

Abstract
Binding of a small molecule to a macromolecular target reduces its conformational freedom, resulting in a negative entropy change that opposes the binding. The goal of this study is to estimate the configurational entropy change of two minor-groove-binding ligands, netropsin and distamycin, upon binding to the DNA duplex d(CGCGAAAAACGCG)·d(CGCGTTTTTCGCG). Configurational entropy upper bounds based on 10-ns molecular dynamics simulations of netropsin and distamycin in solution and in complex with DNA in solution were estimated using the covariance matrix of atom-positional fluctuations. The results suggest that netropsin and distamycin lose a significant amount of configurational entropy upon binding to the DNA minor groove. The estimated changes in configurational entropy for netropsin and distamycin are −127 J K−1 mol−1 and −104 J K−1 mol−1, respectively. Estimates of the configurational entropy contributions of parts of the ligands are presented, showing that the loss of configurational entropy is comparatively more pronounced for the flexible tails than for the relatively rigid central body.
INTRODUCTION
The thermodynamics of binding of small molecules to DNA double helices has been extensively investigated using experimental (1–7) and computational (8–13) approaches. Understanding the favorable and unfavorable contributions to binding free energies from computer simulations provides fundamental insight not directly accessible through experiments and complements high-resolution x-ray crystallographic (14–17) and nuclear magnetic resonance (NMR) (18–22) experiments. Small molecules that bind in the minor groove of DNA are known to interfere with gene expression at the level of transcription and replication and are of great interest in the discovery of novel antibacterial molecules (23–25). In the rational design of new therapeutic agents with improved binding affinity and specificity, understanding the thermodynamics of DNA-drug interactions is one of the key issues (26).
Free energies together with the corresponding enthalpies and entropies of binding have been measured for a large number of DNA-ligand complexes (2,4,6,7,27). However, experimental studies usually give access only to the total change in enthalpy and entropy associated with a given process, but no specific information on the enthalpy and entropy change of the ligand. To analyze the free energy changes that accompany a binding process, investigation of binding enthalpy and entropy contributions is needed, because entropy-enthalpy compensation effects may cause binding events to exhibit very similar binding free energies, although the binding process is driven by different thermodynamic forces (28–31).
Molecular dynamics (MD) simulations are well suited to investigate the structural, dynamic, and thermodynamic properties of macromolecules (32–34). To capture the functioning of complex biomolecules at a molecular level, a static representation provides limited insight, and dynamical information on a sufficiently long timescale is a fundamental prerequisite (35). Significant progress in the development of empirical potential energy functions (force fields) and increasing computer power currently allow MD simulations on the nanosecond timescale for relatively large systems. Thus, simulations provide an extent of sampling of the configurational space that may be sufficient to describe the thermodynamic properties of these systems at equilibrium conditions. In particular, MD simulations of nucleic acids have been reported by several groups, demonstrating results that reproduce the solution NMR data reasonably well (36–38). However, theoretical studies of nucleic acids are still a challenging problem. The reasons are that 1), nucleic acids are highly charged systems, so an accurate treatment of electrostatic (long-range) interactions in computer simulations of these systems is essential (34,39); and 2), their structure and dynamics are largely influenced by the specific nature and concentration of the counterions and by the solvent properties. Consequently, simulations of nucleic acids are sensitive not only to the quality of the force-field parameters, but also to the simulation setup.
Netropsin and distamycin are two naturally occurring oligopeptides that bind noncovalently to domains of the DNA minor groove that are rich in adenine-thymine (AT) base pairs (40,41). Both ligands possess a cationic propylamidinium tail and a rigid body that is constituted of amide groups and methylpyrrole rings. In the case of distamycin, the rigid part is larger and the molecule terminates with a neutral formamide tail, whereas the body of netropsin ends with a (likely more flexible) cationic guanidinum tail (see Fig. 1 for chemical structures). Experiments by means of x-ray crystallography (14,15,17,42,43) and NMR (18,19) have been reported that provide information on the modes of interaction of netropsin and distamycin with the DNA minor groove. By a combination of circular dichroism spectroscopy, ultraviolet-absorption spectroscopy, and isothermal titration calorimetry (1,4,7,44,45), and through theoretical studies (8,12,46–48), the binding thermodynamics of the two ligands were investigated. It has been shown that the thermodynamics of binding depends strongly on the sequence of the base pairs in the binding site, and that the binding of netropsin and distamycin to the minor groove of DNA is either enthalpy- or entropy-driven (28). Furthermore, it has been shown that the binding affinities of netropsin and distamycin for a specific DNA sequence can be considerably different, despite their small structural differences (7). Depending on the specific DNA sequence, the experimental values for standard enthalpies of binding (ΔH°) of netropsin and distamycin range from −67.4 kJ/mol to −36.0 kJ/mol and the standard entropies of binding (ΔS°) range from −78.6 J K−1 mol−1 to 60.3 J K−1 mol−1 (7). The interpretation of experimental thermodynamic binding profiles of minor-groove binders usually assumes that the contributions to the binding free energy arising from conformational changes (of both DNA and ligands) are negligible compared to other forces driving ligand-DNA complexation (restructuring of the solvent, counter ion release, DNA-ligand interactions, and restriction of the rotational and translational degrees of freedom) (26). The motivation for this assumption in the case of (1:1) DNA minor-groove binding is that 1), the double helix is not considerably distorted; and 2), the structure of the ligand is basically unaltered, as observed from x-ray crystallographic studies. Thus, the binding of a ligand to the minor groove of DNA is usually treated as a rigid-body association, with the unfavorable entropy contributions from the loss of rotational and translational degrees of freedom estimated as 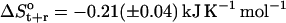 (49,50). However, the appropriate estimate of the
(49,50). However, the appropriate estimate of the 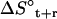 term is debated in the current literature (2,3,7,51), and recent experiments suggest that netropsin and distamycin may lose different amounts of rotational, translational, and configurational entropy upon formation of the DNA-drug complexes (7). Neglecting the configurational contribution seems reasonable for small and rigid binders, but not for more flexible ligands. Calculation of the configurational entropy change of DNA is currently not feasible computationally due to the size of the double helix. In the following text, we therefore only consider the entropy change due to the change in ligand flexibility.
term is debated in the current literature (2,3,7,51), and recent experiments suggest that netropsin and distamycin may lose different amounts of rotational, translational, and configurational entropy upon formation of the DNA-drug complexes (7). Neglecting the configurational contribution seems reasonable for small and rigid binders, but not for more flexible ligands. Calculation of the configurational entropy change of DNA is currently not feasible computationally due to the size of the double helix. In the following text, we therefore only consider the entropy change due to the change in ligand flexibility.
FIGURE 1.

(A) Snapshot of the complex of netropsin with a d((CG)2A5(CG)2)·d((CG)2T5(CG)2) DNA duplex after ∼1 ns of MD simulation. (B) Chemical structure of netropsin (upper) and distamycin (lower) molecules. Rectangular boxes define the atom subgroups used to estimate configurational entropies: tail 1 (t1), body (b), and tail 2 (t2). Netropsin and distamycin molecules possess identical body (b) and tail 1 (t1) moieties (red rectangles), but differ in tail 2 (t2) (blue rectangles). The atoms of the central peptide bond (code 4) are labeled with a star. Reference codes for the entropy calculations are summarized in Table 1.
During the past decades, the calculation of accurate free-energy differences from molecular simulations has become possible in practice (52–59). In contrast, the reliable estimation of entropies and entropy differences from such simulations is still a difficult task (60–72). The possibility to estimate configurational entropy from MD trajectories was first proposed (using impractical internal coordinates) by Karplus and Kushick under a quasiharmonic assumption (60). Some years later, Schlitter introduced a heuristic formula, based on Cartesian coordinates, which provides an easily applicable approach to compute an approximate (71) upper bound to the absolute entropy of a nondiffusive system from a simulation trajectory (63). Recently, Andricioaei and Karplus revised the quasiharmonic approach to enable the use of Cartesian coordinates (67). The alternative formulations proposed by Schlitter (63) and by Andricioaei and Karplus (67) result in very similar entropy estimates (71,73–75). In the first case, the diagonalization of the (mass-weighted) covariance matrix is substituted by a determinant calculation and the formula for the entropy of a quantum-mechanical oscillator is replaced by an approximate heuristic expression (which slightly overestimates the entropy upper bound (71)). This is computationally less expensive, which is why it is used in this work. Both methods provide approximate configurational entropies because the accuracy depends on 1), how harmonic and 2), how uncorrelated the normal modes of the simulated molecule are. An analysis of the quasiharmonic assumption and corrections for the anharmonicity and second-order correlation effects has recently been reported (71).
