

Abstract
AMPA-type glutamate receptors are tetrameric ion channels that mediate fast excitatory synaptic transmission in the mammalian brain. When agonists occupy the binding domain of individual receptor subunits, this domain closes, triggering rearrangements that couple agonist binding to channel opening. Here we compare the kinetic behavior of GluR2 channels activated by four different ligands, glutamate, AMPA, quisqualate, and 2-Me-Tet-AMPA, full agonists that vary in potency by up to two orders of magnitude. After reduction of desensitization with cyclothiazide, deactivation decays were strongly agonist dependent. The time constants of decay increased with potency, and slow components in the multiexponential decays became more prominent. The desensitization decays of agonist-activated currents also contained multiple exponential components, but they were similar for the four agonists. The time course of recovery from desensitization produced by each agonist was described by two sigmoid components, and the speed of recovery varied substantially. Recovery was fastest for glutamate and slowest for 2-Me-Tet-AMPA, and the amplitude of the slow component of recovery increased with agonist potency. The multiple kinetic components appear to arise from closed-state transitions that precede channel gating. Stargazin increases the slow kinetic components, and they likely contribute to the biexponential decay of excitatory postsynaptic currents.
INTRODUCTION
Glutamate is the main excitatory transmitter in the mammalian brain, and members of three subfamilies of glutamate receptors (GluRs), 2-amino-3-(3-hydroxy-5-methyl-4-isoxazole) propionic acid (AMPA), kainate, and N-methyl-d-aspartate (NMDA), are ligand-gated ion channels that are found at synapses throughout the central nervous system (1,2). Structures of the isolated ligand-binding core from the AMPA receptor (AMPAR) subunit GluR2 (GluR2-S1S2) show that glutamate and other agonists bind at the base of a deep cleft between two globular domains (1 and 2), causing the translation and rotation of domain 2 such that the cleft closes (3–5). Comparison of the structure of GluR2-S1S2 in complex with various ligands has shown that the ligand-binding core can adopt multiple agonist-dependent conformations and that differences in agonist efficacy correlate with different amounts of cleft closure (5–7).
The closed-cleft conformation is stabilized by interactions between ligands and residues in domains 1 and 2 as well as by direct and water-mediated hydrogen bonds that occur only after the binding cleft closes. The relationship between the extent of cleft closure and channel function has been the focus of several studies, but much less is known about how the stability of the closed-cleft conformation impacts channel properties. Early GluR2-S1S2 crystal structures indicate that binding domain closure renders the entire binding cleft inaccessible to solvent, likely trapping agonists in the cleft (3). This raised the possibility that the rate at which agonists dissociate, a major determinant of receptor affinity, may primarily be determined by the rate at which the binding domain opens (3,8). Consistent with this view, binding cleft mutations predicted to destabilize the closed-cleft conformation increased the apparent rate of agonist dissociation and also altered the relative efficacies of glutamate and quisqualate (9).
To explore further the relationships among agonist potency, the stability of the closed binding cleft, and AMPAR function, we have characterized the kinetic behavior of GluR2 channels when activated by glutamate, AMPA, quisqualate, or (S)-2-amino-3-[3-hydroxy-5-(2-methyl-2H-5-tetrazolyl)-4-isoxazolyl]propionic acid (2-Me-Tet-AMPA (10,11)). These four ligands differ substantially in potency but are all full agonists at wild-type AMPARs and produce similar amounts of binding domain closure (3,8,12). The results reveal kinetic behavior that may reflect differences in the stability of the closed-cleft conformation, which we suggest could contribute to the kinetics of excitatory postsynaptic currents (EPSCs).
METHODS
The tsA201 cells were maintained and transfected as described (13). The GluR2 plasmid used in this study was the flip splice variant and encoded a glutamine at the Q/R site (kindly provided by Mark Mayer, NIH). The stargazin plasmid was a generous gift from Susumu Tomita (UCSF). GluR4 and stargazin plasmids were cotransfected at a 1:2 ratio. Recordings from outside-out patches were performed 24 to 48 h posttransfection at room temperature (14). The holding potential was −80 mV, and series resistance compensation was set at 60–80%. The external solution was (in mM): 150 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, 10 HEPES, pH 7.4. Patch pipettes (open tip resistance 3 to 5 MΩ) were filled with a solution containing (in mM): 135 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, 11 EGTA, 10 HEPES, pH 7.4. Glutamate, AMPA, quisqualate, 2-Me-Tet-AMPA, and cyclothiazide were added to the external solution and applied with θ-pipettes mounted on a piezoelectric bimorph (13,14). At the end of some recordings, junction currents evoked by changes in open-tip potentials were measured. The 10–90% risetimes of these currents gave solution exchange times of 100–200 μs. At high agonist concentrations, the 10–90% risetimes of agonist-evoked currents were typically 400–500 μs.
Agonist-evoked currents were analog low-pass filtered at 3 kHz, sampled at 20–50 kHz, and written directly to the hard drive of the computer. For deactivation and the onset of desensitization, 20 to 30 consecutive responses were averaged, and exponential functions were fitted to the decays of the mean currents with Igor software (Wavemetrics, Lake Oswego, OR). The number of exponential components required was determined from inspection of the residual traces and nested t-tests (15) as described in detail before (14). Concentration–response data were normalized (see Results), and the mean normalized results were fitted with Hill-type functions to obtain EC50 values and values for the Hill coefficient. Recovery data were obtained from two-pulse protocols. The peak amplitude of the current evoked by the second pulse was expressed as a fraction of the response to the paired first pulse. Mean results from several patches were fitted with the equation:
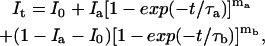 |
where It is the peak current at a given interpulse interval t, Ia and Ib are the limiting amplitudes of the fast and slow components of recovery, I0 is the current at zero time (the relative amplitude of the plateau current at the end of the first pulse), and τa,τb and ma,mb are the respective time constants and exponents of each component of recovery.
