCA); the Na+/K+ ATPase; and the sarcolemmal Ca2+ pump. The ATP-dependence of the kinetics of these pumps is detailed in the following sections.
Rice and collaborators (15) adopted the Shannon et al. (20) formulation of SERCA kinetics. This equation (Eq. 6) incorporates the forward and reverse modes of Ca2+ transport across the sarcoplasmic reticulum (SR) membrane. We modified the original rate expression to account for ATP-dependence of SERCA, in accordance with experimental data obtained in vitro by Sakamoto and Tonomura (21) using a fragmented SR preparation. These studies demonstrated a Michaelis-Menten dependence of enzyme activity with respect to ATP and mixed-type inhibition of the enzyme by ADP under conditions of high Mg2+ and low Ca2+. The experimental data (21) were also incorporated by Jeneson et al. (22) in their model of energy cycling in human skeletal muscle. Equation 6 represents the rate of the SERCA, explicitly describing its ATP and ADP dependence. The factor that incorporates [ATP]i and [ADP]i in the denominator has been adopted from Jeneson et al. (22) with the parameter values determined by Sakamoto and Tonomura (21),
(6)
being
and
where represents the affinity of SERCA for ATP, and Ki,up and K′i,up are the ADP inhibition constants (Table 7). As shown experimentally by Sakamoto and Tonomura (21), Eq. 6 indicates that ADP inhibits the pump both by competitive (accomplished in the term containing Ki,up) and noncompetitive (accounted for by K′i,up) mechanisms. We simulated the behavior of SERCA as a function of Ca2+ concentrations in the myoplasm and inside the SR network (NSR). The introduction of ATP dependence did not significantly change pump rate as a function of Ca2+ in the physiological range of ATPi levels (not shown).
Actomyosin ATPase (AM ATPase)
The data of Regnier et al. (23), concerning mechanisms involved in the regulation of force generation by cardiac muscle, indicate little sensitivity of developed muscle tension to large variations in ATP (from 0.5 to 5 mM). Additionally, Karatzaferi et al. (24) demonstrated that ADP analogs bind to myosin heads, thereby potentiating tension. Accordingly, we assume that developed force is only indirectly sensitive to the ratio of ATP to ADP (see Eqs A96–A121) through the effect of the latter on the SERCA pump, resulting in reduced sarcoplasmic reticulum Ca2+ load leading to reduced developed force. On the other hand, the rate of ATP hydrolysis associated with force generation through actomyosin ATPase (Eq. 7) depends explicitly on both ATP and ADP, as previously demonstrated (25). We have developed a Michaelis-Menten kinetic expression to account for ATP activation of the actomyosin ATPase associated with force generation, together with a competitive ADP inhibition:
(7)
Equation 7 contains a factor accounting for dependence of the AM ATPase on normalized rate of cross-bridge formation through their rate of formation, as denoted by f01 [P0], f12 [P1, and f23 [P2] and the rate constants f01, f12, and f23 (Eqs. A102–A104).
Na+/K+ ATPase
The original expression for the Na+/K+ ATPase corresponds to that introduced by Luo and Rudy in their phase II ventricular cell model (26). This model was able to account for experimental observations performed by Nakao and Gadsby (27) regarding the dependence of the current on membrane potential at different external Na+ concentrations. The Na+/K+ current expression has been modified based on kinetic studies of Sakamoto and Tonomura (21) to account for the dependence of Na+/K+ ATPase activity on ATP, as well as the competitive inhibition exerted by ADP,
(8)
being
with
Sarcolemmal Ca2+ ATPase
Equation 9 gives the modified rate expression incorporating the ATP-dependence of pump activity. Sarcolemmal Ca2+ ATPase rate (Eq. 9) exhibits two different K0.5 values for ATP (). ADP exerts competitive inhibition of the Ca2+ pump by affecting the high-affinity component of the ATP interaction through the inhibition constant, . We adopted the K0.5 values described in Echarte et al. (28), who found two K0.5 values for ATP in their study of the enzyme reaction mechanism involving the formation of an intermediate phosphorylated enzyme state. The affinity constants for ATP suggested the presence of two nucleotide regulatory sites in the sarcolemmal Ca2+ ATPase (29):
(9)
ATP/ADP dependence of the kinetics of the pumps
Fig. 2 displays the behavior of the different pumps introduced in the previous sections as a function of [ATP]i. To compare the modeling results to experimental data obtained in vitro, the simulations employed in vitro Vmax values. These differed from the Vmax values used in the integrated cell model because adjustments were required to simulate the reported currents or fluxes measured in intact cardiac myocytes.
ATP/ADP dependence of cytoplasmic and sarcolemmal pumps. (A and B) The dependence of the SERCA flux (Jup) on cytoplasmic ATP. Panel A shows experimental data redrawn from Sakamoto and Tonomura (21) and panel B shows model simulations using the SERCA rate equation (Eq. 6). The curves labeled 0, 0.3, 0.6, 1.2, and 2.0 mM illustrate the inhibitory effect of increasing ADP concentration. The parameters were set to: Vmax f = 60 nmol mg−1 protein min−1; Vmax r = 64 nmol mg−1 protein min−1; [Ca2+]i = 0.5 mM; and [Ca2+]NSR = 0.3 mM. All the other parameters are as indicated in Table 7. (C and D) The dependence of the myofibrillar adenosine triphosphatase (VAM) on cytoplasmic ATP. Panel C shows experimental data redrawn from Karatzaferi et al. (24) and panel D shows model simulations obtained with Eq. 7 (also Eq. A122). The competitive inhibition by ADP is shown for ADP concentrations of 0.5 and 2.0 mM ADP. The plots were produced with a maximal rate of the myofibrillar ATPase, , of 2.88 s−1, and P1 = 0.2, P2 = 0.7, and P3 = 0.1. Otherwise, the parameters used in the plot are as indicated in Table 11. (E and F) The dependence of the Na+, K+ adenosine triphosphatase activity on ATP. Experimental data (E, from (21)) and model simulations (F) using the rate expression of Eq. 8. Competitive inhibition by ADP at 0, 0.5, 1.0, and 2.0 mM concentrations is shown. The parameters used were Vmax = 3.93 μmol mg−1 min−1, the membrane potential is −80 mV, and the intracellular Na+ is 7.0 mM. The other parameters are as indicated in Table 3. (G and H) The dependence of the sarcolemmal Ca2+ adenosine triphosphatase activity on ATP. Experimental data (G, from (29)) and model simulations (H) obtained with Eq. 9, which corresponds to a typical Michaelis-Menten profile. The following parameter values were used in the simulation: Vmax = 2.55 μmol mg−1 min−1; [Ca2+]i = 2 μM; and ADP = 50 μM. The rest of the parameter values are as indicated in Table 6. Panels A and E are reproduced from Sakamoto and Tonomura (21) by permission of the Japanese Biochemical Society and Oxford University Press. Panel C is from Karatzaferi et al. (24) used with permission from the American Physiological Society. Panel G is reproduced from Pasa et al. (29) with permission from Elsevier.
The experimental results describing the behavior of the sarcoplasmic reticulum Ca2+ pump (Fig. 2 A) (from (21)) were simulated using the rate expression (Eq. 6) depicted in Fig. 2 B for the range of ADP concentrations employed by Sakamoto and Tonomura (21). Only the forward and reverse Vmax values had to be adjusted in the model (see Table 7) to reproduce the SERCA pump fluxes obtained experimentally (20).
The dependence of the AM ATPase rate on ATP, measured by Karatzaferi et al. (24) (Fig. 2 C), could be reproduced using the rate expression of Eq. 7 (Fig. 2 D). As a result of the high affinity of the ATPase for the nucleotide, the rate of ATP consumption by myofibrils will be saturated with ATP in the physiological concentration range (not shown).
Fig. 2 E shows the current carried by the Na+/K+ ATPase, measured experimentally by Sakamoto and Tonomura (21), and simulated using the rate expression shown in Eq. 8 (Fig. 2 F). The expression displays a typical hyperbolic dependence on ATPi concentration that saturates at ATP > 4.0 mM.
The rate of the sarcolemmal Ca2+ pump studied as a function of ATP concentration also exhibits a hyperbolic shape with saturation at high ATP levels (Fig. 2, G and H). As in the case of the AM ATPase, and in the normal physiological range of adenine nucleotide levels, the flux through the sarcolemmal Ca2+ pump will be nearly ATP-independent.
The absolute rates of ATP consumption in the integrated model were calculated based on the absolute fluxes or currents sustained by the ATP-consuming processes multiplied by the stoichiometry of such processes, e.g., 1 ATP is hydrolyzed by SERCA when transporting one Ca2+ from the cytosolic into the sarcoplasmic reticulum compartment. The resulting fluxes of ATP consumption were compared with available experimental evidence by Ebus and Stienen (30) and validated by showing that simulated consumption fluxes were in the correct range.
Computational methods
Rate expressions for each individual model component have been analyzed for their kinetic properties as a module. We used MatLab 6.5 (The MathWorks, Natick, MA) to study each rate expression separately from the assembled model and to check for the adequacy of the rate law and the parameter set being used. Once the behavior of each rate function proved satisfactory, the code of the ECME model, written in C++, was assembled as a system of 50 ordinary differential equations (ODEs) in the development environment of Microsoft Visual Studio (Microsoft, Redmond, WA). Equations were integrated numerically using CVODE, a stiff ODEs solver in C, developed by Cohen and Hindmarsh (31), that uses variable-coefficient Adams and BDF methods (http://citeseer.ist.psu.edu/1230.html).