The aim of this study was to investigate configurational entropy changes of netropsin and distamycin upon binding to the minor groove of the DNA duplex d(CGCGAAAAACGCG)·d(CGCGTTTTTCGCG) in a 1:1 binding mode. We used the approach based on the covariance matrix of atomic mass-weighted fluctuations, because it allows not only the calculation of the configurational entropy of the entire chain but also, within a certain approximation, the calculation of the configurational entropy for different subsets of atoms or degrees of freedom. The same system was the subject of a previous study on relative binding free energies of netropsin and distamycin binding to DNA, which were estimated from up to 2 ns of molecular dynamics simulations (12). Here, to reach sufficient sampling to estimate configurational entropies, the MD simulations of netropsin and distamycin free in solution and of their complexes with DNA were extended to 10 ns. Configurational entropies of the ligands and parts thereof in their free and bound states are estimated. The configurational entropy changes that netropsin and distamycin undergo upon binding to the minor groove of DNA are compared and discussed. Comparison with experimental changes in enthalpy and entropy has limited value, because experimental values include more than the internal contributions (see Table 1 of Baron et al. (75)). On the other hand, estimating entropies of diffusive degrees of freedom is still a computational challenge (69). However, configurational entropy contributions offer an important insight into the binding process at the atomic level.
TABLE 1.
Code definitions of the atom sets used to estimate configurational entropy
| Code | Description |
|---|---|
| type | |
| i | Internal configurational entropy |
| ip | Internal configurational entropy per particle |
| fit and cov | |
| all | All atoms of the ligand |
| 4 | Four atoms of the peptide bond in the central body (N, H, C, O) |
| nh | Nonhydrogen atoms of the ligand |
| DNA | Nonhydrogen atoms of the central GAAAAAC/GTTTTTC segment |
| t1 | Tail 1 |
| t2 | Tail 2 |
| t | Tails (atoms of tail 1 and of tail 2) |
| b | Body |
Reference codes are defined for the type of entropy calculation (type), for the subsets of atoms used to perform the structural superposition (fit), and for the atoms included in the mass-weighted covariance matrix (cov). See Materials and Methods section and Fig. 1 for definitions.
MATERIALS AND METHODS
Molecular dynamics simulations
Four 10-ns MD simulations were performed for netropsin and distamycin, when free in solution and when bound to DNA. Starting from a model-built canonical B-DNA duplex d((CG)2A5(CG)2)·d((CG)2T5(CG)2) (INSIGHTII, Accelrys, San Diego, CA), initial coordinates of a netropsin-DNA and a distamycin-DNA complex were generated employing the structures of netropsin and distamycin molecules from Protein Data Bank (PDB) crystal structures 101D (15,76) and 267D (76,77), with similar (but not identical) DNA sequences. The complexes were solvated in periodic boxes (truncated octahedra) containing 11,034 simple-point-charge (SPC) water molecules (78), and 20 Cl− and 43 Na+ ions, which correspond to an experimental salt concentration of 110 mM NaCl. Similarly, each ligand molecule (free in solution) was solvated in 3225 SPC water molecules and 6 Na+ and 7 Cl− ions. All simulations were carried out using the GROMOS96 simulation package (79,80) and the GROMOS96 45A4 force field, including recently improved nucleic acid parameters (38). The SHAKE algorithm (81) was employed to keep all the bonds constrained to their ideal values, permitting a 2-fs time step for integration of the equations of motion using the leap-frog algorithm (82). For the calculation of nonbonded interactions a triple-range cut-off scheme was used. Interactions within a short-range cut-off of 0.8 nm were calculated at every time step from a pair list that was generated every five steps. At these time points, interactions between 0.8 and 1.4 nm were also calculated and kept constant between updates. The electrostatic interactions outside the outer 1.4 nm cutoff were approximated with a reaction-field contribution (83) using a relative permittivity of 61 (84). To maintain constant temperature (300 K) and pressure (1 atm) a Berendsen thermostat and barostat were employed (85). For details on system setup, force-field parameters, initial equilibration, and MD simulation protocols, we refer to our previous work (12).
Entropy calculations
Configurational entropy calculations were performed following the formulation by Schlitter (63), which provides an approximate (71) upper bound to the absolute entropy S:
 |
(1) |
where kB is Boltzmann's constant, T the absolute temperature, e Euler's number, ℏ Planck's constant divided by 2π, M the 3N-dimensional diagonal matrix containing the N atomic masses of the solute atoms for which the entropy is calculated, and 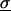 the covariance matrix of atom-positional fluctuations with the elements:
the covariance matrix of atom-positional fluctuations with the elements:
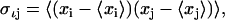 |
(2) |
where xi are the Cartesian coordinates of the atoms considered in the entropy calculation after a least-squares fit of the trajectory configurations using a particular subset of atoms. As an additional test, configurational entropies were alternatively calculated following the procedure of Andricioaei and Karplus (67) (data not reported). Resulting estimates of the configurational entropy from the two formulations differ from each other by <2%, similar to what has been observed in the case of reversibly folding peptides in solution (71), for flexible hydrocarbon chains (75), lipids (74), and rigid organic molecules in water (73). Entropy calculations were performed on trajectory structures saved every 5 ps.
To evaluate the configurational entropies, molecular configurations were superimposed via a translational superposition of centers of mass and a rotational least-squares fit (86), thus excluding overall translational and rotational motion from the calculation of the configurational entropy (64). This yields an internal configurational entropy (code i) or an internal configurational entropy per particle (code ip) (the former divided by the number N of particles used to calculate the covariance matrix defined in Eq. 2). Three different sets of atoms were used to remove overall translational and rotational degrees of freedom of the solute (Table 1), to verify the influence of the subsets of atoms used for fitting on the final entropy estimates.
All nonhydrogen atoms of the ligand under consideration (code nh).
Four atoms (N, H, C, and O) of the peptide bond in the central body of the ligand molecules (code 4). In Fig. 1, the corresponding atoms are marked with an asterisk.
Nonhydrogen atoms of the central GAAAAAC/GTTTTTC segment of the corresponding d(CGCGAAAAACGCG)·d(CGCGTTTTTCGCG) DNA duplex (code DNA).
Next to the configurational entropies of the ligands, configurational entropies of subsets of atoms denoted as tail 1 (t1), tail 2 (t2), tails (t), and body (b) (see Fig. 1) were also calculated. The subset of atoms named tails (t) includes all the atoms of tail 1 and tail 2.
Estimated configurational entropies are referenced using the notation 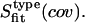 The code cov refers to the atoms for which the covariance matrix is calculated, and thus defines the atoms for which an upper bound to the entropy is calculated (nh, t1, t2, t, b). The code fit indicates the atoms for which the center of mass superposition and least-squares fit of the configurations of the trajectory is performed (nh, 4, DNA). The code type refers to the type of entropy calculated (i, ip). For code definitions, see Table 1.
The code cov refers to the atoms for which the covariance matrix is calculated, and thus defines the atoms for which an upper bound to the entropy is calculated (nh, t1, t2, t, b). The code fit indicates the atoms for which the center of mass superposition and least-squares fit of the configurations of the trajectory is performed (nh, 4, DNA). The code type refers to the type of entropy calculated (i, ip). For code definitions, see Table 1.
The decrease in entropy due to correlation in the motions of two subsets of atoms—for example, those represented by the body (b) and tails (t)—can be estimated (65) as
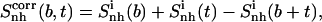 |
(3) |
where the entropy 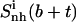 (i.e.,
(i.e., 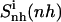 ) includes all correlations between the atoms in the subsets b and t, and the type and fit used are the same in the calculations of the three terms.
) includes all correlations between the atoms in the subsets b and t, and the type and fit used are the same in the calculations of the three terms.
Entropy differences between bound and free states for each ligand were estimated, for example, for nonhydrogen atoms (nh) as
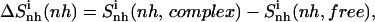 |
(4) |
and represent a change in internal entropy of the ligand upon binding to DNA. The codes complex and free refer to the bound and free simulations of the ligand, respectively.