Monte Carlo simulations and kinetic modeling were done with Channel Lab software (Synaptosoft, Decatur, GA). Details of the procedures have been published previously (9,13,16) and are given in the Supplementary Material.
RESULTS
Crystallographic structures of GluR2-S1S2 and the kinetics of glutamate binding to the isolated AMPAR binding core suggest that the rate at which the binding cleft opens may be a major determinant of agonist affinity (3,8,12,17). For glutamate and quisqualate, recent patch-clamp studies of wild-type and mutant GluR2 channels suggested that the stability of the closed-cleft conformation can also influence the efficacy of agonists (9). Here we compare the kinetic properties of GluR2 channels activated by four agonists that display large differences in potency (and likely different stabilities of the closed-cleft conformation).
Agonist potency and the kinetics of deactivation
Fig. 1 a shows concentration–response curves for glutamate, AMPA, quisqualate, and 2-Me-Tet-AMPA that were obtained by measuring steady-state currents after reduction of desensitization with 100 μM cyclothiazide (CTZ). Currents from individual outside-out patches were normalized to the size of the current evoked in the same patch by a near-saturating concentration of agonist. Hill-type fits to the mean results gave EC50 values of 296 μM, 66.2 μM, 16.3 μM, and 3.4 μM for glutamate, AMPA, quisqualate, and 2-Me-Tet-AMPA, respectively (Hill coefficients: 1.09–1.34). Although the EC50 values are larger by about three orders of magnitude than the equilibrium dissociation constants estimated for binding of each agonist to the isolated GluR2 binding core, the relative differences in agonist potency agree well with the corresponding differences in affinity determined in binding experiments (Table 1).
FIGURE 1.

Deactivation shows agonist-dependent components that do not depend on receptor occupancy. (a) Mean concentration–response data for steady-state currents (in 100 μM CTZ). The smooth curves are Hill-type fits. Current amplitudes were normalized to currents evoked in the same patch by near-saturating concentrations. Error bars show mean ± SE values, which in most cases were less than half the symbol size. (b) Currents evoked in individual patches by 2-ms applications (arrowheads) of 10 mM glutamate, 0.5 mM AMPA, 0.2 mM quisqualate, and 0.1 mM 2-Me-Tet-AMPA (in CTZ). The decays of the currents were fitted with multiexponential functions (solid curves; individual components shown as dashed lines). (c) For each of the four agonists, weighted mean time constants are plotted against the EC50 ratio (glutamate EC50/agonist EC50). (d) Currents evoked by different concentrations of 2-Me-Tet-AMPA (left). In the panels to the right, the individual currents were scaled to have the same peak amplitude, and the slow components obtained from the biexponential fits to the decays are shown as solid curves.
TABLE 1.
Properties of GluR2 currents evoked by the four agonists
| Deactivation*
|
Desensitization†
|
Recovery‡
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| τ1 (ms) | τ2 (ms) | τ3 (ms) | Fast component (%) | τf (ms) | τs (ms) | Slow component (%) | τa (ms) | τb (ms) | ma | mb | Slow component (%) | EC50 (μM) | KD§ (nM) | |
| Glu | 1.3 ± 0.2 | 17 ± 3.5 | 99 ± 0.5 | 4.6 ± 0.5 | 15 ± 2.1 | 14 ± 2.3 | 17 | 82 | 2.3 | 2.0 | 13 | 296 ± 19.2 | 89–460 (3,46,47) | |
| AMPA | 6.3 ± 0.5 | 27 ± 4.2 | 78 ± 6.6 | 3.8 ± 0.4 | 11 ± 1.0 | 24 ± 2.4 | 130 | 582 | 1.4 | 1.1 | 32 | 66.2 ± 2.3 | 11–25 (3,12,46,47) | |
| Quis | 8.5 ± 0.6 | 32 ± 4.7 | 73 ± 5.3 | 3.7 ± 0.3 | 15 ± 3.2 | 10 ± 3.8 | 99 | 460 | 1.8 | 1.4 | 37 | 16.3 ± 0.5 | 7.6–12 (12,46) | |
| 2-Me-Tet-AMPA | 9.1 ± 1.4 | 54 ± 2.7 | 156 ± 8.4 | 19 ± 5.0 | 8.4 ± 0.9 | 46 ± 7.0 | 9.0 ± 4.0 | 824 | 3474 | 1.6 | 1.5 | 58 | 3.4 ± 0.8 | 3.8 (8) |
Mean (± SE) values from multiexponential fits to the current decays during 2-ms applications of agonist in 100 μM CTZ (n = 4 to 6 patches per agonist).
Mean (± SE) values from biexponential fits to the current decays during 100-ms applications of agonist (n = 4 to 9 patches per agonist).
Values from double Hodgkin-Huxley-like fits to the mean data shown in Fig. 6 (n = 6 to 8 patches per agonist).
Values for binding to GluR2-S1S2 (from indicated references). When IC50 values were measured, we converted them to KD values with the equation: KDI = IC50/(1 + [L]/KDL), where KDI is the KD of the competing ligand, and [L] and KDL are the concentration of radioligand and its KD value.
To determine how differences in potency correlated with the rate of deactivation, we next compared the time course with which the currents decayed on termination of 2-ms applications of each agonist (in CTZ). Representative results are shown in Fig. 1 b. The deactivation decays were fitted adequately with two exponential components for glutamate, AMPA, and quisqualate, but three exponential components were consistently observed in the decays of currents evoked by 2-Me-Tet-AMPA. The mean time constants and relative amplitudes obtained from multiexponential fits to the deactivation decays are given in Table 1.