Before starting to run any specific protocol, we simulated the behavior of the system without stimulation to reach a steady state. Subsequently, the model was run for ∼800 s with pacing at 0.25 Hz. The state attained after 800 s is, in fact, a point in a steady trajectory of state variables. In such a trajectory it may be verified that most of the EC coupling related variables (e.g., V, [Ca2+]i, and the level of Ca2+ in the dyadic subspace: [Ca2+]SS) change periodically as a result of the periodic stimulus (illustrated in Supplementary Material, Fig. S1). Since the trajectories described by the variables are constant, this state will be described as a pseudo-steady state, which defined the initial condition for all runs in a series of experiments. The different protocols tested were programmed as a subroutine in the main file of the program code. Changes in the pacing frequency were programmed according to the experimental protocol of Brandes and Bers ((17,32); see their article, section called Time-Dependent Changes in Workload), but were entered manually for the pseudo-steady state studies of the force-frequency relations. During simulation, the results output is stored as text files that are analyzed later with the assistance of Microcal Origin 6.0 (Microcal Software, Northampton, MA).
Experimental methods
Muscle preparation and experimental setup
Male LBN-F1 rats weighing 175–250 g (Harlan Sprague Dawley; Indianapolis, IN) were anesthetized by intraperitoneal injection of 1.0 ml/kg pentobarbital sodium (360 mg/ml). After intracardiac heparinization, the hearts were rapidly excised and placed in Krebs-Henseleit buffer containing (in mM): 120 NaCl, 5 KCl, 2 MgSO4, 1.2 NaH2PO4, 20 NaHCO3, 0.25 Ca2+, and 10 glucose (pH 7.4). Additionally, 20 mM 2,3-butanedione monoxime (BDM) was added to the dissection buffer to prevent cutting injury. The effects of BDM after brief exposure have been found to be reversible (33). Hearts were cannulated via the ascending aorta and retrogradely perfused with the same buffer equilibrated with 95% O2–5% CO2. A flat, uniform, unbranched trabecula from the right ventricle was carefully dissected, leaving a block of tissue at one end from the right ventricle free wall and a small part of the valve at the other end to facilitate mounting. The dimensions of the muscles were measured using a calibration reticle in the ocular of the dissection microscope. The muscles have an oval cross-sectional profile with an average width of 0.29 ± 0.05 mm and a thickness of 0.097 ± 0.028 mm. This small muscle dimension assures an adequate supply of oxygen to the whole tissue volume. The cross-sectional area, calculated assuming an ellipsoid shape, averaged 0.022 mm2. All experimental protocols conformed to Johns Hopkins University institutional guidelines regarding the use and care of animals.
Trabeculae were mounted between a basket-shaped extension of the force transducer and a hooklike extension of a micromanipulator screw with the aid of a dissection microscope. This attachment and mounting procedure was chosen because it proved to minimize damage to the muscle preparation (34). The muscles were stretched until a further increase in length would cause the resting and developed tensions to increase in parallel, corresponding to a sarcomere length (2.2 μm) that is slightly below the maximal developed tension (34).
To monitor the contractile parameters during the experiment, each twitch was analyzed on-line with a subroutine developed for LabVIEW (Ver. 6.1, National Instruments, Austin, TX) that acquires and processes the raw data from the force transducer. The forces developed by the rat trabeculae were computed in mN.mm−2. These were displayed as relative changes in force to compare them with the simulations, since the model was based on guinea-pig EC coupling properties, which produces different absolute tensions.
Respiration measurements
We used a fiber-optic oxygen sensor (Ocean Optics, Dunedin, FL) to measure muscle respiration. This probe works as a spectrometer-coupled chemical sensor for full spectral analysis of dissolved oxygen pressure. The principle of detection is the fluorescence emission at 600 nm of a ruthenium compound that is quenched by O2. At the tip of the fiber optic probe (Ø = 300 μm), the O2-sensitive compound is embedded in a resin. The fluorescence emitted is a function of the O2 dissolved in the surroundings of the probe.
The respiratory flux was measured with the fiber-optic device monitoring the O2 levels close to the surface of contracting trabeculae (within 200 μm) upon interruption of the perfusion flow during a 45-s to 1-min interval. Under these conditions, we recorded a linear change in the fluorescent signal whose slope is proportional to the respiratory flux. As expected, the respiratory rate increases with the frequency of stimulation and the Ca2+ levels in the buffer bathing the muscle. The measurements were referred to the maximal levels of respiration attained under the experimental conditions described in the presence of the uncoupler FCCP (1 μM). The respiratory flux was completely inhibited in the presence of NaCN (5 mM). All our experimental recordings were performed at 22°C.
RESULTS
Force-frequency relations
Fundamental cardiac muscle properties were explored by examining the energetic and inotropic responses to an increase in stimulation frequency. This force-frequency relation is muscle type- (atrial or ventricular) and species-specific (35,36). The force-frequency relation has been studied under pseudo steady-state and transient conditions (see next section).
Pseudo steady-state studies
The force versus frequency studies under pseudo-steady state in silico conditions were performed by varying the frequency of pacing and analyzing the model behavior after ∼500–1000 s (8–15 min) of simulation time at each frequency.
Systolic (maximal) and diastolic (minimal) forces were computed from the behavior during one beat immediately after 800 s of simulation under each parametric condition. Developed force was computed as the difference between the systolic and the diastolic force. Fig. 3 A shows dependence of developed force as a function of frequency at three different levels of extracellular Ca2+, [Ca2+]o. In the range of [Ca2+]o studied, force increased linearly with frequency, attaining a value of 32 mN mm−2 at 2.0 Hz in 2.0 mM Ca2+. The slope of the force-frequency relationship increased with increased [Ca2+]o. The increment in developed tension correlates with the increase in the amplitude of the Ca2+ transient in the cytosol (see Supplementary Material, Fig. S2). This “positive staircase” results from enhanced time-averaged Ca2+ entry and SR Ca2+ loading and is, in part, indirectly influenced by intracellular Na+ concentration, which modulates SR Ca2+ load by shifting the balance of Ca2+ fluxes through the actions of the sarcolemmal Na+/Ca2+ exchanger.
Force-frequency relations. Developed force simulated by the ECME model (A) and measured in experiments on rat ventricular trabeculae (B) as a function of the pacing frequency at various extracellular Ca2+ levels. The simulations were run for 1500 s from the initial condition indicated in Table 16. The parameters used are as described in Tables 1–15 except for the extracellular Ca2+ concentration, which is indicated in the legend. The developed force was calculated after the model reached a pseudo-steady state from the difference between the maximum and the minimum forces during a contraction. The experiment data point shown for 1.5 mM Ca2+ at 0.25 Hz corresponded to a developed tension of 45 mN mm−2.
Simulated force-frequency relations (Fig. 3 A) qualitatively resembled those obtained experimentally in the cardiac rat trabeculae (Fig. 3 B). However, quantitative differences were found between experimental data and model predictions, especially for values obtained at low Ca2+ concentration. The slope of the force versus frequency relationship at 1.0 mM extracellular Ca2+ was much larger in the experiments than in the model (Fig. 3). In the simulations, force values changed by 30-fold when stimulation frequency increased from 0.25 to 2.0 Hz, whereas in rat trabeculae, force increased only approximately threefold in the presence of 1.0 mM extracellular Ca2+ between 0.25 and 2 Hz.
The bioenergetic performance of the system, monitored through the rate of oxygen consumption (VO2), was also studied using the same experimental setup for isometric force measurements in rat ventricular trabeculae (see Experimental Methods). In this way, we could not only directly compare experimental contractile activity with in silico data but also measure VO2 as a function of the pacing frequency. The value VO2 increased with the frequency of stimulation in the model under pseudo steady-state conditions, depending on [Ca2+]o (Fig. 4 A). At high [Ca2+]o, the frequency-VO2 relationship displays a close to linear dependence, which was also reflected in simulations of the NADH levels (not shown). In the model, an increase in frequency will increase the levels of cytosolic Ca2+ and ADP, which will be translated into higher mitochondrial [Ca2+]m and [ADP]m (not shown) to activate two main mechanisms for activating oxidative phosphorylation. [Ca2+]m will stimulate the TCA cycle dehydrogenases, whereas [ADP]m will activate respiration through respiratory control. The close-to-linear increase in respiration with frequency is likely due to a larger effect of the TCA dehydrogenases stimulation by Ca2+ than the corresponding ADP stimulation of respiration as also suggested by the NADH profile (not shown).
Respiration associated with force-frequency relations. O2 consumption simulated by the ECME model (A) and measured in rat ventricular trabeculae (B) as a function of the pacing frequency at various extracellular Ca2+ levels. The plots in panel A were obtained from the same simulations reported in Fig. 3. The experimentally determined rate of respiration, VO2, is expressed relative to the maximum observed when the muscle was exposed to the uncoupler FCCP. Treatment with 5 mM NaCN completely inhibited the respiratory flux. The simulated VO2 was averaged after the model reached a pseudo-steady state. Under steady-state resting conditions, VO2 averaged 0.08 mM s−1.
Model simulations were qualitatively in line with the experimental data obtained in rat ventricular trabeculae (Fig. 4 B). Representing the combined data from Figs. 3 and 4 as the respiratory rate versus developed tension shows the proportionality between respiration and mechanical work that has been reported previously in working hearts (37) (see Supplementary Material, Fig. S3). The quantitative differences observed in Fig. 4 are, at least in part, due to the fact that the model has been designed and the parameters adjusted for the dynamics of guinea-pig ventricular myocytes, while the experimental system corresponds to rat ventricular trabeculae (see Discussion).
Time-dependent behavior of mitochondrial energetics after changes in workload
We explored the time-dependent behavior of the model with a protocol in which the stimulus frequency was raised from 0.25 to 2.0 Hz and then returned to 0.25 Hz. This protocol has been previously used experimentally in a preparation of ventricular rat trabeculae while mitochondrial energetic and EC coupling variables were monitored (17,32).
The increase in pacing frequency produces a drop in the level of NADH (undershoot, phase I in Fig. 5 A) followed by a recovery phase (phase II in Fig. 5 A) until a new steady NADH level is reached. A subsequent decrease in the pacing frequency causes an overshoot in the NADH concentration (phase III in Fig. 5 A), which is later followed by a relaxation toward the baseline NADH level. The ECME simulation of the same pacing protocol used by Brandes and Bers (17,32) qualitatively reproduces the profile of NADH and recorded experimentally (Fig. 5, A and B). The magnitude of the NADH decrease during the undershoot is somewhat lower than that observed experimentally (3% against 10%, respectively).