RESULTS AND DISCUSSION
Conformational analysis of DNA
Fig. 2 shows the time series of Watson-Crick hydrogen bonds between the base pairs for both netropsin-DNA and distamycin-DNA complexes and the resulting cumulative occurrences. In the first complex (upper panel), the hydrogen bonds between pairs of bases are well preserved over the whole binding site. During 10 ns of this simulation, the bases of the binding site remain hydrogen-bonded >70% of the time. In the case of the distamycin-DNA complex (lower panel), Watson-Crick hydrogen bonds at the termini of the double helix are distributed differently along the bases, reflecting the structural difference of this second ligand. In the part of the DNA binding site where the structure of tail 2 of distamycin differs from the structure of tail 2 of netropsin (see Fig. 1), some of the Watson-Crick hydrogen bonds occur <50% of the simulation time. The adenine bases in the AT base pairs near tail 2 of distamycin tend to move slightly outward from the minor groove without fulfilling the hydrogen-bond criterion. Nevertheless, the MD trajectories show that the DNA double-helix geometry is well-preserved for both ligand-DNA complexes. As is reported in other studies (38,87), the central part of the DNA double helix is found to be more stable than the termini. Interestingly, for the first CG base pair in the netropsin-DNA complex and for the last GC base pair in the distamycin-DNA complex, the corresponding time series show reversible hydrogen-bonding along the 10 ns of simulation. Time series of the Watson-Crick hydrogen bonds systematically show that the 45A4 GROMOS force field captures the correct hydrogen-bond formation along the simulation.
FIGURE 2.

Watson-Crick hydrogen bonds along the sequence of the primary DNA strand in netropsin-DNA (upper panel) and distamycin-DNA (lower panel). Time series of their occurrence and the corresponding cumulative values are displayed based on 10-ns MD simulations. Hydrogen bonds close to tail 2 of netropsin and distamycin (schematically represented) are in gray. Hydrogen bonds are defined to have a maximum hydrogen–acceptor distance of 0.25 nm and a minimum donor-hydrogen-acceptor angle of 135°.
The atom-positional root-mean-square deviations (RMSD) of the nonhydrogen atoms in the central GAAAAAC/GTTTTTC segment from the initial DNA structure remain in the range 0.2–0.4 nm (the highest values of 0.38 nm and 0.34 nm were for netropsin and distamycin, respectively, complexed with DNA; data not shown), which are reasonable deviations considering the size of the DNA molecules. For the base pair atoms these values are reduced to 0.26 nm in the netropsin-DNA complex and to 0.22 nm in the distamycin-DNA complex. The backbone atoms deviate slightly more from the starting structure (i.e., 0.39 nm in the netropsin-DNA complex and 0.36 nm in the distamycin-DNA complex). However, no large-scale changes in the conformation of the DNA double helix, particularly in the geometry of the minor groove, were observed for either of the complexes, demonstrating suitability of the simulated trajectories for the estimation of the configurational entropy changes of ligands upon their binding to the DNA minor groove.
Configurational entropy of netropsin and distamycin
For netropsin and distamycin free in solution and complexed to DNA, Fig. 3 shows the convergence properties of 1), internal configurational entropy 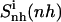 and
and 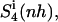 and 2), the relative motions between ligand and DNA
and 2), the relative motions between ligand and DNA 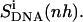 Most (99%) of the final internal configurational entropy estimate
Most (99%) of the final internal configurational entropy estimate 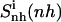 was collected within 83% of the simulation time for the netropsin-DNA complex and within 45% of the simulation time for the distamycin-DNA complex. For the ligands in their free state, 99% of
was collected within 83% of the simulation time for the netropsin-DNA complex and within 45% of the simulation time for the distamycin-DNA complex. For the ligands in their free state, 99% of 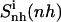 was reached faster, i.e., within 56% of the simulation time for netropsin and within 31% of the simulation time for distamycin. All curves are characterized by rapid increases in the build-up corresponding to structural changes of the ligands. These stepwise increases are more pronounced for distamycin than for netropsin. The corresponding structural changes are reflected in the atom-positional RMSD of the ligand from the starting structure along the DNA-distamycin simulation (Fig. 4).
was reached faster, i.e., within 56% of the simulation time for netropsin and within 31% of the simulation time for distamycin. All curves are characterized by rapid increases in the build-up corresponding to structural changes of the ligands. These stepwise increases are more pronounced for distamycin than for netropsin. The corresponding structural changes are reflected in the atom-positional RMSD of the ligand from the starting structure along the DNA-distamycin simulation (Fig. 4).
FIGURE 3.

Configurational entropy per atom of netropsin (A) and distamycin (B) free in solution (thin lines) and when bound to DNA (thick lines) calculated for nonhydrogen atoms. Configurational entropy was estimated for each ligand after removal of overall translation and rotation using all nonhydrogen atoms (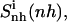 solid line) or using only four atoms of the central CO-NH peptide bond (
solid line) or using only four atoms of the central CO-NH peptide bond (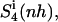 dashed line), and after a translational superposition of centers of mass and a rotational least-squares fit using the nonhydrogen atoms of the central GAAAAAC/GTTTTTC segment (
dashed line), and after a translational superposition of centers of mass and a rotational least-squares fit using the nonhydrogen atoms of the central GAAAAAC/GTTTTTC segment (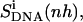 dot-dashed line) of the DNA duplex. The arrows point to the first and second rapid increases in entropy for distamycin.
dot-dashed line) of the DNA duplex. The arrows point to the first and second rapid increases in entropy for distamycin.
FIGURE 4.

Atom-positional RMSD of ligand trajectory structures from the initial structures along 10-ns simulations of the ligand-DNA complexes. Time series are calculated using all nonhydrogen atoms and atom subgroups for tail 1, tail 2, and the body (see Fig. 1).
Fig. 4 shows the nonhydrogen atom-positional RMSD for 1), the entire netropsin and distamycin molecules when bound to DNA, 2), their bodies, and 3), both of their tails. It can be seen that during the simulation no large structural changes occur in the netropsin molecule, whereas the main structural changes in distamycin appear in the formamide tail 2 of this molecule. During the simulation, the torsion angle between tail 2 and the body of distamycin fluctuates so that the plane of pyrrole ring of tail 2 moves out of the plane formed by the two pyrrole rings in the body of this ligand. It is obvious that the large conformational changes observed in the RMSD of distamycin correlate with the jumps in entropy build-up in Fig. 3. The configurations of the distamycin molecule that correspond to the increases in RMSD and configurational entropy are shown in Fig. 5. The changes in distamycin tail 2 also slightly affect the configuration of its body and can be correlated with the structural changes in the binding site of the DNA double helix in the complex, as observed in the analysis of Watson-Crick hydrogen bonds (see Fig. 2). For both ligands, two characteristic orientations of tail 1 can be observed. In particular, tails of netropsin and distamycin flip between two configurations with almost perpendicular relative orientation of the terminal propylamidine group (see also Fig. 5). Similar behavior can be observed for tail 2 of netropsin.
FIGURE 5.

Relative motion of distamycin inside the DNA minor groove. Three representative snapshots of distamycin from the 10-ns simulation of the distamycin-DNA complex are shown superimposed after least-square fitting: initial conformation (black), and conformations corresponding to the first (blue) and second (red) rapid increase in configurational entropy (see arrows in Figs. 3 and 6).
Configurational entropy estimates for the free and bound simulations of netropsin and distamycin are reported in Table 2. The internal configurational entropy 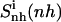 of netropsin free in solution is 862 J K−1 mol−1 (28 J K−1 mol−1 per atom) and is reduced to 735 J K−1 mol−1 (24 J K−1 mol−1 per atom) upon binding. Correspondingly, for distamycin, the internal configurational entropy
of netropsin free in solution is 862 J K−1 mol−1 (28 J K−1 mol−1 per atom) and is reduced to 735 J K−1 mol−1 (24 J K−1 mol−1 per atom) upon binding. Correspondingly, for distamycin, the internal configurational entropy 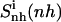 amounts to 902 J K−1 mol−1 (26 J K−1 mol−1 per atom) and is reduced to 798 J K−1 mol−1 (23 J K−1 mol−1 per atom) upon binding to the minor groove of DNA. The change in internal configurational entropy
amounts to 902 J K−1 mol−1 (26 J K−1 mol−1 per atom) and is reduced to 798 J K−1 mol−1 (23 J K−1 mol−1 per atom) upon binding to the minor groove of DNA. The change in internal configurational entropy 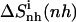 for the netropsin molecule thus amounts to −127 J K−1 mol−1 (−4 J K−1 mol−1 per atom). In the case of distamycin, the internal configurational entropy change is slightly smaller, i.e., −104 J K−1 mol−1 (−3 J K−1 mol−1 per atom).
for the netropsin molecule thus amounts to −127 J K−1 mol−1 (−4 J K−1 mol−1 per atom). In the case of distamycin, the internal configurational entropy change is slightly smaller, i.e., −104 J K−1 mol−1 (−3 J K−1 mol−1 per atom).