The deactivation decays were strongly agonist dependent, being fastest for glutamate and slowest for 2-Me-Tet-AMPA. The time constants for the corresponding components increased with agonist potency, and the slow components of decay became increasingly prominent. Although the fast component of decay dominated the time course seen with glutamate (99.1%), the relative amplitude of the fast component was only 19% with 2-Me-Tet-AMPA. As an index of the overall speed of deactivation, mean time constants were calculated for each patch, where the time constants of each component obtained from the multiexponential fits were weighted by their relative amplitudes. In Fig. 1 c, the weighted mean time constants for each agonist are plotted against the corresponding fold differences in potency. The correlation between these two parameters was excellent (p < 0.001; linear regression analysis).
AMPARs are tetrameric assemblies in which each of the four subunits contains a binding site for glutamate, and individual AMPARs open to increasingly large conductance levels as the number of subunits occupied by agonist increases (18,19). At the concentrations used in the deactivation protocols illustrated in Fig. 1 b, most receptors are likely fully occupied at the end of the application, and multiple agonist molecules must dissociate before the channel is no longer able to open. The probability that channels will open after free agonist is removed will increase as the rate of dissociation slows, and if partially occupied channels have different gating kinetics, this might give rise to multiexponential decays. To test this possibility, we compared the deactivation kinetics at agonist concentrations that gave substantially different steady-state currents. As illustrated for 2-Me-Tet-AMPA in Fig. 1 d, for each of the agonists, deactivation decays did not vary significantly with concentration. These results argue against the idea that the multiple deactivation components detected with each agonist reflect occupancy-dependent differences in the kinetics of gating.
Desensitization is biexponential
We next examined the kinetics of the channels with desensitization intact. We noted previously that the desensitization decays of glutamate-activated currents through GluR1 and GluR4 homomeric channels were biexponential (14,20). The desensitization decays of GluR2-mediated currents were likewise consistently biexponential. This is illustrated in Fig. 2 a, where the decay of a typical current elicited by a sustained application of 10 mM glutamate has been fitted with one or two exponential components. Although single exponential fits were not obviously poor, the fits deviated systematically during the early and late phases of decay, as evidenced by inspection of the residual trace (the point-by-point difference between the data and the fit). In contrast, biexponential fits gave flat residual traces and were significantly better as assessed by nested t-tests for currents evoked in each of nine patches (p < 0.01 (15)). Similar results were obtained for currents activated by lower concentrations of glutamate (Fig. 2 b), and comparison of the mean results for 500 μM and 10 or 20 mM glutamate demonstrated that the desensitization decays did not depend significantly on receptor occupancy.
FIGURE 2.

Glutamate-induced desensitization shows multiple kinetic components that do not depend on receptor occupancy. (a) Current evoked by 100-ms applications of 10 mM glutamate. Left and right panels show one- and two-exponential fits (solid curves) to the decays. The residuals are also shown (res; obtained by subtracting the data from the fit). Insets are the same currents and fits on an expanded timescale (dashed lines show individual components). (b) Currents evoked by sustained applications of two different concentrations of glutamate (left). In the panels to the right, the individual currents were scaled to have the same peak amplitude. The individual components obtained from the biexponential fits to the decays are shown as dashed curves.
The desensitization decays of currents elicited by AMPA, quisqualate, and 2-Me-Tet-AMPA were also consistently fitted better by two exponential components (as assessed by inspection of the residuals and nested t-tests). As for glutamate, inclusion of a third component did not significantly improve the fits. Examples of currents evoked by each of the four agonists are shown in Fig. 3. In contrast to the deactivation decays in CTZ, the time course of desensitization did not correlate with agonist potency. The mean time constants and relative amplitude of the slow component of decay are given in Table 1. The mean (± SD) steady-state currents (expressed as a percentage of the peak current) were 2.1 ± 0.4%, 1.4 ± 0.2%, 0.9 ± 0.1%, and 0.8 ± 0.3% for glutamate, AMPA, quisqualate, and 2-Me-Tet-AMPA, respectively.
FIGURE 3.

Desensitization decays are similar for each agonist. Currents evoked by 100-ms applications of (a) 10 mM glutamate, (b) 0.5 mM AMPA, (c) 0.2 mM quisqualate, and (d) 0.1 mM 2-Me-Tet-AMPA. The decays of the currents were fitted with biexponential functions (solid curves; dashed lines show individual components).
The results show that agonist potency does not significantly influence the rate of entry into desensitization or the relative amplitudes of the fast and slow components. For AMPA receptors, many published reports indicate that desensitization and channel opening primarily occur in parallel from the same closed states. As a result, time constants of desensitization reflect the ratio of the rate constants for channel opening and entry into desensitization (β and δ in Fig. 4), which determine the average number of openings per channel before they desensitize, as well as the rate constant for channel closing, α, the reciprocal of which determines the duration of each opening. The simplest explanation for the similar desensitization time constants obtained with each agonist is that these three rate constants are similar for all four agonists. The similar amplitudes of the slow components of desensitization seen with each agonist also suggest that the states that give rise to the multiple deactivation components in CTZ are populated slowly relative to desensitization.
FIGURE 4.

Possible kinetic mechanisms. In model 1, one cleft closure step is followed by gating transitions to three different open states. In models 2 and 3, gating transitions are preceded by three cleft-closure steps that occur in parallel or sequentially. Only a single subunit is shown.