Mitochondrial energetics in response to changes in workload. Model simulations are shown for a change in the myocyte pacing frequency from 0.25 Hz (0–100 s) to 2.0 Hz (100–300 s) and back to 0.25 Hz (300–500 s). The parameters and initial conditions used in the simulations are as indicated in Tables 1–16. The NADH time course (A) and mitochondrial matrix Ca2+ concentrations (B) are plotted. Regions I, II, and III indicate the different phases in the temporal evolution of the NADH signal. Panel C shows the profile of [ATP]i and [CrP]i obtained in the same simulation.
The ATP and CrP levels of the pool related to EC coupling during the transient is shown in Fig. 5 C. The decrease in ATP after the increase in pacing frequency is barely noticeable, because of the efficient buffering by CrP, whose levels decrease ∼3 mM. This result may be compared with the changes associated with an increase in heart rate in ferret hearts, whose phosphorylated energy intermediates were monitored over time with 31P NMR (38). Our simulations reproduce the profile in CrP when glycolysis is not operating. Moreover, the model behavior also simulates the lack of change in the total ATP observed during stimulation at 2 Hz (38).
Ca2+- and ADP-mediated regulatory mechanisms involved in mitochondrial energy supply
Once validated, an advantage of the integrated model is that it may be used to test competing hypotheses reported in the literature concerning the various mechanisms involved in the regulation of energy supply and demand in the heart. A similar approach was followed in the case of the isolated mitochondrial model (16).
Two basic control mechanisms are incorporated into the model, so it was essential to determine the independent contributions of each in the transient responses to the perturbation of workload. First, respiratory control by ATP consumption leading to increased ADP concentrations in both the cytoplasm and the mitochondrial matrix was eliminated by clamping cytosolic ATP and ADP to a constant levels. Second, the Ca2+-dependence of the activation of the Krebs cycle dehydrogenases was adjusted to abolish the upstream effect of Ca2+ to increase NADH production. This was accomplished by eliminating the effects of Ca2+ on the IDH and KGDH rates (Eqs. A147 and A149). Both tests were performed with the same parameter set and frequency range used in the control simulation shown in Fig. 5 A.
As shown in Fig. 6, when ATPi is clamped, the dynamics of the simulated mitochondrial NADH signal are completely altered. Upon an increase in pacing frequency, there is no undershoot of the NADH signal and the NADH increases monotonically with a rate similar to that observed during the recovery phase of the transient in the earlier simulations (phase II, Fig. 5 A). The NADH overshoot (phase III) is also eliminated when ATPi is clamped (Fig. 6 A).
Contribution of Ca2+ activation of mitochondrial dehydrogenases and respiratory control by ADP to mitochondrial NADH dynamics. (A) The profile of NADH in simulations run with the pacing protocol indicated in Fig. 5, with myoplasmic ATP (both ATPi and ATPic) clamped. (B) The NADH profile when the factor accounting for the stimulation by Ca2+ of IDH and KGDH is eliminated from the corresponding equations (see Eqs. A145 and A148).
Removing Ca2+-dependent activation of the dehydrogenases (Fig. 6 B) reveals the underlying effect of the stimulation of respiration by ADP. When pacing frequency is increased, the concentration of NADH drops to lower levels than in the control (compare phases I, in Figs. 5 A and 6 B) and does not recover (compare phases II, in Figs. 5 A and 6 B). Upon return to the low pacing frequency, NADH increases monotonically in time (Fig. 6 B).
These results illustrate how the interplay between ADP-dependent stimulation of respiration and Ca2+-mediated activation of the Krebs cycle dehydrogenases accounts for the transient kinetics of the NADH response during changes in workload. Changes in force are rapidly manifested as changes in the NADH oxidation rate by respiration, leading to a net decrease, or increase, in the NADH signal when workload is increased, or decreased, respectively. The somewhat slower response time of mitochondrial Ca2+ dynamics and activation of NADH production by the Ca2+ stimulation of the TCA cycle dehydrogenases contributes to the recovery of the NADH signal during the undershoot and to the NADH overshoot when workload decreases. Briefly, the NADH initial undershoot is linked to the sudden increase in respiratory rate not counterbalanced by an increased NADH production by mitochondrial dehydrogenases. Later, during the recovery phase, NADH production is activated by Ca2+ stimulation of the TCA cycle dehydrogenases. These remain activated even after the sudden drop in respiration that occurs when the pacing rate is decreased. This transient imbalance explains the overshoot in NADH that slowly relaxes to a lower NADH level when the mitochondrial Ca2+ levels decrease and dehydrogenases activation ceases.
The simultaneous regulatory effects of Ca2+ and ADP were further explored in the simulations shown in Fig. 7. When the rate of ATP hydrolysis by the myofibrillar ATPase was halved there was a decrease in the amplitude of the NADH transients (undershoot and overshoot) in accord with the smaller changes in ATP and ADP (Fig. 7 A: compare the solid trace at lower maximal ATPase rate, with the shaded control profile).
Effect of changing the myofibrillar ATPase and mitochondrial Ca2+ uniporter on the ECME model energetic behavior after changes in workload. (A) The behavior of NADH in simulations with the pacing protocol and parametric conditions as described in the legend of Fig. 5, except that the maximal rate of ATP hydrolysis by the AM-ATPase was decreased to one-half (solid trace; mM ms−1) of the control value (shaded trace). (B) The profile of average ATPi for changes in workload under control conditions (shaded trace) or with decreased myofibrillar ATPase activity (solid trace). (C) The NADH profile in model simulations with the same pacing protocol and parameters as described in the legend of Fig. 5, but with the maximal rate of the mitochondrial Ca2+ uniporter decreased to 1/10th (solid trace; mM ms−1) of the control value (shaded trace). (D) The profile of [Ca2+]m accumulation for the low uniporter condition described in panel C (solid trace) as compared to the control (shaded trace).
In contrast, if the maximal rate of the mitochondrial Ca2+ uniporter is decreased to 1/10th of the control value, the NADH profile resembles that obtained in the absence of stimulation of Krebs cycle dehydrogenases by Ca2+ (compare Figs. 7 C and 6 B). The lack of a recovery phase (evident in the solid trace of Fig. 7 C) is attributable to the extremely low levels of [Ca2+]m attained during this protocol (Fig. 7 D).
Effects of stimulation frequency on transient bioenergetic and contractile behavior
To subject the ECME model to a more comprehensive and stringent test, we monitored the dynamics of mitochondrial energetics (VO2, ADPm, NADH) and contractile work (force), as well as the two main effectors, Ca2+m and ADPm, while the stimulatory frequency was varied over a wide range. The pacing frequency was increased from a baseline of 0.25 Hz to a higher frequency, e.g., 0.5, 1.0, 1.5, or 2.0 Hz, and returned to 0.25 Hz as indicated in the diagram at the top of Fig. 8. This in silico experiment emulates the experimental protocol reported by Brandes and Bers ((32) Fig. 8 B) and tests how changes in the average force and Ca2+ transport work contribute to the different phases of the mitochondrial NADH transient. ECME model simulations of the average force and the transient kinetics of the NADH profile qualitatively reproduce the experimental results (Fig. 8 A). The contractile activity drives changes in the adenine nucleotide pool and the respiratory flux (Fig. 8 C), in close correlation with the average force (Fig. 8 A). Fig. 8 D shows how the responses of ADPm and can account for the larger undershoot/overshoot and recovery phases of the NADH signal as the frequency increases.
Inotropic and bioenergetic transient behavior of the ECME model as a function of the pacing frequency. (A and B) The NADH and average developed force profiles during transients in workload at various frequencies in simulations (A) and experiments (B) (from (32)). The simulation protocol consists of abrupt changes in pacing frequency from 0.25 Hz to 0.5, 1.0, 1.5, and 2.0 Hz, as indicated in the bar diagram at the top of panel A. (C) The average oxygen consumption rate (in mM s−1) with the NADH profile shown for reference. (D) The time course of the two effectors of the bioenergetic machinery (average ADPm and ) that are involved in the energetic response to a change in workload. Parameters and initial conditions are the same as those used in Figs. 3–5 Panel B was reproduced from Brandes and Bers (32) by copyright permission of the Biophysical Society.
DISCUSSION
A mathematical model was developed in which the electrical, Ca2+ handling and contractile properties of the cardiac myocyte were integrated with mitochondrial bioenergetics. The ECME model is able to reproduce the pseudo-steady state and, importantly, the dynamic profile of changes in essential physiological variables such as ADP, NADH, and Ca2+ in cardiac myocytes subjected to changes in stimulation frequency and/or external Ca2+. Interactions between sarcolemmal and mitochondrial ion transport mechanisms, ATP consumption, and oxidative phosphorylation were sufficiently robust to simulate the matching of energy supply and demand over a range of workloads.
Excitation-contraction coupling/mitochondrial energetics model description
The excitation-contraction coupling/mitochondrial energetics (ECME) model is the first to integrate detailed biophysical descriptions of the activity of cardiac ion channels, sarcolemmal and mitochondrial ion transport pathways, excitation-contraction coupling, and myofilament activation with a model of mitochondrial oxidative phosphorylation. This allowed us to explore the interrelationship between the principal energy-consuming processes of the cell (i.e., contraction and ion transport) and the energy-generating system. Signaling between the cytosolic processes and mitochondrial bioenergetics was accomplished by 1), coupling the pools of high-energy phosphate intermediates in the cytoplasmic and mitochondrial compartments; and 2), linking Ca2+ levels in the two compartments via the mitochondrial Ca2+ uniporter and Na+/Ca2+ exchanger. The key to accurately simulating the dynamic and steady-state responses of bioenergetics to changes in workload lies in the balance between stimulation of the respiratory chain by enhanced ATP hydrolysis and stimulation of the Krebs cycle dehydrogenases. The differences in the relative response times of these two processes can account for the transient overshoot and undershoot behavior of mitochondrial NADH. Experimental evidence supporting a direct link in the response time of Krebs cycle dehydrogenases activation by mitochondrial Ca2+ and the recovery kinetics of NADH was provided by Brandes and Bers (17).