TABLE 2.
Configurational entropies of netropsin and distamycin when free in solution and when bound to the minor groove of the DNA duplex, and corresponding configurational entropy changes upon binding
| Free in solution
|
In complex with DNA
|
Binding
|
|||||
|---|---|---|---|---|---|---|---|
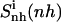 |
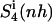 |
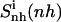 |
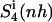 |
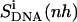 |
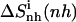 |
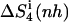 |
|
| Netropsin | 862 (28) | 985 (32) | 735 (24) | 886 (29) | 1014 (33) | −127 (−4) | −99 (−3) |
| Distamycin | 902 (26) | 1036 (29) | 798 (23) | 953 (27) | 1133 (34) | −104 (−3) | −83 (−2) |
Calculated type of entropy, and subsets of atoms used in the entropy calculations and in the least-squares fitting procedures, are referenced using the codes defined in Table 1. The configurational entropy differences between the free and bound forms of the ligands are calculated using Eq. 4. Per-atom entropies are given in parentheses. All values are in J K−1 mol−1.
To capture the rotational motions of the ligand complexed to the DNA minor groove, the mass-weighted covariance matrix of atom-positional fluctuations was calculated after fitting only four atoms (of the central peptide bond in the ligand; code 4) of the trajectory structures. This procedure yields estimates of the configurational entropy 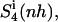 which contains contributions from the relative rotation of the ligand with respect to the initial structure. Most (99%) of the final entropy estimate
which contains contributions from the relative rotation of the ligand with respect to the initial structure. Most (99%) of the final entropy estimate 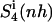 was reached within 79% of the simulation time for the netropsin-DNA complex and within 43% of the simulation time for the distamycin-DNA complex. For netropsin and distamycin free in solution, the corresponding values were reached within 48% and 36% of the simulation time, respectively. The values of
was reached within 79% of the simulation time for the netropsin-DNA complex and within 43% of the simulation time for the distamycin-DNA complex. For netropsin and distamycin free in solution, the corresponding values were reached within 48% and 36% of the simulation time, respectively. The values of 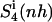 (see Table 2) are expected (and found) to be comparatively higher than those for the internal configurational entropy
(see Table 2) are expected (and found) to be comparatively higher than those for the internal configurational entropy 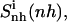 because the rotation of the ligand is partially sampled in the entropy calculations. The value of
because the rotation of the ligand is partially sampled in the entropy calculations. The value of 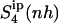 for netropsin when free in solution is 32 J K−1 mol−1 and is reduced to 29 J K−1 mol−1 upon binding of the ligand to DNA. In the case of distamycin, the resulting values of
for netropsin when free in solution is 32 J K−1 mol−1 and is reduced to 29 J K−1 mol−1 upon binding of the ligand to DNA. In the case of distamycin, the resulting values of 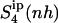 are slightly lower, i.e., 29 J K−1 mol−1 for distamycin free in solution and 27 J K−1 mol−1 for distamycin in complex with DNA. The ranking of absolute configurational entropies and relative entropies of binding thus remains unchanged, and the contribution of rotational motion seems to influence the two ligands similarly.
are slightly lower, i.e., 29 J K−1 mol−1 for distamycin free in solution and 27 J K−1 mol−1 for distamycin in complex with DNA. The ranking of absolute configurational entropies and relative entropies of binding thus remains unchanged, and the contribution of rotational motion seems to influence the two ligands similarly.
Relative motions of the ligands with respect to DNA may be captured from the calculations of the mass-weighted covariance matrix after a configurational superposition procedure based on nonhydrogen atoms of the central bases GAAAAAC/GTTTTTC of the DNA duplex (code DNA). Resulting values 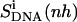 reported in Table 2 are higher than the internal configurational entropies
reported in Table 2 are higher than the internal configurational entropies 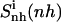 in which the nonhydrogen atoms of the ligands were used in the fitting procedures. Most (99%) of the final entropy estimate
in which the nonhydrogen atoms of the ligands were used in the fitting procedures. Most (99%) of the final entropy estimate 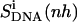 was reached within 99% of the simulation time for netropsin-DNA and within 41% of the simulation time for the distamycin-DNA complex. The corresponding time series (Fig. 3) display evident stepwise increases, particularly rapid in the case of distamycin bound to DNA, which samples repeatedly new regions of its conformational space in the first part of the simulation. Similar conclusions can be drawn for internal configurational entropy estimates of distamycin bound to DNA when sampled using the fitting of nonhydrogen atoms of the ligand (
was reached within 99% of the simulation time for netropsin-DNA and within 41% of the simulation time for the distamycin-DNA complex. The corresponding time series (Fig. 3) display evident stepwise increases, particularly rapid in the case of distamycin bound to DNA, which samples repeatedly new regions of its conformational space in the first part of the simulation. Similar conclusions can be drawn for internal configurational entropy estimates of distamycin bound to DNA when sampled using the fitting of nonhydrogen atoms of the ligand (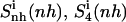 ).
).
The changes in configurational entropy of the ligands upon binding to the minor groove of the DNA duplex (CGCGAAAAACGCG)·d(CGCGTTTTTCGCG) show that netropsin loses more internal configurational entropy than distamycin upon binding. The calculated differences (Eq. 4) are in the range of estimated rotational and translational entropy differences reported in the literature (i.e., 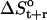 = −0.21(± 0.04) kJ K−1 mol−1) (49,50). The magnitude of these contributions is significant when compared to the total binding free energies accompanying minor groove binding. Recently reported standard free energies of binding of netropsin and distamycin to various DNA sequences obtained from ultraviolet melting and isothermal titration calorimetry experiments range from −39.7 kJ mol−1 for binding of netropsin to the 5′-AAGTT-3′ binding site to −54.0 kJ mol−1 for binding of netropsin to the 5′-AAAAA-3′ binding site (7). Larger configurational entropic cost in the case of netropsin binding to DNA may be the consequence of stronger electrostatic and van der Waals interactions holding netropsin, as compared to distamycin, more tightly in the minor groove. Additionally, we note that netropsin contains more rotatable bonds than distamycin, which may lead to a larger reduction of conformational freedom upon binding to the DNA minor groove. We note, however, that 1), experimentally the configurational entropy loss is sequence-specific and may significantly vary depending on the DNA base pair sequence; 2), this study does not attempt to calculate configurational entropy (and its differences) for the DNA double helix (this would require significantly longer simulations); 3), the entropy (and its differences) of the diffusive solvent water molecules were not examined in this study due to the intrinsic limitation of the Schlitter and quasiharmonic approaches to nondiffusive systems (63,64,71); and 4), the configurational entropies estimated are upper bounds to the true entropy of the simulated system (63,71).
= −0.21(± 0.04) kJ K−1 mol−1) (49,50). The magnitude of these contributions is significant when compared to the total binding free energies accompanying minor groove binding. Recently reported standard free energies of binding of netropsin and distamycin to various DNA sequences obtained from ultraviolet melting and isothermal titration calorimetry experiments range from −39.7 kJ mol−1 for binding of netropsin to the 5′-AAGTT-3′ binding site to −54.0 kJ mol−1 for binding of netropsin to the 5′-AAAAA-3′ binding site (7). Larger configurational entropic cost in the case of netropsin binding to DNA may be the consequence of stronger electrostatic and van der Waals interactions holding netropsin, as compared to distamycin, more tightly in the minor groove. Additionally, we note that netropsin contains more rotatable bonds than distamycin, which may lead to a larger reduction of conformational freedom upon binding to the DNA minor groove. We note, however, that 1), experimentally the configurational entropy loss is sequence-specific and may significantly vary depending on the DNA base pair sequence; 2), this study does not attempt to calculate configurational entropy (and its differences) for the DNA double helix (this would require significantly longer simulations); 3), the entropy (and its differences) of the diffusive solvent water molecules were not examined in this study due to the intrinsic limitation of the Schlitter and quasiharmonic approaches to nondiffusive systems (63,64,71); and 4), the configurational entropies estimated are upper bounds to the true entropy of the simulated system (63,71).