GluR2 kinetics can be modeled by sequential, but not parallel, conformational changes
To gain insight into the source of the kinetic behavior described above, we performed Monte Carlo simulations of agonist-evoked currents. Previous kinetic mechanisms we proposed accounted for the tetrameric structure of AMPA receptors, with each subunit providing a binding site for agonists (9,13,16). Our kinetic models also included multiple desensitization transitions to account for the two-step nature of recovery from desensitization, which likely reflects the dimer-of-dimers organization of the channels (13,21). However, the results reported here were obtained at near-saturating concentrations, and the kinetics of the current decays did not depend significantly on receptor occupancy (Figs. 1 d and 2 b). In addition, our previous work showed that occupancy of a single subunit was sufficient to desensitize GluR1 and GluR4 channels (13). Therefore the multiple kinetic components described above likely reflect channel behavior that is independent of the number of subunits contributing to the currents. We therefore treated the receptor as though it were a single subunit, which greatly simplified the modeling and is unlikely to substantially impact the general conclusions. Open and closed transitions in our simplified models would result in steps between adjacent conductance levels in tetrameric channels.
The models we examined are shown in Fig. 4. Each model contains three types of transitions (governed by the rate constants in parentheses): a), transitions that correspond to association and dissociation of agonist to the open-cleft conformation of the binding domain (k+1, k−1); b), transitions that correspond to closing and opening of the binding domain or stabilization and destabilization of closed-cleft conformations (CC, CO and CS, CD, respectively); and c), gating transitions that correspond to conformational changes that would lead to step changes in unitary conductance (β, α) or desensitization and recovery from it (δ, γ). The multiple open states in Fig. 4 have identical occupancies and unitary conductances and do not correspond to the multiple open states in our previous articles (9,13).
In model 1, we assumed that the multiple deactivation and desensitization components arise from different kinetics of activation gating. Once the binding cleft closes, the channel can enter three kinetically distinct open states that have different opening and closing rate constants. In model 2, we assumed that the binding cleft could adopt one of three closed-cleft conformations and that the rate constants governing these and subsequent gating steps differed. In model 3, activation gating and desensitization occur from each of three different closed-cleft conformations that are adopted sequentially. Here the multiple components arise from the kinetics of transitions between the closed states, not from the kinetics of the open states (which are identical).
Models 1 and 2 invariably reproduced the experimental results poorly. For example, β/α and CC/CO, CS/CD values that reproduced the multiple deactivation components gave activation time courses that were slower than those observed and peak current amplitudes that depended on the duration of exposure to agonist. In addition, the desensitization decays could be reproduced only by setting the rate constant for entry into desensitization (δ) to values substantially larger than β, which gave low peak popen values and steady-state currents that were too small. In contrast, activation, deactivation, and desensitization were all reproduced well by model 3 (see Supplementary Material for details).
The speed of deactivation depends on the duration of exposure to agonist
In model 3, the second and third cleft-closure steps equilibrate slowly (CS and CD ≪ β and δ). The values of CC, β, α, and δ are assumed to be identical or similar for each agonist, ensuring rapid activation and similar desensitization decays (supplemental Table 2, Supplementary Material), and in the absence of CTZ, most channels will desensitize before they reach the RG*2 and RG*3 states. A main feature of model 3 is that the rate constant for binding cleft opening, CO, and the steady-state occupancies of the RG* states differ for the different agonists. With glutamate, most channels reside in state RG*1, whereas with 2-Me-Tet-AMPA, the channels are primarily in states RG*2 and RG*3. For the deactivation protocols in CTZ, the slow components arise because channels in RG*3 and RG*2 must pass through states RG*2 and RG*1 before the binding cleft can open. Gating will continue during these sojourns, and the duration of this activity will depend on how quickly the channels return to state RG.
Because model 3 assumes that sequential occupancy of the states RG*2 and RG*3 occurs slowly, the model predicts that the speed of deactivation should depend on the duration of the agonist applications. To test this prediction, we applied each agonist for different times and analyzed the decays of the currents at the end of the applications. Examples of the results obtained with short and long applications are shown in Fig. 5 a. For each agonist, and in all patches examined, deactivation decays became slower as the length of the application was increased, reflecting both an increase in the time constants for the various decay components and an increase in the relative amplitude of the slow components of decay. As predicted from Monte Carlo simulations using model 3, the weighted mean time constants of deactivation increased as a function of application time. Plots of the weighted time constants versus application duration were approximately exponential, and the time constants of exponential fits to the results increased with agonist potency (glutamate < AMPA < quisqualate < 2-Me-Tet-AMPA). The experimental results obtained with glutamate and 2-Me-Tet-AMPA are presented in Fig. 5 b.
FIGURE 5.

Deactivation depends on the duration of exposure to agonist and is faster for T686S mutant channels. (a) Decays of currents at the end of 2-ms (dotted traces) and 500-ms (solid traces) agonist applications (in CTZ). (b) Plot of weighted mean time constants of deactivation versus application time (log scale) for glutamate and 2-Me-Tet-AMPA. The curves are single-exponential fits. (c) Currents at the end of 50-ms applications of 0.2 mM quisqualate for GluR2-WT (solid trace) and GluR2-T686S (dotted curve). (d) The time constants of deactivation were faster and the relative amplitude of the slow component was smaller for the T686S mutant (mean ± SE, four to six patches).
Recently we concluded that mutations at threonine 686 in GluR2 speed the rate of glutamate dissociation by destabilizing the closed-cleft conformation and increasing the rate at which the binding cleft opens (9). If the deactivation time course primarily reflects the stability of the closed-cleft conformation, and the slow components arise from increasingly stable closed-cleft conformations (as in model 3), then the T686 mutations should decrease the deactivation time constants and decrease the relative amplitude of the slow decay components. We therefore compared the deactivation decays seen with quisqualate for GluR2 and GluR2(T686S) homomers. As shown in Fig. 5, c and d, the T686S mutation increased the rate of deactivation and decreased the relative amplitude of the slow component of decay.