As in our previous model of the isolated mitochondrion (16), we have sought to reproduce the dynamic response to an abrupt change in workload as the most stringent test of the integrated model. In this regard, there have been few studies in which mitochondrial responses have been recorded in parallel with force measurements, so we have relied heavily on the data of Brandes and Bers (17), who recorded NADH transients, mitochondrial Ca2+ accumulation, and force in a unbranched rat cardiac trabecula preparation. Whether this preparation accurately reproduces the time course of change of the same parameters in an in vivo setting will require comparable recordings of all the variables of interest in the intact heart.
Nevertheless, the model results demonstrate that for changes in workload induced by increasing or decreasing stimulation frequency, the NADH dynamics of the muscle can be described by the combination of ADP-dependent and Ca2+-dependent regulatory mechanisms. Small changes in ATPi, and consequently in the levels of ADP in the cytoplasmic and mitochondrial compartments, mediate rapid changes in the mitochondrial NADH/NAD+ redox state. When ATPi was clamped, the rapid decrease in NADH upon an increase in work was eliminated, as was the overshoot when work was decreased. In this condition, NADH increased and decreased monotonically as workload was varied, primarily reflecting the effects of Ca2+ on the rate of NADH production by the Krebs cycle. In the model, the Ca2+-sensitive dehydrogenases IDH and KGDH exert the most control over mitochondrial NADH levels when ATP is clamped (16).
When the Ca2+ sensitivity of the Krebs cycle dehydrogenases was eliminated in the model, the effects of mitochondrial ADP on NADH oxidation were revealed clearly. An acute increase in average force is rapidly translated via the adenine nucleotide pool to an increase in respiration and, conversely, a decrease in work quickly results in a decrease in the rate of NADH oxidation by the respiratory chain. This raises the important question of how to model the high-energy phosphate transfer system between the sites of ATP consumption in the cytoplasm and ATP production by the mitochondria. This is a subject of ongoing debate (7) that must take into consideration the local spatial organization of the cardiomyocyte and the kinetics of the buffering reactions mediated by creatine kinase (and, ideally, other buffers like adenylate kinase as well). From first-principles, at high work load, mitochondrial ATP production has to match the approximately eightfold increase in the rate of ATP hydrolysis by the actomyosin ATPase. Hence, ADP influx across the mitochondrial inner membrane must increase in parallel. In cardiac muscle, the creatine kinase system can act as both an ATP buffer and a high-energy phosphate shuttle (39). This presents a problem in a model that does not formally incorporate spatial aspects with respect to incorporating the ability of the CK reaction to buffer the bulk cytosolic ATP concentration, while still being able to describe rapid ADP and Pi delivery to the mitochondrial adenine nucleotide and phosphate transporters, which are thought to be in a complex with the mitochondrial ATP synthase (40). As a compromise, we have included two nucleotide pools in the model, a bulk cytosolic pool that is largely buffered by the CK reaction and a pool with minimal buffering associated with the excitation-contraction coupling processes. The latter pool allows the mitochondria to rapidly and directly sense changes in energetic demand via mitochondrial ADP concentration. This representation should not be viewed as a literal description of compartmentation in the physical system, but as a construct that allowed us to simulate rapid phosphoryl (i.e., high energy phospho-group) transfer to and from the mitochondria without creating a computationally intractable spatial model of the cell (requiring partial differential equations).
Comparison of model and experimental results
The incorporation of both the ADP- and Ca2+-dependent regulatory mechanisms was required to adequately describe the experimental data of Brandes and Bers (32) with regard to the different effects of changes in work associated with contractile activity and the work associated with changes in Ca2+ handling. Contractile activity can be varied in the absence of a change in average cytosolic Ca2+ by changing resting sarcomere length, while Ca2+ homeostasis can be varied by changing stimulation frequency, external Ca2+, or inotropic intervention (e.g., β-adrenergic receptor stimulation). In response to an increased pacing rate, mitochondrial NADH transiently decreases (phase I) and then recovers (phase II; Fig. 5 A) (32). The phase II recovery of NADH depends on the average cytosolic (and consequently mitochondrial) Ca2+, and the net redox state attained after equilibration to the new workload is strongly affected by the sarcomere length. As the sarcomere length approaches the optimum of the Frank-Starling curve (i.e., Lmax), average force for any given Ca2+ is increased and the total energetic demand is increased, thus leading to a net oxidation of the NADH/NAD+ redox pool. Our simulations show that ADPm, which reflects changes in cytosolic ADP, accounts for the initial rapid decline of NADH at the onset of high frequency stimulation (Figs. 6 B and 7 A, and Fig. 8, A and D). The rapid decrease in ADPm as the workload is lowered also underlies the transient overshoot of NADH during the protocol. At this point, NADH production, which was accelerated by Ca2+ stimulation of the dehydrogenases, remains activated, while NADH oxidation decreases more rapidly. Again, the balance between the relaxation times of the demand-led and Krebs cycle processes determines the overall profile.
The effect of increasing the pacing rate in ferret hearts results in unchanged ATP levels and an initial decrease of CrP followed by a recovery to steady-state levels (if glycolysis is operating) (38). In the absence of glycolytic flux, the recovery phase of CrP is not observed and upon return to the low pacing frequency CrP level increases to reach the original steady-state levels (38). The model simulations reproduce the condition in the absence of glycolysis (Fig. 5 C) since according to the model, only mitochondrial ATP synthesis can replenish CrP. The change in Pi could not be directly computed because the operation of the adenine nucleotide translocator is carrying phosphoryl groups into the cytoplasm whose flux is counterbalanced by the phosphate carrier (not accounted for by the model). However, the main determinant of Pi dynamics in the cytosol is CrP, thus the Pi should mirror the CrP concentration profile when subjected to changes in pacing frequency (not shown).
Another agreement between our simulations and the experimental results concern the cytosolic levels of metabolites: [ATP], [NADH], [CrP], and [Ca2+], which are very close to the physiological values observed in ventricular myocytes of various species. As a matter of fact, NADH is highly reduced in the mitochondrial matrix (∼75%) (17). Since the total concentration of mitochondrial pyrimidine nucleotides is 10 mM in our model, an 80% reduction will correspond to 8 mM NADH (Fig. 5 A), which resembles the value reported (17). ATP was reported to be in the order of 7.5 mM; CrP is ∼21 mM at low pacing frequency, and can decrease up to 15 mM (38). Ca2+ resting level is ∼100–200 nM whereas the systolic concentration can peak from 400 nM to 1 μM (41).
A sensitivity analysis (data not shown) performed at 0.25, 1.0, and 2 Hz under the same parametric conditions as those used in the experiments shown in Figs. 3–5 revealed that the steps to which respiratory or ATP synthesis fluxes were most sensitive were localized downstream of NADH. This is in accordance with our earlier metabolic control analysis carried out for the isolated mitochondrial model (16). In the case of the integrated model, the processes that influence the rate of ATP hydrolysis are now explicitly described in a detailed physiological context, thus leading to important interdependencies between the steps that strongly influence E-C coupling and energetics.
It should be noted that Balaban and co-workers (1) have failed to observe NADH transients in intact hearts of the type reported by Brandes and Bers, leading to the notion that energy supply is rapidly increased to meet energetic demand through fast parallel activation of the respiratory chain and Krebs cycle dehydrogenases by a cytoplasmic messenger. Potential candidates for the active messenger include Ca2+ (12) or Pi (13), while ADP was thought to play a diminished role in the process. In isolated mitochondria, respiration was shown to respond rapidly (within hundreds of milliseconds) to changes in both Ca2+ and ADP (12). However, van Beek and co-workers (42) stated that the transient changes in energy fluxes and intermediates do not necessarily occur simultaneously and are altered upon inhibition of creatine kinase and/or glycolytic flux (43). The transient time was found to be much shorter for adenine nucleotides than for respiration, arguing against simultaneous parallel activation of energy production and consumption by a single, common effector (42). Further investigation into the transient dynamics of respiration, Ca2+, force, NADH, and ATP turnover in response to changes in cardiac workload will be required to resolve these issues. Our results indicate that Ca2+ and ADP, affecting mitochondrial energetics with different response times, are sufficient to reproduce available experimental data. Mixed regulation of mitochondrial energetics would potentially confer more flexibility to the heart in responding to different physiological challenges, for example, as could occur when changes in workload are elicited by variations in ADP with or without associated changes in the average cytosolic Ca2+ (32,44).
Directions for further model development
In taking on the challenge of incorporating most of the fundamental functional properties of the cardiac myocyte, the present integrated model is able to qualitatively reproduce the pseudo-steady state and transient responses of mitochondrial oxidative phosphorylation to changes in cardiac work. Semiquantitative agreement with the force-respiration relationship obtained in isolated cardiac trabeculae subjected to various pacing frequencies and external Ca2+ concentrations was also achieved. Quantitative differences between our simulations and available experimental data can, in part, be explained by differences in the E-C coupling properties of the rat (which we choose for experiments due to the extensive validation of this preparation in our laboratory and others) and the Guinea-pig (which serves as a well established model representative of the E-C coupling properties of larger mammals) (41). The relative contributions of SERCA and the Na+/Ca2+ exchanger to the reuptake of cytoplasmic Ca2+ after the peak are different in rats and guinea-pigs. As a consequence, the fraction of Ca2+ that is recycled is much larger in rats than in guinea-pigs. The rat trabeculae experiments were carried out at 22°C whereas the model parameters were adjusted for currents and fluxes operating at 37°C. This is another source of variation and further experimentation with different species and temperature will be required to determine their contribution to quantitative discrepancies with the model.