Classical molecular dynamics force fields are often based on atomic models, in which each atom is represented by one interaction site, with the exception of aliphatic groups, for which the C-atom and bound H-atoms are treated as one interaction site (38,79). This united-atom simplification has been shown to reproduce the properties of n-alkanes as accurately as all-atom (i.e., including explicit aliphatic H-atoms) force fields (88). In this study, the aliphatic hydrogen atoms of the ligands were treated with the united atom model, whereas all remaining atoms were treated explicitly. To investigate the effect of hydrogen atoms on entropy estimates, the calculations have been repeated alternatively including nonaliphatic hydrogen atoms (16 for netropsin out of 47 total; 15 for distamycin out of 50 total). This leads to slightly larger values of internal configurational entropies (i.e., 997 and 1022 J K−1 mol−1 for netropsin and distamycin, respectively, free in solution, and 853 and 903 J K−1 mol−1 for netropsin and distamycin, respectively, in complex with DNA). Of course, the per-atom weighted values slightly decrease (the contribution of nonaliphatic hydrogen atoms to the configurational entropy is 16% for netropsin and 13% for distamycin both free in solution and when bound to DNA).
Configurational entropies of the subgroups
The flexibility of the tails of minor groove binders is an important element of ligand-DNA recognition (48). To investigate this aspect, the atoms of the ligands were divided into three subgroups, the body (b), tail 1 (t1) and tail 2 (t2). For each subset, the internal configurational entropies were estimated. The entropy contributions from the subgroups, as well as the entropy of the entire ligands, are presented in Fig. 6 for netropsin and distamycin. The corresponding results are reported in Table 3. Most (99%) of the final entropy estimates for tail 1 and tail 2 of the ligands complexed to DNA were reached in 85% and 75% of the simulation time for netropsin and 50% and 38% for distamycin. The corresponding values for the ligands free in solution are considerably lower (i.e., 31% and 13% of the simulation time for tail 1 and tail 2 of netropsin, and 32% and 10% of the simulation time for tail 1 and tail 2 of distamycin). For the more rigid body of the ligands in their free and bound forms, 99% of the final entropy estimate was always collected within 50% of the simulation time (i.e., within 30% and 37% for netropsin and distamycin in complex with the DNA, and within 44% and 15% for the ligands free in solution). Estimates of configurational entropy obtained for different subgroups range from 21 J K−1mol−1 to 47 J K−1mol−1, reflecting diverse flexibility of the subgroups. The configurational entropy of the body of both ligands 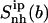 in their free states amounts to 24 J K−1 mol−1 and is reduced upon binding to 21 J K−1 mol−1 for netropsin and to 22 J K−1 mol−1 for distamycin. The body of both ligands is expected (and found) to be considerably more rigid than the corresponding tails. Configurational entropies of tail 1 and tail 2,
in their free states amounts to 24 J K−1 mol−1 and is reduced upon binding to 21 J K−1 mol−1 for netropsin and to 22 J K−1 mol−1 for distamycin. The body of both ligands is expected (and found) to be considerably more rigid than the corresponding tails. Configurational entropies of tail 1 and tail 2, 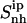 (t1) and
(t1) and 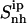 (t2), of netropsin free in solution are 45 and 42 J K−1 mol−1, respectively. In the case of distamycin, the corresponding values are 47 and 30 J K−1 mol−1, indicating the difference in flexibility of tail 2 of the investigated molecules. Upon binding, the per-atom configurational entropies of tail 1 and tail 2 of netropsin are both reduced to 36 J K−1 mol−1. For distamycin, the configurational entropy is reduced to 40 J K−1 mol−1 for tail 1 and to 27 J K−1 mol−1 for tail 2. The entropy changes of specific subgroups upon binding to the minor groove can be calculated (see Materials and Methods). Tail 1 of netropsin loses 9 J K−1 mol−1 of internal configurational entropy per atom and tail 2 loses 6 J K−1 mol−1 per atom upon binding. Tail 1 of distamycin loses 7 J K−1 mol−1 per atom and tail 2 loses 3 J K−1 mol−1 per atom, respectively. The internal entropic cost for the body of the ligand molecule
(t2), of netropsin free in solution are 45 and 42 J K−1 mol−1, respectively. In the case of distamycin, the corresponding values are 47 and 30 J K−1 mol−1, indicating the difference in flexibility of tail 2 of the investigated molecules. Upon binding, the per-atom configurational entropies of tail 1 and tail 2 of netropsin are both reduced to 36 J K−1 mol−1. For distamycin, the configurational entropy is reduced to 40 J K−1 mol−1 for tail 1 and to 27 J K−1 mol−1 for tail 2. The entropy changes of specific subgroups upon binding to the minor groove can be calculated (see Materials and Methods). Tail 1 of netropsin loses 9 J K−1 mol−1 of internal configurational entropy per atom and tail 2 loses 6 J K−1 mol−1 per atom upon binding. Tail 1 of distamycin loses 7 J K−1 mol−1 per atom and tail 2 loses 3 J K−1 mol−1 per atom, respectively. The internal entropic cost for the body of the ligand molecule 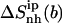 upon binding to DNA is 3 and 2 J K−1 mol−1 for netropsin and distamycin, respectively. Comparison of entropy changes in tails and body of both ligands reveals that the highest contributions to the entropy of binding come from the restriction in the flexibility of the ligand tails. The loss of internal configurational entropy for the (structurally equal) body and tail 1 of the ligands is comparable for both ligands, whereas the entropic loss of tail 2 is higher for the more flexible tail of netropsin. Furthermore, in the build-up of the entropy curves for distamycin bound to DNA (Fig. 6), the stepwise increases in the internal configurational entropy of tail 2 corresponding to the already mentioned structural changes in the ligand (Figs. 4 and 5) can again be observed.
upon binding to DNA is 3 and 2 J K−1 mol−1 for netropsin and distamycin, respectively. Comparison of entropy changes in tails and body of both ligands reveals that the highest contributions to the entropy of binding come from the restriction in the flexibility of the ligand tails. The loss of internal configurational entropy for the (structurally equal) body and tail 1 of the ligands is comparable for both ligands, whereas the entropic loss of tail 2 is higher for the more flexible tail of netropsin. Furthermore, in the build-up of the entropy curves for distamycin bound to DNA (Fig. 6), the stepwise increases in the internal configurational entropy of tail 2 corresponding to the already mentioned structural changes in the ligand (Figs. 4 and 5) can again be observed.
FIGURE 6.

Internal entropy per atom calculated for nonhydrogen atoms of the entire ligand molecules (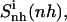 solid line), and of the subgroups tail 1 (
solid line), and of the subgroups tail 1 (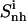 (t1), dotted line), tail 2 (
(t1), dotted line), tail 2 (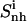 (t2), dashed line), and body (
(t2), dashed line), and body (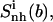 dot-dashed line) for netropsin free in solution (A) and when bound to DNA (B), and for distamycin free in solution (C) and when bound to DNA (D). The arrows point to the first and second rapid increases in entropy for distamycin (compare to Fig. 3).
dot-dashed line) for netropsin free in solution (A) and when bound to DNA (B), and for distamycin free in solution (C) and when bound to DNA (D). The arrows point to the first and second rapid increases in entropy for distamycin (compare to Fig. 3).
TABLE 3.