Recovery from desensitization exhibits multiple agonist-dependent components
With glutamate, AMPARs do not recover from desensitization along a simple exponential time course, but rather, this time course appears to be governed by two rate-determining steps (13,21). We proposed that these steps correspond to reassembly of the monomer–monomer interface in first one dimer and then the other, and that the rate at which the interface reassembled was in part determined by the stability of the closed conformation of the binding domain (9). With glutamate, it has also been noted that a small proportion of the channels recover much more slowly (13,22,23).
To investigate further the relationship between the kinetics of recovery and agonist potency, we measured recovery from desensitization for each agonist with standard two-pulse protocols. The left panels in Fig. 6, a–d, show representative recovery results for glutamate, AMPA, quisqualate, and 2-Me-Tet-AMPA (note different timescales). Mean data for each agonist from several patches are presented in the middle panels, where the results have been fitted with the sum of two Hodgkin-Huxley–type components (significantly better than one-component fits, p < 0.01). The right panels show the data for AMPA and 2-Met-Tet-AMPA on a faster timescale to illustrate the sigmoid shape of the early phase of recovery.
FIGURE 6.

Recovery from desensitization shows fast and slow components. Left panels are examples of the results from two-pulse protocols to measure recovery from desensitization with 10 mM glutamate, 0.5 mM AMPA, 0.2 mM quisqualate, and 0.1 mM 2-Me-Tet-AMPA. Other panels show mean (± SE) data from six to eight patches. The data were fitted with two Hodgkin-Huxley–type components. In b and d, the right panels show the early phase of recovery.
In contrast to entry into desensitization, the time constants of recovery and the relative amplitude of the slow component became larger as agonist potency increased. The results suggest that, as in deactivation, the recovery components equilibrate slowly, and this equilibration continues after the channels desensitize. Whether the fast and slow components of deactivation and the fast and slow components of recovery arise from a common mechanism is unclear. If this were true, it might be expected that agonist-dependent differences in the respective time constants and relative amplitudes would be similar, which does not appear to be the case (Table 1). For example, the difference in the recovery time constants for 2-Met-Tet-AMPA and those for the other agonists is much bigger than the corresponding differences for deactivation. This discrepancy would be partially resolved, however, if we missed a small fast component of recovery for 2-Met-Tet-AMPA and the two recovery components detected correspond to gating modes that give rise to the second and third components of deactivation.
The m values obtained from the recovery fits varied for the four agonists but were typically substantially greater than 1, and for any given agonist, the m values were similar for the two components. In total, the results suggest that both components of recovery consist of two rate-determining transitions and that the molecular determinants of the rate of recovery are qualitatively similar for each recovery component and for each agonist. We proposed previously that the slow component of recovery detected for glutamate arose from slow dissociation of the last bound glutamate from desensitized channels (13). This proposal was never attractive from a structural standpoint, and our present results suggest it is incorrect.
The effect of stargazin on AMPAR kinetics
For the GluR2 channels studied here, the slow components detected in the decays of glutamate-activated currents are small, likely too small to impact the shape of synaptic currents significantly. However, native AMPARs interact with various auxiliary proteins that localize the receptors at postsynaptic densities. One such protein is stargazin, a member of the transmembrane AMPAR regulatory protein (TARP) family that was shown to be required for the localization of AMPARs at mossy fiber/granule cell synapses in the cerebellum (24–27). In addition to its effects on receptor targeting, recent studies have shown that stargazin also alters AMPAR gating, slowing both deactivation and desensitization (20,28,29). Interactions between stargazin and GluRs also increase the slow component of desensitization for glutamate-activated currents and slow the decay of AMPAR synaptic currents in neurons (20). The latter results suggest that the multiple gating modes implicit in our results may impact the shape of synaptic currents, especially if association with stargazin increases the slow deactivation component observed with glutamate.
To characterize further the effect of stargazin on the deactivation kinetics of glutamate-activated AMPAR currents, we compared the kinetics of these currents with and without coexpression of stargazin. To compare the results with our previous work (20), we chose GluR4 for these experiments. In both the absence and presence of CTZ, coexpression of stargazin significantly slowed the deactivation time constants and increased the relative amplitude of the slow deactivation component seen with glutamate (Fig. 7). Thus, as shown previously for desensitization (20), coexpression of stargazin results in a significant slow component in the decays of currents evoked with glutamate. Because most native AMPA-receptor complexes likely contain stargazin (or a related TARP), the kinetic behavior that gives rise to the slow deactivation and desensitization components may contribute to the time course of the decay of EPSCs.
FIGURE 7.

Stargazin increases the slow component of deactivation. (a and b) Currents evoked by 2-ms applications of 10 mM glutamate (no CTZ) in patches containing GluR4 (a) or GluR4 and stargazin (b). The slow components from biexponential fits to the decays are shown (solid curves). Peak currents are off-scale. (c) Currents evoked by 2-ms applications of 10 mM glutamate (in CTZ) in patches containing GluR4 (dotted trace) and GluR4/stargazin (solid trace) channels. (d) Mean time constants for the fast and slow deactivation components (τ1, τ2: left and middle panels) and the mean relative amplitude of the slow component (right) without and with coexpression of stargazin in the absence and presence of CTZ. Stargazin increased both deactivation time constants and the relative amplitude of the slow deactivation component seen with and without CTZ (mean ± SE, data from four and six patches).
DISCUSSION
Our results demonstrate that AMPAR channels show kinetic behavior that gives rise to multiple components in both deactivation and desensitization decays. The multiple deactivation components are most evident when desensitization is reduced with CTZ and, under these conditions, the slow deactivation components become larger as agonist potency increases. The time constants of the individual components likewise increase, and the overall speed of deactivation correlates well with agonist potency. The similarity in the rates of desensitization for the four agonists suggests that the multiple components detected in CTZ arise from entry into states that equilibrate slowly, a conclusion supported by the dependence of the speed of deactivation on the duration of exposure to agonist. Because activation and desensitization occur largely in parallel (13,30–32), the similar desensitization decays also suggest that the rate constants for channel opening, closing, and entry into desensitization (β, α, and δ in Fig. 4) are similar for the four agonists, a conclusion consistent with our Monte Carlo simulations (supplemental Table 2, Supplementary Material).