The feasibility of ADP and Ca2+ as important mediators of energy supply and demand matching has been demonstrated in the present model, but this work does not preclude the possibility that other mechanisms/mediators might be involved. For example, compelling evidence exists that various steps in the electron transport chain may be regulated by allosteric mechanisms (45), which would be replicated in our model by changing the concentration of respiratory carriers (ρres) in response to changes in work. Since the rate of the mitochondrial ATP synthase is governed by the protonmotive force and the availability of ADP and Pi, independently of the rate of respiration, we have observed in some simulations that ATP synthesis outpaces the increase in respiration, resulting an increase in the P/O ratio at high work (P/O ratio refers to the quotient of the ATP synthesis rate over the respiratory rate and represents a measure of the efficiency of oxidative phosphorylation). This changing P/O ratio differs from experimental results obtained in isolated rat hearts (46), perhaps suggesting an avenue for further refinement and validation of the model.
Cytosolic Ca2+ handling in the E-C coupling module has been previously validated by simulating the short-term interval force relations of cardiac muscle (15,47). The experimentally observed behavior of restitution and post-extrasystolic potentiation (48) are also appropriately reproduced in the integrated model (see Supplementary Material, Fig. S4). According to the model, Na+ levels determine, to a certain extent, the level of Ca2+ and the rate of Ca2+ removal through the Na+,Ca2+ exchanger activity. This is reflected by the accumulation of Na+ as frequency increases (see Supplementary Material, Fig. S2). Nevertheless, we have not accounted yet for important processes influencing cytosolic Na+ levels such as the Na+,H+ exchanger, which may render a more accurate picture of this cation.
A more controversial issue is to what extent the mitochondria will influence the cytosolic Ca2+ transient and how rapidly Ca2+ is taken up by the mitochondria (49). Indeed, we (50) and others (49,51) have obtained evidence that Ca2+ may be taken up rapidly by the mitochondria during the Ca2+ transient. This again raises the question of the spatial organization of the components of the model, as mentioned above when considering high-energy phosphate transfer rates. This is another area that will likely require further refinement of the model and experimentation. As formulated in this study, the mitochondria responded to the bulk cytoplasmic Ca2+ concentration and did not substantially alter the Ca2+-induced Ca2+ release process of the junctional space.
Comparison with other mathematical models
There have been few previous attempts to develop computational models linking electrophysiology, ion homeostasis, Ca2+ handling, ATP consumption, and mitochondrial energetics. Magnus and Keizer (52) developed a model that incorporated plasmalemmal ion channels, Ca2+ handling, and ATP production by glycolysis and mitochondria to simulate the bursting behavior of pancreatic β-cells. Elements of that model were used as components of our previous isolated cardiac mitochondrial model (16), with extensive modifications including the addition of NADH as a state variable, dependent on the activity of the Krebs cycle. In the present model, both Ca2+ and ADP are now variables that depend on the sum total of physiological processes associated with E-C coupling and mitochondrial function. The only remaining important fixed influx is the supply of Acetyl CoA to the Krebs cycle, which could be refined in the future by incorporating established models of glycolysis and β-oxidation of fatty acids. This could account for potential limitations of higher-order substrate supply, which could be relevant to the simulation of pathological conditions.
Another recent model based on a sound electrophysiological formulation focused on the interactions between the mitochondrial and cytoplasm space with Ca2+ as the sole signaling molecule (53). Presumably owing to the lack of significant respiratory control in the mitochondrial component of this model (54), the interrelationship between a change in E-C coupling and bioenergetics is not well described.
Saks and collaborators (7,9,55,56) have also modeled the energetic processes linked to muscle contraction, but they did not take into account the electrophysiology of the cell. Their model was more oriented to processes occurring in the cytoplasm and mitochondrial intermembrane space, including creatine kinase and adenylate kinase and was intended to address the some of the spatial aspects of phosphoryl transfer that we have discussed above. Force generation in this model was an adjustable function used to calculate its contribution to energy consumption rather than as a biophysical description of contraction.
Concluding remarks
We have developed an integrated model of the electrophysiology, E-C coupling properties, and mitochondrial bioenergetic responses of the cardiac myocyte. The model reproduces the pseudo steady-state force-respiration relationship and is also capable of describing the transient energetic responses to abrupt changes in workload. The ECME model can be used to explore the important interactions between the electrical, contractile metabolic functions of the heart, which will lead to an improved quantitative understanding of cardiac physiology under normal or pathological conditions.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors are grateful to Dr. Linda B. Stull and Michelle Leppo for their help with the rat trabeculae experiments.
This work was supported by grants from Falk Medical Trust No. P020032 (R.L.W.), National Institutes of Health No. R37HL54598 (B.O'R.), and No. P01HL081427 (B.O'R., R.L.W., E.M.).
Adjusted from the value reported in Rice et al. (15) to reproduce the values of the developed force by ventricular guinea-pig muscles, as described in Ruf et al. (62).
Adjusted from the value reported in Rice et al. (15) to reproduce the values of the developed force by ventricular guinea-pig muscles, as described in Ruf et al. (62).
The forward rate constants of the CK reaction were adjusted to reproduce the range of values from Clark et al. (67).
†
The equilibrium constant was averaged from that reported by Selivanov et al. (62) and the one by Saupe et al. (66) including the proton concentration [H+] inside the equilibrium constant.
‡
Calculated according to the 25% of the resting energy being used for macromolecular turnover as reported by Ingwall and Weiss (65).
The kinetic constants of all the TCA cycle enzyme steps have been multiplied by a factor of 1.5–4 with respect to those indicated in Cortassa et al. (16) to match the fluxes of the TCA cycle (68) in the integrated model that faces additional restrictions than those of the isolated mitochondrial model.
†
The values of the isocitrate dehydrogenase activation constants by ADP and Ca2+ have been modified to reproduce appropriately the kinetics reported by Rutter and Denton (68).
The respiratory complex II carriers concentration was adjusted with respect to the value in Cortassa et al. (16) to match the reported range of oxygen consumption rates (0.04–0.5 mM s−1 = 5–60 μMol O2/min/mg) (1).
†
The total nucleotide level and inorganic phosphate were corrected with respect to previous publication (16) to follow reported experimental evidence (1,69).
The maximal rate of the Ca2+ uniporter had to be adjusted to meet the transport fluxes experimentally determined (70,71) in the new integrated model structure in which the Ca2+ levels in the cytoplasm are no longer steady as they were in the isolated mitochondrial model (16).
TABLE 16.
States' variables initial values
Symbol
Description
Value
V
Sarcolemmal membrane potential.
−84.78
mNa
Sodium channel activation gate.
0.03282
nNa
Sodium channel inactivation gate.
0.98415
jNa
Sodium channel slow inactivation gate.
0.99003
X
Potassium channel activation gate.
1.616 × 10−4
[Na+]i
Intracellular Na+ concentration.
7.001
[K+]i
Intracellular K+ concentration.
138.3
[Ca2+]i
Intracellular Ca2+ concentration.
5.52 × 10−5
[Ca2+]NSR
Network SR Ca2+ concentration.
0.1998
[Ca2+]JSR
Junctional SR Ca2+ concentration.
0.1998
[Ca2+]SS
Ca2+ concentration in the subspace.
5.857 × 10−5
[Ca2+]m
Mitochondrial free Ca2+ concentration.
9.76 × 10−5
PC1
Fraction of RyR channels in PC1 state.
0.9299
PC2
Fraction of RyR channels in PC2 state.
0.0700
PO2
Fraction of RyR channels in PO2 state.
3.51 × 10−11
PO1
Fraction of RyR channels in PO1 state.
8.309 × 10−5
C0
L-type Ca2+ channel closed-mode normal.
0.9991
C1
L-type Ca2+ channel closed-mode normal.
8.175 × 10−5
C2
L-type Ca2+ channel closed-mode normal.
2.508 × 10−9
C3
L-type Ca2+ channel closed-mode normal.
3.421 × 10−14
C4
L-type Ca2+ channel closed-mode normal.
1.749 × 10−19
O
L-type Ca2+ channel open-mode normal.
2.624 × 10−20
CCa0
L-type Ca2+ channel closed-mode Ca.
8.28 × 10−4
CCa1
L-type Ca2+ channel closed-mode Ca.
2.71 × 10−7
CCa2
L-type Ca2+ channel closed-mode Ca.
3.326 × 10−11
CCa3
L-type Ca2+ channel closed-mode Ca.
1.815 × 10−15
CCa4
L-type Ca2+ channel closed-mode Ca.
3.712 × 10−20
OCa
L-type Ca2+ channel open-mode Ca.
0
Y
ICa inactivation gate.
0.979
[LTRPNCa]
Ca2+ bound to low-affinity troponin sites.
0.00849
[HTRPNCa]
Ca2+ bound to high-affinity troponin sites.
0.1321
[N0]
Nonpermissive tropomyosin with zero crossbridges.
0.9998
[N1]
Nonpermissive tropomyosin with one crossbridge.
2.243 × 10−5
[P0]
Permissive tropomyosin with zero crossbridges.
2.601 × 10−5
[P1]
Permissive tropomyosin with one crossbridge.
2.248 × 10−5
[P2]
Permissive tropomyosin with two crossbridges.
4.199 × 10−5
[P3]
Permissive tropomyosin with three crossbridges.
3.657 × 10−5
[ATP]i
EC coupling linked ATP concentration.
7.977
[ADP]i
EC coupling linked ADP concentration.
0.023
[ATP]ic
Cytosolic ATP concentration not linked to EC coupling.
7.972
[ADP]ic
Cytosolic ADP concentration not linked to EC coupling.