Internal configurational entropies of atom subgroups of netropsin and distamycin free in solution and bound to DNA, and correlations between their body and tails
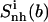 |
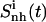 |
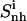 (t1) (t1) |
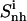 (t2) (t2) |
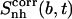 |
|
|---|---|---|---|---|---|
| Netropsin (free) | 384 (24) | 623 (41) | 363 (45) | 297 (42) | 157 |
| Netropsin (bound) | 333 (21) | 513 (34) | 290 (36) | 252 (36) | 113 |
| Δ (bound − free) | −51 (−3) | −110 (−7) | −73 (−9) | −45 (−6) | −44 |
| Distamycin (free) | 393 (24) | 651 (34) | 372 (47) | 328 (30) | 145 |
| Distamycin (bound) | 354 (22) | 568 (30) | 316 (40) | 296 (27) | 122 |
| Δ (bound − free) | −39 (−2) | −83 (−4) | −56 (−7) | −32 (−3) | −23 |
Corresponding changes upon binding are also reported (Δ). The body and tails of the ligands are represented in Fig. 1; Table 1 reports the reference codes. Only nonhydrogen atoms were used in the calculations. Least-squares superposition of structures was done using all nonhydrogen atoms. Per-atom entropies are given in parentheses. Correlation entropy 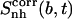 was calculated using Eq. 3. All values are in J K−1 mol−1.
was calculated using Eq. 3. All values are in J K−1 mol−1.
Correlation effects
It is evident that the internal configurational entropies calculated for the subsets of atoms of a ligand do not add up to the total entropy of the ligand (see Tables 2 and 3). The correlation between the motion of the body and the tails, 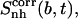 of the ligands can thus be obtained (Eq. 3). The differences in entropy due to correlation in the motion between the tails and the body for netropsin and distamycin in their bound and free states are reported in the last column of Table 3. The value of
of the ligands can thus be obtained (Eq. 3). The differences in entropy due to correlation in the motion between the tails and the body for netropsin and distamycin in their bound and free states are reported in the last column of Table 3. The value of 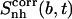 upon binding reduces from 157 J K−1 mol−1 to 113 J K−1 mol−1 (netropsin) and from 145 to 122 J K−1 mol−1 (distamycin). The difference in correlation between the tails and the central part of netropsin when bound to DNA and when free in solution amounts to -44 J K−1 mol−1. In the case of distamycin, the corresponding difference is smaller (i.e., −23 J K−1 mol−1), which is a consequence of greater flexibility of netropsin when compared to distamycin. Thus, in the latter case, the change in correlation upon binding is smaller.
upon binding reduces from 157 J K−1 mol−1 to 113 J K−1 mol−1 (netropsin) and from 145 to 122 J K−1 mol−1 (distamycin). The difference in correlation between the tails and the central part of netropsin when bound to DNA and when free in solution amounts to -44 J K−1 mol−1. In the case of distamycin, the corresponding difference is smaller (i.e., −23 J K−1 mol−1), which is a consequence of greater flexibility of netropsin when compared to distamycin. Thus, in the latter case, the change in correlation upon binding is smaller.
CONCLUSION
Upon binding of a ligand to the minor groove of DNA, the translational, rotational, and internal motion of the ligand is reduced. The entropic cost the ligand pays depends on the specific chemical characteristics of the ligand itself and of the DNA binding sequence. Here, the changes in configurational entropy of netropsin and distamycin upon complex formation with the DNA duplex d(CGCGAAAAACGCG)·d(CGCGTTTTTCGCG) were estimated. The contribution of internal configurational entropy loss in the ligand is generally omitted in the analysis of the experimental binding data, since minor groove binding does not require significant changes in DNA or ligand conformation. This study shows that netropsin and distamycin ligands lose a considerable amount of internal configurational entropy upon binding to the minor groove. In particular, the number of conformations that are available to the tails of the ligands becomes small upon complex formation, consequently lowering the corresponding configurational entropy upper bounds. It is found that netropsin loses more entropy upon binding than distamycin. We have shown that internal entropy changes that occur upon binding of netropsin and distamycin to the DNA minor groove can be estimated on a 10-ns timescale using Schlitter's approximation and the GROMOS 45A4 force field. The configurational entropy changes calculated in this work can be used in the interpretation of minor-groove binding phenomena and can improve the thermodynamic description and understanding of the binding of small ligands to the minor groove of DNA.
Acknowledgments
J.D. thanks the Federal Commission for Scholarships for Foreign Students (FCS) of the Swiss Government for a fellowship during the academic year 2004/05 and the Slovenian Ministry of Education, Science and Sports (grant no. P1-0201). Financial support from the National Center of Competence in Research (NCCR) in Structural Biology of the Swiss National Science Foundation (SNSF) is gratefully acknowledged.
References
- 1.Rentzeperis, D., and L. A. Marky. 1995. Interaction of minor groove ligands to an AAATT/AATTT site: correlation of thermodynamic characterization and solution structure. Biochemistry. 34:2937–2945. [DOI] [PubMed] [Google Scholar]
- 2.Haq, I., J. E. Ladbury, B. Z. Chowdhry, T. C. Jenkins, and J. B. Chaires. 1997. Specific binding of Hoechst 33258 to the d(CGCAAATTTGCG)2 duplex: calorimetric and spectroscopic studies. J. Mol. Biol. 271:244–257. [DOI] [PubMed] [Google Scholar]
- 3.Ren, J., T. C. Jenkins, and J. B. Chaires. 2000. Energetics of DNA intercalation reactions. Biochemistry. 39:8439–8447. [DOI] [PubMed] [Google Scholar]
- 4.Lah, J., and G. Vesnaver. 2000. Binding of distamycin A and netropsin to the 12mer DNA duplexes containing mixed AT-GC sequences with at most five or three successive AT base pairs. Biochemistry. 39:9317–9327. [DOI] [PubMed] [Google Scholar]
- 5.Wang, L., A. Kumar, D. W. Boykin, C. Bailly, and W. D. Wilson. 2002. Comparative thermodynamics for monomer and dimer sequence-dependent binding of a heterocyclic dication in the DNA minor groove. J. Mol. Biol. 317:361–374. [DOI] [PubMed] [Google Scholar]
- 6.Leng, F., J. B. Chaires, and J. Waring. 2003. Energetics of echinomycin binding to DNA. Nucleic Acids Res. 31:6191–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lah, J., and G. Vesnaver. 2004. Energetic diversity of DNA minor-groove recognition by small molecules displayed through some model ligand-DNA systems. J. Mol. Biol. 342:73–89. [DOI] [PubMed] [Google Scholar]
- 8.Singh, S. B., and P. A. Kollman. 1999. Calculating the absolute free energy of association of netropsin and DNA. J. Am. Chem. Soc. 121:3267–3271. [Google Scholar]
- 9.Harris, S. A., E. Gavathiotis, M. S. Searle, M. Orozco, and C. A. Laughton. 2001. Cooperativity in drug-DNA recognition: a molecular dynamics study. J. Am. Chem. Soc. 123:12658–12663. [DOI] [PubMed] [Google Scholar]
- 10.Špačková, N., T. E. Cheatham III, F. Ryjáček, F. Lankaš, L. van Meervelt, P. Hobza, and J. Šponer. 2003. Molecular dynamics simulations and thermodynamics analysis of DNA-drug complexes. Minor groove binding between 4′,6-diamidino-2-phenylindole and DNA duplexes in solution. J. Am. Chem. Soc. 125:1759–1769. [DOI] [PubMed] [Google Scholar]
- 11.Korolev, N., A. P. Lyubartsev, A. Laaksonen, and L. Nordenskiöld. 2004. Molecular dynamics simulation study of oriented polyamine- and Na-DNA: sequence specific interactions and effects on DNA structure. Biopolymers. 73:542–555. [DOI] [PubMed] [Google Scholar]
- 12.Dolenc, J., C. Oostenbrink, J. Koller, and W. F. van Gunsteren. 2005. Molecular dynamics simulations and free energy calculations of netropsin and distamycin binding to an AAAAA DNA binding site. Nucleic Acids Res. 33:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge, W., B. Schneider, and W. Olson. 2005. Knowledge-based elastic potentials for docking drugs or proteins with nucleic acids. Biophys. J. 88:1166–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, X., B. Ramakrishnan, S. T. Rao, and M. Sundaralingam. 1994. Binding of two distamycin A molecules in the minor groove of an alternating B-DNA duplex. Nat. Struct. Biol. 1:169–175. [DOI] [PubMed] [Google Scholar]