Binding domain closure and agonist potency
As noted, substantial evidence indicates that agonist binding to AMPA receptors is accompanied by closure of the binding cleft between domains 1 and 2, and the extent to which the binding cleft closes is a major determinant of agonist efficacy (3–7). The available evidence strongly suggests that binding domain closure also leads to rearrangement of the dimer interface that uncouples domain closure from activation and results in desensitization (32). The similar rates at which the four full agonists studied here desensitize, all of which result in similar amounts of binding domain closure in GluR2-S1S2 crystal structures (3,8,12), are consistent with these previous conclusions.
Closure of the binding domain traps agonists in the cleft, and the stability of the closed-cleft conformation therefore substantially impacts the rate at which they dissociate and the kinetics of deactivation (4,8,9). The results presented here are consistent with model 3, where the multiple kinetic components arise from closed-cleft conformations that differ in stability and where increases in agonist potency correlate with increased occupancy of more stable conformations.
Crystal structures of the GluR2-S1S2 binding core in complex with each of the four agonists reveal differences in the detailed interactions within the cleft that might be expected to influence the stability of the closed-cleft conformation (3,8,12). For example, there are significant differences in the interactions made by the substituents corresponding to the γ-carboxylate group of glutamate. The γ-carboxyl oxygen of glutamate interacts with the domain 2 residues S654 and T655 in GluR2 and forms a water-mediated hydrogen bond with the backbone amide of E705 (3). In the AMPA- and quisqualate-bound structures, the water-mediated hydrogen bond with E705 is replaced by direct ligand–protein hydrogen bonds formed with the AMPA isoxazole nitrogen and the 5-carbonyl oxygen of quisqualate (3,12). As another example, both AMPA and 2-Me-Tet-AMPA contain an isoxazole ring, but this shared moiety adopts different positions within the binding cleft, resulting in two additional direct ligand–protein interactions in the 2-Me-Tet-AMPA structure (3,8). In addition, the 2-methyltetrazole group of 2-Me-Tet-AMPA fills a partially hydrophobic pocket formed by several residues in domains 1 and 2 more completely than the 5-methyl group of AMPA, and these hydrophobic interactions are absent for glutamate and quisqualate (3,8,12).
Key residues in the binding pocket also adopt different conformations in the different agonist-bound GluR2-S1S2 structures. For example, the peptide bond between the domain 2 residues D651 and S652 assumes a cis orientation in all AMPA and 2-Met-Tet-AMPA protomers (3,12). This peptide bond “flip,” which results in two additional hydrogen bonds with backbone atoms on residues in domain 1, was observed in four of five quisqualate-bound structures but only one of three glutamate-bound protomers (3,12). There are also differences in the rotamer orientations of the side chains of L650 and M708 among the four agonists (3,8,12). In total, the noted differences support the view that the strength of interdomain interactions and the stability of binding cleft closure are positively correlated with agonist potency, and, given the complexity of the interactions involved, it seems reasonable that multiple agonist-bound conformations might be adopted that differ in relative frequency and stability.
The contribution of closed-state transitions to AMPAR kinetics
Our Monte Carlo simulations suggest that the multiple components of deactivation, desensitization, and recovery we describe here result primarily from a series of slowly equilibrating closed states, not from differences in open-state kinetics. In model 3, we assume that the sequential closed-state transitions correspond to conformational changes within the binding cleft, but we have no direct evidence for this, and the RG*2 and RG*3 states may result from rearrangements within other portions of the protein. We also have no evidence that the putative multiple conformations are the same for each agonist (as implied in model 3); this is merely a simple explanation for our results. All model 3 requires is that the binding cleft remains closed during transitions between the RG* states, preventing agonist dissociation and allowing both activation gating and desensitization. Although they need not be identical, it is also implicit in model 3 that the β and α values be similar for the different RG* states (and the different agonists) because the different deactivation and desensitization components arise not from large differences in the kinetics of transitions governed by these rate constants but from the kinetics of the sequential closed-cleft transitions.
Although the detailed molecular interactions underlying the multiple kinetic components remain to be resolved, one clear result from our studies is that the multiple deactivation components seen with each full agonist show a strong dependence on agonist potency. The EC50 values measured in CTZ are linearly correlated with the weighted time constants of deactivation (Fig. 1 c), which reflect both increased time constants and increased amplitudes of the slower components as agonist potency increases. The relative potencies of the four agonists also agree well with the relative KD values obtained in binding experiments with the isolated GluR2-S1S2 binding core (Table 1), consistent with the idea that the multiple kinetic components seen in our experiments arise from multiple conformations of the binding domain. It seems likely that the large disparity between EC50 and KD values for individual agonists in Table 1 results because domain 2 is tethered to the first and second transmembrane helices in the intact channel, which may, in general, destabilize the closed-cleft conformation. In binding experiments on full-length receptors, KD values are strongly skewed by desensitization, which results from rearrangement of the dimer interface (32) and is accompanied by stabilization of the closed-cleft conformation of the binding domain (13). In binding experiments, the similarity between the KD values for full-length receptors and the corresponding values obtained with the isolated binding core likely arises because in both cases the strain imposed by the open state of the channel is relieved, albeit for different reasons, resulting in increased stability of the closed-cleft conformation.
The effect of stargazin
In addition to its effects on receptor trafficking, recent reports show that the auxiliary subunit stargazin influences AMPA receptor gating (20,28,29). Single-channel studies demonstrated that stargazin increases the relative frequency of large-conductance openings and causes a twofold increase in the duration of bursts (20). The increased burst durations reflect an increased number of openings per burst without alterations in the length of individual openings, showing that one primary effect of stargazin is to increase the rate constant for channel opening (β). The effects of stargazin to slow both deactivation and desensitization decays are consistent with the single-channel results.