1.Balaban, R. S. 2002. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J. Mol. Cell. Cardiol. 34:1259–1271. [DOI] [PubMed] [Google Scholar]
2.Evans, L., and Y. Matsuoka. 1915. The effect of various mechanical conditions on the gaseous metabolism and efficiency of the mammalian heart. J. Physiol. (Lond.). 49:378–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
3.Brown, G. C. 1992. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J. 284:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
4.Neely, J. R., H. Liebermeister, E. J. Battersby, and H. E. Morgan. 1967. Effect of pressure development on oxygen consumption by isolated rat heart. Am. J. Physiol. 212:804–814. [DOI] [PubMed] [Google Scholar]
5.Williamson, J. R., C. Ford, J. Illingworth, and B. Safer. 1976. Coordination of citric acid cycle activity with electron transport flux. Circ. Res. 38:I39–I51. [PubMed] [Google Scholar]
6.Heineman, F. W., and R. S. Balaban. 1990. Control of mitochondrial respiration in the heart in vivo. Annu. Rev. Physiol. 52:523–542. [DOI] [PubMed] [Google Scholar]
7.Saks, V. A., A. V. Kuznetsov, M. Vendelin, K. Guerrero, L. Kay, and E. K. Seppet. 2004. Functional coupling as a basic mechanism of feedback regulation of cardiac energy metabolism. Mol. Cell. Biochem. 256–257:185–199. [DOI] [PubMed] [Google Scholar]
8.Chance, B., and G. R. Williams. 1955. A method for the localization of sites for oxidative phosphorylation. Nature. 176:250–254. [DOI] [PubMed] [Google Scholar]
9.Saks, V. A., T. Kaambre, P. Sikk, M. Eimre, E. Orlova, K. Paju, A. Piirsoo, F. Appaix, L. Kay, V. Regitz-Zagrosek, E. Fleck, and E. Seppet. 2001. Intracellular energetic units in red muscle cells. Biochem. J. 356:643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
10.Kaasik, A., V. Veksler, E. Boehm, M. Novotova, A. Minajeva, and R. Ventura-Clapier. 2001. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ. Res. 89:153–159. [DOI] [PubMed] [Google Scholar]
11.Joubert, F., P. Mateo, B. Gillet, J. C. Beloeil, J. L. Mazet, and J. A. Hoerter. 2004. CK flux or direct ATP transfer: versatility of energy transfer pathways evidenced by NMR in the perfused heart. Mol. Cell. Biochem. 256–257:43–58. [DOI] [PubMed] [Google Scholar]
12.Territo, P. R., S. A. French, M. C. Dunleavy, F. J. Evans, and R. S. Balaban. 2001. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, and light scattering. J. Biol. Chem. 276:2586–2599. [DOI] [PubMed] [Google Scholar]
13.Bose, S., S. French, F. J. Evans, F. Joubert, and R. S. Balaban. 2003. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J. Biol. Chem. 278:39155–39165. [DOI] [PubMed] [Google Scholar]
14.Jafri, M. S., J. J. Rice, and R. L. Winslow. 1998. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 74:1149–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
15.Rice, J. J., M. S. Jafri, and R. L. Winslow. 2000. Modeling short-term interval-force relations in cardiac muscle. Am. J. Physiol. Heart Circ. Physiol. 278:H913–H931. [DOI] [PubMed] [Google Scholar]
16.Cortassa, S., M. A. Aon, E. Marban, R. L. Winslow, and B. O'Rourke. 2003. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 84:2734–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
17.Brandes, R., and D. M. Bers. 2002. Simultaneous measurements of mitochondrial NADH and Ca2+ during increased work in intact rat heart trabeculae. Biophys. J. 83:587–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
18.Hoerter, J. A., R. Ventura-Clapier, and A. Kuznetsov. 1994. Compartmentation of creatine kinases during perinatal development of mammalian heart. Mol. Cell. Biochem. 133–134:277–286. [DOI] [PubMed] [Google Scholar]
19.Saks, V. A., R. Ventura-Clapier, and M. K. Aliev. 1996. Metabolic control and metabolic capacity: two aspects of creatine kinase functioning in the cells. Biochim. Biophys. Acta. 1274:81–88. [DOI] [PubMed] [Google Scholar]
20.Shannon, T. R., K. S. Ginsburg, and D. M. Bers. 2000. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys. J. 78:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
21.Sakamoto, J., and Y. Tonomura. 1980. Order of release of ADP and Pi from phosphoenzyme with bound ADP of Ca2+-dependent ATPase from sarcoplasmic reticulum and of Na+, K+-dependent ATPase studied by ADP-inhibition patterns. J. Biochem. (Tokyo). 87:1721–1727. [DOI] [PubMed] [Google Scholar]
22.Jeneson, J. A., H. V. Westerhoff, and M. J. Kushmerick. 2000. A metabolic control analysis of kinetic controls in ATP free energy metabolism in contracting skeletal muscle. Am. J. Physiol. Cell Physiol. 279:C813–C832. [DOI] [PubMed] [Google Scholar]
23.Regnier, M., D. A. Martyn, and P. B. Chase. 1998. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophys. J. 74:2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
24.Karatzaferi, C., K. H. Myburgh, M. K. Chinn, K. Franks-Skiba, and R. Cooke. 2003. Effect of an ADP analog on isometric force and ATPase activity of active muscle fibers. Am. J. Physiol. Cell Physiol. 284:C816–C825. [DOI] [PubMed] [Google Scholar]
25.Pate, E., and R. Cooke. 1989. A model of crossbridge action: the effects of ATP, ADP and Pi. J. Muscle Res. Cell Motil. 10:181–196. [DOI] [PubMed] [Google Scholar]
26.Luo, C. H., and Y. Rudy. 1994. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74:1071–1096. [DOI] [PubMed] [Google Scholar]
27.Nakao, M., and D. C. Gadsby. 1989. [Na] and [K] dependence of the Na/K pump current-voltage relationship in Guinea-pig ventricular myocytes. J. Gen. Physiol. 94:539–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
28.Echarte, M. M., V. Levi, A. M. Villamil, R. C. Rossi, and J. P. Rossi. 2001. Quantitation of plasma membrane calcium pump phosphorylated intermediates by electrophoresis. Anal. Biochem. 289:267–273. [DOI] [PubMed] [Google Scholar]
29.Pasa, T. C., A. S. Otero, H. Barrabin, and H. M. Scofano. 1992. Regulation of the nucleotide dependence of the cardiac sarcolemma Ca2+-ATPase. J. Mol. Cell. Cardiol. 24:233–242. [DOI] [PubMed] [Google Scholar]
30.Ebus, J. P., and G. J. Stienen. 1996. Origin of concurrent ATPase activities in skinned cardiac trabeculae from rat. J. Physiol. 492:675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
31.Cohen, S. D., and A. C. Hindmarsh. 1994. CVODE User Guide. In Lawrence Livermore National Laboratory report. Livermore, CA.
32.Brandes, R., and D. M. Bers. 1999. Analysis of the mechanisms of mitochondrial NADH regulation in cardiac trabeculae. Biophys. J. 77:1666–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
33.Mulieri, L. A., G. Hasenfuss, F. Ittleman, E. M. Blanchard, and N. R. Alpert. 1989. Protection of human left ventricular myocardium from cutting injury with 2,3-butanedione monoxime. Circ. Res. 65:1441–1449. [DOI] [PubMed] [Google Scholar]
34.Janssen, P. M., L. B. Stull, and E. Marban. 2002. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. Am. J. Physiol. Heart Circ. Physiol. 282:H499–H507. [DOI] [PubMed] [Google Scholar]
35.Szigligeti, P., C. Pankucsi, T. Banyasz, A. Varro, and P. P. Nanasi. 1996. Action potential duration and force-frequency relationship in isolated rabbit, Guinea-pig and rat cardiac muscle. J. Comp. Physiol. [B]. 166:150–155. [DOI] [PubMed] [Google Scholar]
36.Maier, L. S., D. M. Bers, and B. Pieske. 2000. Differences in Ca2+-handling and sarcoplasmic reticulum Ca2+-content in isolated rat and rabbit myocardium. J. Mol. Cell. Cardiol. 32:2249–2258. [DOI] [PubMed] [Google Scholar]
37.Neely, J. R., M. Whitmer, and S. Mochizuki. 1976. Effects of mechanical activity and hormones on myocardial glucose and fatty acid utilization. Circ. Res. 38:I22–I30. [PubMed] [Google Scholar]
38.Elliott, A. C., G. L. Smith, and D. G. Allen. 1994. The metabolic consequences of an increase in the frequency of stimulation in isolated ferret hearts. J. Physiol. 474:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
39.Jacobus, W. E. 1985. Respiratory control and the integration of heart high-energy phosphate metabolism by mitochondrial creatine kinase. Annu. Rev. Physiol. 47:707–725. [DOI] [PubMed] [Google Scholar]
40.Chen, C., Y. Ko, M. Delannoy, S. J. Ludtke, W. Chiu, and P. L. Pedersen. 2004. Mitochondrial ATP synthasome: three-dimensional structure by electron microscopy of the ATP synthase in complex formation with carriers for Pi and ADP/ATP. J. Biol. Chem. 279:31761–31768. [DOI] [PubMed] [Google Scholar]
41.Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers, Dordrecht, The Netherlands; Boston, MA.
42.Eijgelshoven, M. H., J. H. van Beek, I. Mottet, M. G. Nederhoff, C. J. van Echteld, and N. Westerhof. 1994. Cardiac high-energy phosphates adapt faster than oxygen consumption to changes in heart rate. Circ. Res. 75:751–759. [DOI] [PubMed] [Google Scholar]
43.Harrison, G. J., M. H. van Wijhe, B. de Groot, F. J. Dijk, L. A. Gustafson, and J. H. van Beek. 2003. Glycolytic buffering affects cardiac bioenergetic signaling and contractile reserve similar to creatine kinase. Am. J. Physiol. Heart Circ. Physiol. 285:H883–H890. [DOI] [PubMed] [Google Scholar]
44.Brandes, R., and D. M. Bers. 1997. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ. Res. 80:82–87. [DOI] [PubMed] [Google Scholar]
45.Kadenbach, B. 2003. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim. Biophys. Acta. 1604:77–94. [DOI] [PubMed] [Google Scholar]
46.Bittl, J. A., and J. S. Ingwall. 1985. Reaction rates of creatine kinase and ATP synthesis in the isolated rat heart. A 31P NMR magnetization transfer study. J. Biol. Chem. 260:3512–3517. [PubMed] [Google Scholar]
47.Johnson, E. A. 1979. Force-interval relationship in cardiac muscle. In Handbook of Physiology. The Cardiovascular System. The Heart. American Physiological Society, Bethesda, MD. 475–492.