- 15.Goodsell, D. S., M. L. Kopka, and R. E. Dickerson. 1995. Refinement of netropsin bound to DNA: bias and feedback in electron density map interpretation. Biochemistry. 34:4983–4993. [DOI] [PubMed] [Google Scholar]
- 16.Vlieghe, D., J. Šponer, and L. van Meervelt. 1999. Crystal structure of d(GGCCAATTGG) complexed with DAPI reveals novel binding mode. Biochemistry. 38:16443–16451. [DOI] [PubMed] [Google Scholar]
- 17.Uytterhoeven, K., J. Šponer, and L. van Meervelt. 2002. Two 1:1 binding modes for distamycin in the minor groove of d(GGCCAATTGG). Eur. J. Biochem. 269:2868–2877. [DOI] [PubMed] [Google Scholar]
- 18.Pelton, J. G., and D. E. Wemmer. 1989. Structural characterization of a 2:1 distamycin A-d(CGCAAATTGGC) complex by two-dimensional NMR. Proc. Natl. Acad. Sci. USA. 86:5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelton, J. G., and D. E. Wemmer. 1990. Binding modes of distamycin-A with d(CGCAAATTTGCG)2 determined by 2-dimensional NMR. J. Am. Chem. Soc. 112:1393–1399. [Google Scholar]
- 20.Gavathiotis, E., G. J. Sharman, and M. S. Searle. 2000. Sequence-dependent variation in DNA minor groove width dictates orientational preference of Hoechst 33258 in A-tract recognition: solution NMR structure of the 2:1 complex with d(CTTTTGCAAAAG)2. Nucleic Acids Res. 28:728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urbach, A. R., J. L. Love, S. A. Ross, and P. B. Dervan. 2002. Structure of a β-alanine-linked polyamide bound to a full helical turn of purine tract DNA in a 1:1 motif. J. Mol. Biol. 320:55–71. [DOI] [PubMed] [Google Scholar]
- 22.Anthony, N. G., B. F. Johnston, A. I. Khalaf, S. P. MacKay, J. A. Parkinson, C. J. Suckling, and R. D. Waigh. 2004. Short lexitropsin that recognizes the DNA minor groove at 5′-ACTAGT-3′: understanding the role of isopropyl-thiazole. J. Am. Chem. Soc. 126:11338–11349. [DOI] [PubMed] [Google Scholar]
- 23.Pindur, U., and G. Fischer. 1996. DNA complexing minor groove-binding ligands: perspectives in antitumour and antimicrobial drug design. Curr. Med. Chem. 3:379–406. [Google Scholar]
- 24.Kaizerman, J. A., M. I. Gross, Y. Ge, S. White, W. Hu, J. X. Duan, E. E. Baird, K. W. Johnson, R. D. Tanaka, H. E. Moser, and R. W. Bürli. 2003. DNA binding ligands targeting drug-resistant bacteria: structure, activity and pharmacology. J. Med. Chem. 46:3914–3929. [DOI] [PubMed] [Google Scholar]
- 25.Dias, N., U. Jacquemard, B. Bladeyrou, C. Tardy, A. Lansiaux, P. Colson, F. Tanious, W. D. Wilson, S. Routier, J. Y. Mérour, and C. Bailly. 2004. Targeting DNA with novel diphenylcarbazoles. Biochemistry. 43:15169–15178. [DOI] [PubMed] [Google Scholar]
- 26.Chaires, J. B. 1997. Energetics of drug-DNA interactions. Biopolymers. 44:201–215. [DOI] [PubMed] [Google Scholar]
- 27.Hopkins, H. P., M. Yang, W. D. Wilson, and D. W. Boykin. 1991. Intercalation binding of 6-substituted naphthothiopheneamides to DNA: enthalpy and entropy components. Biopolymers. 31:1105–1114. [DOI] [PubMed] [Google Scholar]
- 28.Breslauer, K. J., D. P. Remeta, W. Y. Chou, R. Ferrante, J. Curry, D. Zaunczkowski, J. G. Snyder, and L. A. Marky. 1987. Enthalpy-entropy compensations in drug-DNA binding studies. Proc. Natl. Acad. Sci. USA. 84:8922–8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallicchio, E., M. M. Kubo, and R. M. Levy. 1998. Entropy-enthalpy compensation in solvation and ligand binding revisited. J. Am. Chem. Soc. 120:4526–4527. [Google Scholar]
- 30.Cooper, A. 1999. Thermodynamic analysis of biomolecular interactions. Curr. Opin. Chem. Biol. 3:557–563. [DOI] [PubMed] [Google Scholar]
- 31.Ladbury, J. E., and M. A. Williams. 2004. The extended interface: measuring non-local effects in biomolecular interactions. Curr. Opin. Struct. Biol. 14:562–569. [DOI] [PubMed] [Google Scholar]
- 32.Hansson, T., C. Oostenbrink, and W. F. van Gunsteren. 2002. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 12:190–196. [DOI] [PubMed] [Google Scholar]
- 33.Karplus, M., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- 34.Cheatham III, T. E. 2004. Simulation and modeling of nucleic acid structure, dynamics and interactions. Curr. Opin. Struct. Biol. 14:360–367. [DOI] [PubMed] [Google Scholar]
- 35.Dyson, H. J., and P. E. Wright. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6:197–208. [DOI] [PubMed] [Google Scholar]
- 36.Arthanari, H., K. J. McConnell, R. Beger, M. A. Young, D. L. Beveridge, and P. H. Bolton. 2003. Assessment of the molecular dynamics structure of DNA in solution based on calculated and observed NMR NOESY volumes and dihedral angles from scalar coupling constants. Biopolymers. 68:3–15. [DOI] [PubMed] [Google Scholar]
- 37.Dixit, S. B., F. Pitici, and D. L. Beveridge. 2004. Structure and axis curvature in two dA6·dT6 DNA oligonucleotides: comparison of molecular dynamics simulations with results from crystallography and NMR spectroscopy. Biopolymers. 75:468–479. [DOI] [PubMed] [Google Scholar]
- 38.Soares, T. A., P. H. Hünenberger, M. A. Kastenholz, V. Kräutler, T. Lenz, R. D. Lins, C. Oostenbrink, and W. F. van Gunsteren. 2005. An improved nucleic acid parameter set for the GROMOS force field. J. Comput. Chem. 26:725–737. [DOI] [PubMed] [Google Scholar]
- 39.Kastenholz, M. A., and P. H. Hünenberger. 2004. Influence of artificial periodicity and ionic strength in molecular dynamics simulations of charged biomolecules employing lattice-sum-methods. J. Phys. Chem. B. 108:774–788. [Google Scholar]
- 40.Wartell, R. M., J. E. Larson, and R. D. Wells. 1974. Netropsin-specific probe for A-T regions of duplex deoxyribonucleic acid. J. Biol. Chem. 249:6719–6731. [PubMed] [Google Scholar]
- 41.van Dyke, M. W., R. P. Hertzberg, and P. B. Dervan. 1982. Map of distamycin, netropsin, and actinomycin binding sites on heterogeneous DNA. DNA cleavage inhibition patterns with methidiumpropyl-EDTA.Fe(II). Proc. Natl. Acad. Sci. USA. 79:5470–5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kopka, M. L., C. Yoon, D. Goodsell, P. Pjura, and R. E. Dickerson. 1985. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc. Natl. Acad. Sci. USA. 82:1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kopka, M. L., C. Yoon, D. Goodsell, P. Pjura, and R. E. Dickerson. 1985. Binding of an antitumor drug to DNA. Netropsin and CGCGAATTBrCGCG. J. Mol. Biol. 183:553–563. [DOI] [PubMed] [Google Scholar]
- 44.Dasgupta, D., P. Parrack, and V. Sasisekharan. 1987. Interaction of synthetic analogues of distamycin with poly(dA-dT): role of the conjugated N-methylpyrrole system. Biochemistry. 26:6381–6386. [DOI] [PubMed] [Google Scholar]
- 45.Ren, J., and J. B. Chaires. 1999. Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry. 38:16067–16075. [DOI] [PubMed] [Google Scholar]
- 46.Singh, S. B., Ajay, D. E. Wemmer, and P. A. Kollman. 1994. Relative binding affinities of distamycin and its analog to d(CGCAAGTTGGC).d(GCCAACTTGCG): comparison of simulation results with experiment. Proc. Natl. Acad. Sci. USA. 91:7673–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wellenzohn, B., R. H. Winger, A. Hallbrucker, E. Mayer, and K. R. Liedl. 2000. Simulation of EcoRI dodecamer netropsin complex confirms class 1 complexation mode. J. Am. Chem. Soc. 122:3927–3931. [Google Scholar]
- 48.Wellenzohn, B., W. Flader, R. H. Winger, A. Hallbrucker, E. Mayer, and K. R. Liedl. 2001. Significance of ligand tails for interaction with the minor groove of B-DNA. Biophys. J. 81:1588–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finkelstein, A. V., and J. Janin. 1989. The price of lost freedom: entropy of bimolecular complex formation. Protein Eng. 3:1–3. [DOI] [PubMed] [Google Scholar]
- 50.Spolar, R. S., and M. T. Record. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science. 263:777–784. [DOI] [PubMed] [Google Scholar]
- 51.Baginski, M., F. Fogolari, and J. M. Briggs. 1997. Electrostatic and non-electrostatic contributions to the binding free energies of anthracycline antibiotics to DNA. J. Mol. Biol. 274:253–267. [DOI] [PubMed] [Google Scholar]
- 52.Beveridge, D. L., and F. M. DiCapua. 1989. Free energy via molecular simulation: applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 18:431–492. [DOI] [PubMed] [Google Scholar]
- 53.Straatsma, T. P., and J. A. McCammon. 1992. Computational alchemy. Annu. Rev. Phys. Chem. 43:407–435. [Google Scholar]
- 54.King, P. M. 1993. Free energy via molecular simulation: a primer. In Computer Simulation of Biomolecular Systems: Theoretical and Experimental Applications, vol. 2. W. F. van Gunsteren, P. K. Weiner, and A. J. Wilkinson, editors. Escom Science, Leiden, The Netherlands. 267–314.