The enhancement of the slow component detected with glutamate in the deactivation decays suggests that stargazin also promotes kinetic behavior seen with more potent agonists, perhaps by shifting the equilibrium toward more stable channel conformations in which the binding cleft remains closed. Interestingly, the effects of stargazin on deactivation decays were similar in the absence and presence of CTZ. Thus, although both stargazin and CTZ slow deactivation, they appear to do so via independent mechanisms.
The substantial slow component of deactivation seen with glutamate after coexpression of stargazin strongly suggests that the kinetic behavior described here contributes to the shape of synaptic currents and therefore to the fidelity of frequency encoding at CNS synapses. Although the amplitudes of the slow deactivation and desensitization components are relatively small, with stargazin, the total charge transfer during the fast and slow components is similar. Because many AMPARs show appreciable calcium permeability, the slow component may also contribute significantly to calcium-mediated signaling.
It is generally accepted that the kinetic behavior of NMDA receptors influences synaptic signaling, and recent single-channel studies of NMDA and glycine receptors indicate that channel opening is preceded by slow conformational changes similar to the closed-cleft transitions in model 3 (33,34). For NMDA receptors, the kinetics of these pregating transitions are such that they impact the size of responses to consecutive glutamate applications and may play a role in short-term changes in synaptic strength. Although many presynaptic and postsynaptic factors potentially influence the rapid kinetics of AMPAR EPSCs (35), relatively little attention has been focused on the contribution of the kinetic behavior of the channels themselves. The biexponential decay of AMPA-receptor EPSCs noted in many previous studies has been suggested to result from heterogeneity in the postsynaptic receptor population, the kinetics of glutamate release and clearance, synapse geometry, or spillover from neighboring release sites (30,36–45). Our results indicate that the intrinsic kinetic properties of AMPARs likely also contribute to the biexponential decays of EPSCs.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
This work was supported by the National Institutes of Health (NS-047712).
Wei Zhang and Antoine Robert contributed equally to this work.
References
- 1.Madden, D. R. 2002. The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci. 3:91–101. [DOI] [PubMed] [Google Scholar]
- 2.Dingledine, R., K. Borges, D. Bowie, and S. F. Traynelis. 1999. The glutamate receptor ion channels. Pharmacol. Rev. 51:7–61. [PubMed] [Google Scholar]
- 3.Armstrong, N., and E. Gouaux. 2000. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 28:165–181. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong, N., Y. Sun, G. Q. Chen, and E. Gouaux. 1998. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 395:913–917. [DOI] [PubMed] [Google Scholar]
- 5.Jin, R., T. G. Banke, M. L. Mayer, S. F. Traynelis, and E. Gouaux. 2003. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 6:803–810. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong, N., M. Mayer, and E. Gouaux. 2003. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc. Natl. Acad. Sci. USA. 100:5736–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin, R., and E. Gouaux. 2003. Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry. 42:5201–5213. [DOI] [PubMed] [Google Scholar]
- 8.Hogner, A., J. S. Kastrup, R. Jin, T. Liljefors, M. L. Mayer, J. Egebjerg, I. K. Larsen, and E. Gouaux. 2002. Structural basis for AMPA receptor activation and ligand selectivity: crystal structures of five agonist complexes with the GluR2 ligand-binding core. J. Mol. Biol. 322:93–109. [DOI] [PubMed] [Google Scholar]
- 9.Robert, A., N. Armstrong, J. E. Gouaux, and J. R. Howe. 2005. AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J. Neurosci. 25:3752–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bang-Andersen, B., S. M. Lenz, N. Skjaerbaek, K. K. Soby, H. O. Hansen, B. Ebert, K. P. Bogeso, and P. Krogsgaard-Larsen. 1997. Heteroaryl analogues of AMPA. Synthesis and quantitative structure-activity relationships. J. Med. Chem. 40:2831–2842. [DOI] [PubMed] [Google Scholar]
- 11.Vogensen, S. B., H. S. Jensen, T. B. Stensbol, K. Frydenvang, B. Bang-Andersen, T. N. Johansen, J. Egebjerg, and P. Krogsgaard-Larsen. 2000. Resolution, configurational assignment, and enantiopharmacology of 2-amino-3-[3-hydroxy-5-(2-methyl-2H- tetrazol-5-yl)isoxazol-4-yl]propionic acid, a potent GluR3- and GluR4-preferring AMPA receptor agonist. Chirality. 12:705–713. [DOI] [PubMed] [Google Scholar]
- 12.Jin, R., M. Horning, M. L. Mayer, and E. Gouaux. 2002. Mechanism of activation and selectivity in a ligand-gated ion channel: structural and functional studies of GluR2 and quisqualate. Biochemistry. 41:15635–15643. [DOI] [PubMed] [Google Scholar]
- 13.Robert, A., and J. R. Howe. 2003. How AMPA receptor desensitization depends on receptor occupancy. J. Neurosci. 23:847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert, A., S. N. Irizarry, T. E. Hughes, and J. R. Howe. 2001. Subunit interactions and AMPA receptor desensitization. J. Neurosci. 21:5574–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horn, R. 1987. Statistical methods for model discrimination. Applications to gating kinetics and permeation of the acetylcholine receptor channel. Biophys. J. 51:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein, R. M., and J. R. Howe. 2004. Effects of the lurcher mutation on GluR1 desensitization and activation kinetics. J. Neurosci. 24:4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abele, R., K. Keinanen, and D. R. Madden. 2000. Agonist-induced isomerization in a glutamate receptor ligand-binding domain. A kinetic and mutagenetic analysis. J. Biol. Chem. 275:21355–21363. [DOI] [PubMed] [Google Scholar]