48.Wier, W. G., and D. T. Yue. 1986. Intracellular calcium transients underlying the short-term force-interval relationship in ferret ventricular myocardium. J. Physiol. 376:507–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
49.Huser, J., L. A. Blatter, and S. S. Sheu. 2000. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J. Bioenerg. Biomembr. 32:27–33. [DOI] [PubMed] [Google Scholar]
50.Maack, C., S. Cortassa, M. A. Aon, A. N. Ganesan, T. Liu, and B. O'Rourke. 2006. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. In press. [DOI] [PMC free article] [PubMed]
51.Pozzan, T., and R. Rizzuto. 2000. The renaissance of mitochondrial calcium transport. Eur. J. Biochem. 267:5269–5273. [DOI] [PubMed] [Google Scholar]
52.Magnus, G., and J. Keizer. 1998. Model of β-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am. J. Physiol. 274:C1158–C1173. [DOI] [PubMed] [Google Scholar]
53.Matsuoka, S., N. Sarai, H. Jo, and A. Noma. 2004. Simulation of ATP metabolism in cardiac excitation-contraction coupling. Prog. Biophys. Mol. Biol. 85:279–299. [DOI] [PubMed] [Google Scholar]
54.Korzeniewski, B., and J. P. Mazat. 1996. Theoretical studies on the control of oxidative phosphorylation in muscle mitochondria: application to mitochondrial deficiencies. Biochem. J. 319:143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
55.Vendelin, M., O. Kongas, and V. Saks. 2000. Regulation of mitochondrial respiration in heart cells analyzed by reaction-diffusion model of energy transfer. Am. J. Physiol. Cell Physiol. 278:C747–C764. [DOI] [PubMed] [Google Scholar]
56.Vendelin, M., M. Lemba, and V. A. Saks. 2004. Analysis of functional coupling: mitochondrial creatine kinase and adenine nucleotide translocase. Biophys. J. 87:696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
57.Page, E. 1978. Quantitative ultrastructural analysis in cardiac membrane physiology. Am. J. Physiol. 235:C147–C158. [DOI] [PubMed] [Google Scholar]
58.Sheu, S. S., and H. A. Fozzard. 1982. Transmembrane Na+ and Ca2+ electrochemical gradients in cardiac muscle and their relationship to force development. J. Gen. Physiol. 80:325–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
59.Sheu, S. S. 1989. Cytosolic sodium concentration regulates contractility of cardiac muscle. Basic Res. Cardiol. 84(Suppl 1):35–45. [DOI] [PubMed] [Google Scholar]
60.Elimban, V., D. Zhao, and N. S. Dhalla. 1987. A comparative study of the rat heart sarcolemmal Ca2+-dependent ATPase and myosin ATPase. Mol. Cell. Biochem. 77:143–152. [DOI] [PubMed] [Google Scholar]
61.Winslow, R. L., J. Rice, S. Jafri, E. Marban, and B. O'Rourke. 1999. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ. Res. 84:571–586. [DOI] [PubMed] [Google Scholar]
62.Ruf, T., S. Hebisch, R. Gross, N. Alpert, H. Just, and C. Holubarsch. 1996. Modulation of myocardial economy and efficiency in mammalian failing and non-failing myocardium by calcium channel activation and β-adrenergic stimulation. Cardiovasc. Res. 32:1047–1055. [DOI] [PubMed] [Google Scholar]
63.Selivanov, V. A., A. E. Alekseev, D. M. Hodgson, P. P. Dzeja, and A. Terzic. 2004. Nucleotide-gated KATP channels integrated with creatine and adenylate kinases: amplification, tuning and sensing of energetic signals in the compartmentalized cellular environment. Mol. Cell. Biochem. 256–257:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
64.Joubert, F., B. Gillet, J. L. Mazet, P. Mateo, J. Beloeil, and J. A. Hoerter. 2000. Evidence for myocardial ATP compartmentation from NMR inversion transfer analysis of creatine kinase fluxes. Biophys. J. 79:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
65.Ingwall, J. S., and R. G. Weiss. 2004. Is the failing heart energy-starved? On using chemical energy to support cardiac function. Circ. Res. 95:135–145. [DOI] [PubMed] [Google Scholar]
66.Saupe, K. W., M. Spindler, J. C. Hopkins, W. Shen, and J. S. Ingwall. 2000. Kinetic, thermodynamic, and developmental consequences of deleting creatine kinase isoenzymes from the heart. Reaction kinetics of the creatine kinase isoenzymes in the intact heart. J. Biol. Chem. 275:19742–19746. [DOI] [PubMed] [Google Scholar]
67.Clark, J. F., A. V. Kuznetsov, and G. K. Radda. 1997. ADP-regenerating enzyme systems in mitochondria of guinea pig myometrium and heart. Am. J. Physiol. 272:C399–C404. [DOI] [PubMed] [Google Scholar]
68.Rutter, G. A., and R. M. Denton. 1988. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios. Biochem. J. 252:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
69.Randle, P. J., and P. K. Tubbs. 1979. Carbohydrate and fatty acid metabolism. In Handbook of Physiology. R. M. Berne, N. Sperelakis, and R. Geiger, editors. American Physiological Society, Bethesda, MD. 805–844.
70.Gunter, K. K., and T. E. Gunter. 1994. Transport of calcium by mitochondria. J. Bioenerg. Biomembr. 26:471–485. [DOI] [PubMed] [Google Scholar]
71.Gunter, T. E., and D. R. Pfeiffer. 1990. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258:C755–C786. [DOI] [PubMed] [Google Scholar]



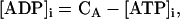
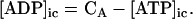
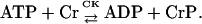
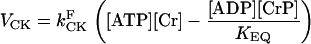
 ) (Eq. A141) and transported to the cytoplasm for its utilization by constitutive energy-consuming processes through
) (Eq. A141) and transported to the cytoplasm for its utilization by constitutive energy-consuming processes through  (Eq. A142). The CK at the cytoplasmic site,
(Eq. A142). The CK at the cytoplasmic site, 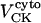 (Eq. A140), catalyzes the phosphorylation of ADP from CrP to generate ATP (CK reaction as written above in the reverse direction) that will be used by the constitutive cytosolic ATPases ruled by
(Eq. A140), catalyzes the phosphorylation of ADP from CrP to generate ATP (CK reaction as written above in the reverse direction) that will be used by the constitutive cytosolic ATPases ruled by 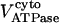 (fixed parameter, Table 12). Thus, the direction of the CK reaction depends on the enzyme localization, i.e., toward CrP synthesis in the mitochondria and toward the synthesis of ATP in the cytoplasm. The creatine metabolite levels in each pool, like the adenine nucleotide concentrations, are linked through conservation relationships as follows:
(fixed parameter, Table 12). Thus, the direction of the CK reaction depends on the enzyme localization, i.e., toward CrP synthesis in the mitochondria and toward the synthesis of ATP in the cytoplasm. The creatine metabolite levels in each pool, like the adenine nucleotide concentrations, are linked through conservation relationships as follows: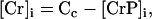
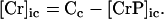
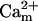 . Thus, mitochondria act as an additional Ca2+ and Na+ sink in our model.
. Thus, mitochondria act as an additional Ca2+ and Na+ sink in our model.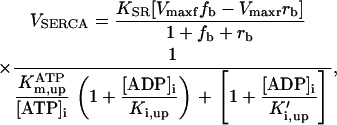
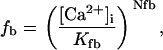
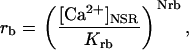
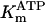 represents the affinity of SERCA for ATP, and Ki,up and K′i,up are the ADP inhibition constants (Table 7). As shown experimentally by Sakamoto and Tonomura (21), Eq. 6 indicates that ADP inhibits the pump both by competitive (accomplished in the term containing Ki,up) and noncompetitive (accounted for by K′i,up) mechanisms. We simulated the behavior of SERCA as a function of Ca2+ concentrations in the myoplasm and inside the SR network (NSR). The introduction of ATP dependence did not significantly change pump rate as a function of Ca2+ in the physiological range of ATPi levels (not shown).