- 55.van Gunsteren, W. F., T. C. Beutler, F. Fraternali, P. M. King, A. E. Mark, and P. E. Smith. 1993. Computation of free energy in practice: choice of approximations and accuracy limiting factors. In Computer Simulation of Biomolecular Systems: Theoretical and Experimental Applications, vol. 2. W. F. van Gunsteren, P. K. Weiner, and A. J. Wilkinson, editors. Escom Science, Leiden, The Netherlands. 315–348.
- 56.Kollman, P. 1993. Free energy calculations: applications to chemical and biochemical phenomena. Chem. Rev. 93:2395–2417. [Google Scholar]
- 57.Straatsma, T. P. 1996. Free energy by molecular simulation. In Reviews in Computational Chemistry, vol. 9. K. B. Lipkowitz and D. B. Boyd, editors. VCH Publishers, New York. 81–127.
- 58.Mark, A. E. 1998. Free energy perturbation (FEP). In Encyclopedia of Computational Chemistry, vol. 2. P. von R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A. Kollman, H. F. Schaefer, and P. R. Schreiner, editors. John Wiley & Sons. 1070–1083.
- 59.Chipot, C., and D. A. Pearlman. 2002. Free energy calculations. The long and winding gilded road. Mol. Sim. 28:1–12. [Google Scholar]
- 60.Karplus, M., and J. Kushick. 1981. Method for estimating the configurational entropy of macromolecules. Macromolecules. 14:325–332. [Google Scholar]
- 61.Edholm, O., and H. J. C. Berendsen. 1984. Entropy estimation from simulations of non-diffusive systems. Mol. Phys. 51:1011–1028. [Google Scholar]
- 62.Di Nola, A., H. J. C. Berendsen, and O. Edholm. 1984. Free energy determination of polypeptide conformations generated by molecular dynamics. Macromolecules. 17:2044–2050. [Google Scholar]
- 63.Schlitter, J. 1993. Estimation of absolute and relative entropies of macromolecules using the covariance-matrix. Chem. Phys. Lett. 215:617–621. [Google Scholar]
- 64.Schäfer, H., A. E. Mark, and W. F. van Gunsteren. 2000. Absolute entropies from molecular dynamics simulation trajectories. J. Chem. Phys. 113:7809–7817. [Google Scholar]
- 65.Schäfer, H., X. Daura, A. E. Mark, and W. F. van Gunsteren. 2001. Entropy calculations on a reversibly folding peptide: changes in solute free energy cannot explain folding behavior. Proteins. 43:45–56.11170213 [Google Scholar]
- 66.Reinhardt, W. P., M. A. Miller, and L. M. Amon. 2001. Why is it so difficult to simulate entropies, free energies, and their differences? Acc. Chem. Res. 34:607–614. [DOI] [PubMed] [Google Scholar]
- 67.Andricioaei, I., and M. Karplus. 2001. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 115:6289–6292. [Google Scholar]
- 68.Lin, S. T., M. Blanco, and W. A. Goddard. 2003. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: validation for the phase diagram of Lennard-Jones fluids. J. Chem. Phys. 119:11792–11805. [Google Scholar]
- 69.Peter, C., C. Oostenbrink, A. van Dorp, and W. F. van Gunsteren. 2004. Estimating entropies from molecular dynamics simulations. J. Chem. Phys. 120:2652–2661. [DOI] [PubMed] [Google Scholar]
- 70.Roccatano, D., A. Di Nola, and A. Amadei. 2004. A theoretical model for the folding/unfolding thermodynamics of single-domain proteins, based on the quasi-Gaussian entropy theory. J. Phys. Chem. B. 108:5756–5762. [Google Scholar]
- 71.Baron, R., W. F. van Gunsteren, and P. H. Hünenberger. 2006. Estimating the configurational entropy from molecular dynamics simulations: anharmonicity and correlation corrections to the quasi-harmonic approximation. Trends. Chem. Phys. In press.
- 72.Chang, C.-E., W. Chen, and M. K. Gilson. 2005. Evaluating the accuracy of the quasiharmonic approximation. J. Chem. Theory Comput. 1:1017–1028. [DOI] [PubMed] [Google Scholar]
- 73.Carlsson, J., and J. Åqvist. 2005. Absolute and relative entropies from computer simulation with applications to ligand binding. J. Phys. Chem. B. 109:6448–6456. [DOI] [PubMed] [Google Scholar]
- 74.Baron, R., A. H. de Vries, P. H. Hünenberger, and W. F. van Gunsteren. 2006. Configurational entropy of lipids in pure and mixed bilayers from atomic-level and coarse-grained molecular dynamics simulations. J. Phys. Chem. B. In press. [DOI] [PubMed]
- 75.Baron, R., A. H. de Vries, P. H. Hünenberger, and W. F. van Gunsteren. 2006. A comparison of atomic-level and coarse-grained models for liquid hydrocarbons from molecular dynamics configurational entropy estimates. J. Phys. Chem. B. 110:8464–8473. [DOI] [PubMed] [Google Scholar]
- 76.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Partridge, B. L., and S. A. Salisbury. 1996. Structural studies on nucleic acids. Protein Data Bank, entry 267D. http://www.rcsb.org/pdb/.
- 78.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, The Netherlands. 331–342.
- 79.van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Vdf Hochschulverlag AG, Zürich.
- 80.Scott, W. R. P., P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, P. Huber, P. Krüger, and W. F. van Gunsteren. 1999. The GROMOS biomolecular simulation program package. J. Phys. Chem. A. 103:3596–3607. [Google Scholar]
- 81.Ryckaert, J.-P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 82.Hockney, R. W. 1970. The potential calculations and some applications. Methods Comput. Phys. 9:136–211. [Google Scholar]
- 83.Tironi, I. G., R. Sperb, P. E. Smith, and W. F. van Gunsteren. 1995. A generalized reaction field method for molecular-dynamics simulations. J. Chem. Phys. 102:5451–5459. [Google Scholar]
- 84.Heinz, T. N., W. F. van Gunsteren, and P. H. Hünenberger. 2001. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys. 115:1125–1136. [Google Scholar]
- 85.Berendsen, H. J. C., J. P. M. Postma, W. F. Van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 86.McLachlan, A. D. 1979. Gene duplications in the structural evolution of chymotrypsin. J. Mol. Biol. 128:49–79. [DOI] [PubMed] [Google Scholar]
- 87.Cheatham III, T. E., and M. A. Young. 2000. Molecular dynamics simulation of nucleic acids: successes, limitations, and promise. Biopolymers. 56:232–256. [DOI] [PubMed] [Google Scholar]
- 88.Schuler, L. D., and W. F. van Gunsteren. 2000. On the choice of dihedral angle potential energy functions for n-alkanes. Mol. Simulat. 25:301–319. [Google Scholar]