- 18.Rosenmund, C., Y. Stern-Bach, and C. F. Stevens. 1998. The tetrameric structure of a glutamate receptor channel. Science. 280:1596–1599. [DOI] [PubMed] [Google Scholar]
- 19.Smith, T. C., and J. R. Howe. 2000. Concentration-dependent substate behavior of native AMPA receptors. Nat. Neurosci. 3:992–997. [DOI] [PubMed] [Google Scholar]
- 20.Tomita, S., H. Adesnik, M. Sekiguchi, W. Zhang, K. Wada, J. R. Howe, R. A. Nicoll, and D. S. Bredt. 2005. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 435:1052–1058. [DOI] [PubMed] [Google Scholar]
- 21.Bowie, D., and G. D. Lange. 2002. Functional stoichiometry of glutamate receptor desensitization. J. Neurosci. 22:3392–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colquhoun, D., P. Jonas, and B. Sakmann. 1992. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hip pocampal slices. J. Physiol. 458:261–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patneau, D. K., and M. L. Mayer. 1991. Kinetic analysis of interactions between kainate and AMPA: evidence for activation of a single receptor in mouse hippocampal neurons. Neuron. 6:785–798. [DOI] [PubMed] [Google Scholar]
- 24.Chen, L., D. M. Chetkovich, R. S. Petralia, N. T. Sweeney, Y. Kawasaki, R. J. Wenthold, D. S. Bredt, and R. A. Nicoll. 2000. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 408:936–943. [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto, K., M. Fukaya, X. Qiao, K. Sakimura, M. Watanabe, and M. Kano. 1999. Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J. Neurosci. 19:6027–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomita, S., L. Chen, Y. Kawasaki, R. S. Petralia, R. J. Wenthold, R. A. Nicoll, and D. S. Bredt. 2003. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J. Cell Biol. 161:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomita, S., M. Fukata, R. A. Nicoll, and D. S. Bredt. 2004. Dynamic interaction of stargazin-like TARPs with cycling AMPA receptors at synapses. Science. 303:1508–1511. [DOI] [PubMed] [Google Scholar]
- 28.Priel, A., A. Kolleker, G. Ayalon, M. Gillor, P. Osten, and Y. Stern-Bach. 2005. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J. Neurosci. 25:2682–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turetsky, D., E. Garringer, and D. K. Patneau. 2005. Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J. Neurosci. 25:7438–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raman, I. M., and L. O. Trussell. 1992. The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron. 9:173–186. [DOI] [PubMed] [Google Scholar]
- 31.Raman, I. M., and L. O. Trussell. 1995. The mechanism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor desensitization after removal of glutamate. Biophys. J. 68:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun, Y., R. Olson, M. Horning, N. Armstrong, M. Mayer, and E. Gouaux. 2002. Mechanism of glutamate receptor desensitization. Nature. 417:245–253. [DOI] [PubMed] [Google Scholar]
- 33.Burzomato, V., M. Beato, P. J. Groot-Kormelink, D. Colquhoun, and L. G. Sivilotti. 2004. Single-channel behavior of heteromeric alpha1beta glycine receptors: an attempt to detect a conformational change before the channel opens. J. Neurosci. 24:10924–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popescu, G., A. Robert, J. R. Howe, and A. Auerbach. 2004. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature. 430:790–793. [DOI] [PubMed] [Google Scholar]
- 35.Jonas, P. 2000. The time course of signaling at central glutamatergic synapses. News Physiol. Sci. 15:83–89. [DOI] [PubMed] [Google Scholar]
- 36.Cathala, L., N. B. Holderith, Z. Nusser, D. A. DiGregorio, and S. G. Cull-Candy. 2005. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat. Neurosci. 8:1310–1318. [DOI] [PubMed] [Google Scholar]
- 37.Diamond, J. S., and C. E. Jahr. 1995. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 15:1097–1107. [DOI] [PubMed] [Google Scholar]
- 38.DiGregorio, D. A., Z. Nusser, and R. A. Silver. 2002. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 35:521–533. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen, T. A., D. A. DiGregorio, and R. A. Silver. 2004. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron. 42:757–771. [DOI] [PubMed] [Google Scholar]
- 40.Patneau, D. K., L. Vyklicky Jr., and M. L. Mayer. 1993. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J. Neurosci. 13:3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silver, R. A., S. G. Cull-Candy, and T. Takahashi. 1996. Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J. Physiol. 494:231–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi, M., Y. Kovalchuk, and D. Attwell. 1995. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J. Neurosci. 15:5693–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taschenberger, H., R. M. Leao, K. C. Rowland, G. A. Spirou, and H. von Gersdorff. 2002. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 36:1127–1143. [DOI] [PubMed] [Google Scholar]
- 44.Taschenberger, H., and H. von Gersdorff. 2000. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J. Neurosci. 20:9162–9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wall, M. J., A. Robert, J. R. Howe, and M. M. Usowicz. 2002. The speeding of EPSC kinetics during maturation of a central synapse. Eur. J. Neurosci. 15:785–797. [DOI] [PubMed] [Google Scholar]
- 46.Arvola, M., and K. Keinanen. 1996. Characterization of the ligand-binding domains of glutamate receptor (GluR)-B and GluR-D subunits expressed in Escherichia coli as periplasmic proteins. J. Biol. Chem. 271:15527–15532. [DOI] [PubMed] [Google Scholar]
- 47.Chen, G. Q., and E. Gouaux. 1997. Overexpression of a glutamate receptor (GluR2) ligand binding domain in Escherichia coli: application of a novel protein folding screen. Proc. Natl. Acad. Sci. USA. 94:13431–13436. [DOI] [PMC free article] [PubMed] [Google Scholar]