represents the affinity of SERCA for ATP, and Ki,up and K′i,up are the ADP inhibition constants (Table 7). As shown experimentally by Sakamoto and Tonomura (21), Eq. 6 indicates that ADP inhibits the pump both by competitive (accomplished in the term containing Ki,up) and noncompetitive (accounted for by K′i,up) mechanisms. We simulated the behavior of SERCA as a function of Ca2+ concentrations in the myoplasm and inside the SR network (NSR). The introduction of ATP dependence did not significantly change pump rate as a function of Ca2+ in the physiological range of ATPi levels (not shown).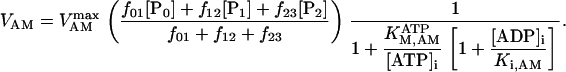
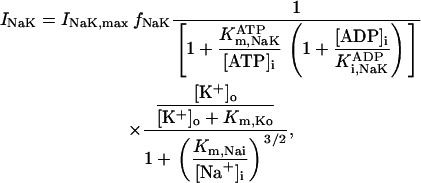
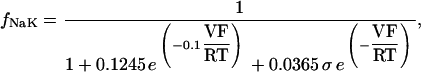
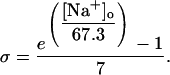
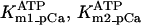 ). ADP exerts competitive inhibition of the Ca2+ pump by affecting the high-affinity component of the ATP interaction through the inhibition constant,
). ADP exerts competitive inhibition of the Ca2+ pump by affecting the high-affinity component of the ATP interaction through the inhibition constant, 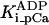 . We adopted the K0.5 values described in Echarte et al. (28), who found two K0.5 values for ATP in their study of the enzyme reaction mechanism involving the formation of an intermediate phosphorylated enzyme state. The affinity constants for ATP suggested the presence of two nucleotide regulatory sites in the sarcolemmal Ca2+ ATPase (29):
. We adopted the K0.5 values described in Echarte et al. (28), who found two K0.5 values for ATP in their study of the enzyme reaction mechanism involving the formation of an intermediate phosphorylated enzyme state. The affinity constants for ATP suggested the presence of two nucleotide regulatory sites in the sarcolemmal Ca2+ ATPase (29):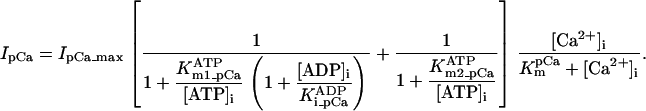

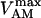 , of 2.88 s−1, and P1 = 0.2, P2 = 0.7, and P3 = 0.1. Otherwise, the parameters used in the plot are as indicated in Table 11. (E and F) The dependence of the Na+, K+ adenosine triphosphatase activity on ATP. Experimental data (E, from (21)) and model simulations (F) using the rate expression of Eq. 8. Competitive inhibition by ADP at 0, 0.5, 1.0, and 2.0 mM concentrations is shown. The parameters used were Vmax = 3.93 μmol mg−1 min−1, the membrane potential is −80 mV, and the intracellular Na+ is 7.0 mM. The other parameters are as indicated in Table 3. (G and H) The dependence of the sarcolemmal Ca2+ adenosine triphosphatase activity on ATP. Experimental data (G, from (29)) and model simulations (H) obtained with Eq. 9, which corresponds to a typical Michaelis-Menten profile. The following parameter values were used in the simulation: Vmax = 2.55 μmol mg−1 min−1; [Ca2+]i = 2 μM; and ADP = 50 μM. The rest of the parameter values are as indicated in Table 6. Panels A and E are reproduced from Sakamoto and Tonomura (21) by permission of the Japanese Biochemical Society and Oxford University Press. Panel C is from Karatzaferi et al. (24) used with permission from the American Physiological Society. Panel G is reproduced from Pasa et al. (29) with permission from Elsevier.
, of 2.88 s−1, and P1 = 0.2, P2 = 0.7, and P3 = 0.1. Otherwise, the parameters used in the plot are as indicated in Table 11. (E and F) The dependence of the Na+, K+ adenosine triphosphatase activity on ATP. Experimental data (E, from (21)) and model simulations (F) using the rate expression of Eq. 8. Competitive inhibition by ADP at 0, 0.5, 1.0, and 2.0 mM concentrations is shown. The parameters used were Vmax = 3.93 μmol mg−1 min−1, the membrane potential is −80 mV, and the intracellular Na+ is 7.0 mM. The other parameters are as indicated in Table 3. (G and H) The dependence of the sarcolemmal Ca2+ adenosine triphosphatase activity on ATP. Experimental data (G, from (29)) and model simulations (H) obtained with Eq. 9, which corresponds to a typical Michaelis-Menten profile. The following parameter values were used in the simulation: Vmax = 2.55 μmol mg−1 min−1; [Ca2+]i = 2 μM; and ADP = 50 μM. The rest of the parameter values are as indicated in Table 6. Panels A and E are reproduced from Sakamoto and Tonomura (21) by permission of the Japanese Biochemical Society and Oxford University Press. Panel C is from Karatzaferi et al. (24) used with permission from the American Physiological Society. Panel G is reproduced from Pasa et al. (29) with permission from Elsevier.

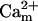 recorded experimentally (Fig. 5, A and B). The magnitude of the NADH decrease during the undershoot is somewhat lower than that observed experimentally (3% against 10%, respectively).
recorded experimentally (Fig. 5, A and B). The magnitude of the NADH decrease during the undershoot is somewhat lower than that observed experimentally (3% against 10%, respectively). 


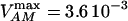 mM ms−1) of the control value (shaded trace). (B) The profile of average ATPi for changes in workload under control conditions (shaded trace) or with decreased myofibrillar ATPase activity (solid trace). (C) The NADH profile in model simulations with the same pacing protocol and parameters as described in the legend of Fig. 5, but with the maximal rate of the mitochondrial Ca2+ uniporter decreased to 1/10th (solid trace;
mM ms−1) of the control value (shaded trace). (B) The profile of average ATPi for changes in workload under control conditions (shaded trace) or with decreased myofibrillar ATPase activity (solid trace). (C) The NADH profile in model simulations with the same pacing protocol and parameters as described in the legend of Fig. 5, but with the maximal rate of the mitochondrial Ca2+ uniporter decreased to 1/10th (solid trace; 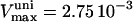 mM ms−1) of the control value (shaded trace). (D) The profile of [Ca2+]m accumulation for the low uniporter condition described in panel C (solid trace) as compared to the control (shaded trace).
mM ms−1) of the control value (shaded trace). (D) The profile of [Ca2+]m accumulation for the low uniporter condition described in panel C (solid trace) as compared to the control (shaded trace).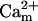 can account for the larger undershoot/overshoot and recovery phases of the NADH signal as the frequency increases.
can account for the larger undershoot/overshoot and recovery phases of the NADH signal as the frequency increases. 
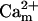 ) that are involved in the energetic response to a change in workload. Parameters and initial conditions are the same as those used in Figs. 3–5 Panel B was reproduced from Brandes and Bers (32) by copyright permission of the Biophysical Society.
) that are involved in the energetic response to a change in workload. Parameters and initial conditions are the same as those used in Figs. 3–5 Panel B was reproduced from Brandes and Bers (32) by copyright permission of the Biophysical Society.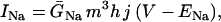
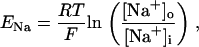
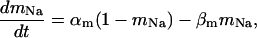
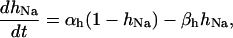
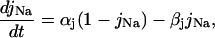
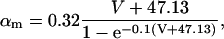
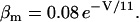
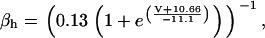
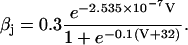
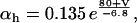
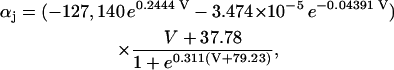
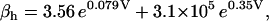
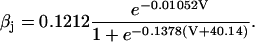
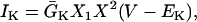
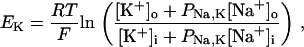
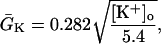
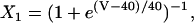
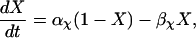
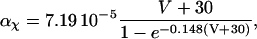
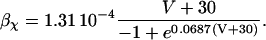
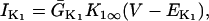
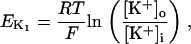
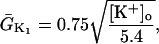
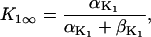
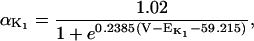
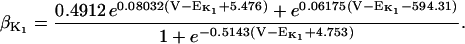
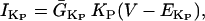
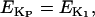
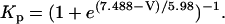
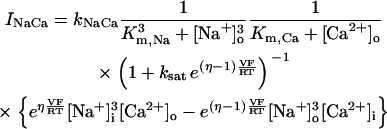
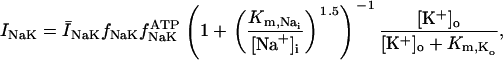
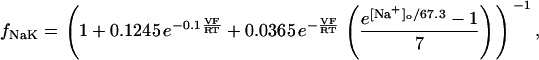
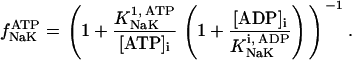
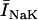
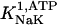
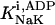
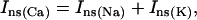
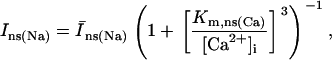
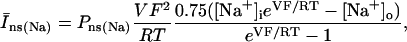
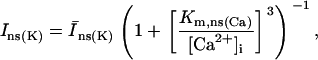
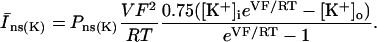
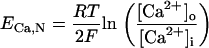
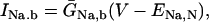
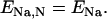
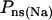
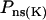
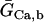
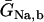
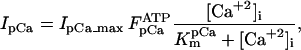
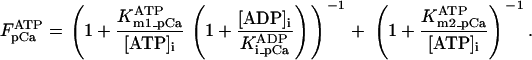
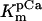
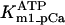
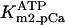
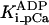
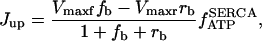
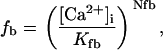
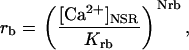
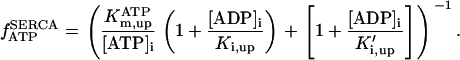
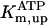
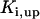
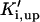
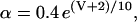
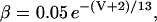
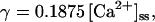
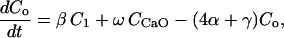
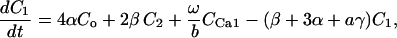
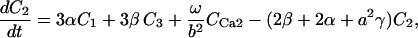
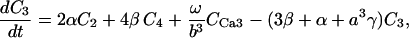
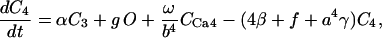
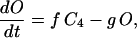
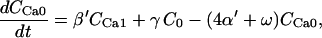
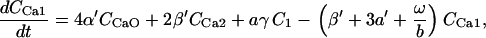
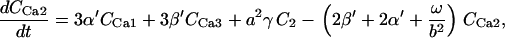
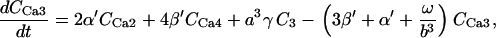
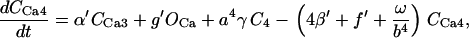
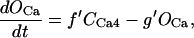
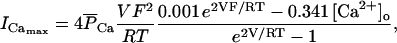
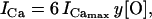
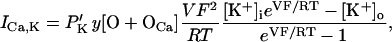
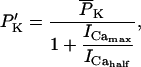
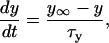
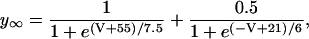
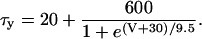
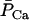
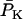
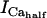
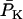 by half.
by half.