

Abstract
Cellular metabolites are moieties defined by their specific binding constants to H+, Mg2+, and K+ or anions without ligands. As a consequence, every biochemical reaction in the cytoplasm has an associated proton stoichiometry that is generally noninteger- and pH-dependent. Therefore, with metabolic flux, pH is altered in a medium with finite buffer capacity. Apparent equilibrium constants and maximum enzyme velocities, which are functions of pH, are also altered. We augmented an earlier mathematical model of skeletal muscle glycogenolysis with pH-dependent enzyme kinetics and reaction equilibria to compute the time course of pH changes. Analysis shows that kinetics and final equilibrium states of the closed system are highly constrained by the pH-dependent parameters. This kinetic model of glycogenolysis, coupled to creatine kinase and adenylate kinase, simulated published experiments made with a cell-free enzyme mixture to reconstitute the network and to synthesize PCr and lactate in vitro. Using the enzyme kinetic and thermodynamic data in the literature, the simulations required minimal adjustments of parameters to describe the data. These results show that incorporation of appropriate physical chemistry of the reactions with accurate kinetic modeling gives a reasonable simulation of experimental data and is necessary for a physically correct representation of the metabolic network. The approach is general for modeling metabolic networks beyond the specific pathway and conditions presented here.
INTRODUCTION
It is well known that pH has significant kinetic and thermodynamic effects on biochemical reactions since H+ reacts rapidly with metabolites forming near-equilibrium mixtures of dissociated anions and undissociated acid forms of the metabolite. The importance of pH in a reaction mixture was recognized by George and Rutman (1,2) when they demonstrated that the large free energy of ATP hydrolysis at pH 7 is due to the extremely favorable proton release as the reaction advanced in a medium of constant pH near neutrality. The same principle of near-equilibrium mixtures holds for other ligands binding to metabolites, e.g., Mg2+ and other metal cations when the free cation concentrations are fixed (3,4). Thus it is recognized that the apparent free energy of a reaction depends on the composition of the reaction medium (pH, ionic strength, and metal ions that have significant binding constants) and its temperature; for a historical review of these developments, see Alberty (5). These effects provide strong constraints on a system of biochemical reactions with the important consequence that the behavior of a simulated metabolic pathway is strongly constrained by its thermodynamics, i.e., by the apparent equilibrium constants of its constituent biochemical reactions, as well as by the details of enzyme kinetics. Both reaction equilibria and kinetics become interdependent when a proton release and uptake in a buffered medium is part of the reaction stoichiometry. A body of literature exists wherein effects of pH, temperature, and Mg2+ concentration on biochemical reaction equilibria were treated both theoretically and experimentally for selected reactions (6–9). However, a comprehensive model that quantifies H+ uptake and release by all biochemical reactions in a metabolic pathway and the effects of the resultant pH change on reaction kinetics and thermodynamics has not been proposed for any realistic cellular condition. This article develops such a model for glycogenolysis and glycolysis, and tests the power of the model to simulate experimental results.
We first define the procedure for computing the pH time course due to biochemical reactions in an environment that has finite buffering approximating cellular conditions beginning from binding equilibria of metabolic species and proton stoichiometry of biochemical reactions. These computations are then used to make a rigorous treatment of biochemical reaction equilibria as a function of pH, temperature, ionic strength, and metal ion concentrations and to incorporate this information into a kinetic model of the reactions. We are not aware of any metabolic model for biochemical reactions in cells that computed pH time course dynamically while incorporating pH effects on biochemical reaction kinetics and thermodynamics. We note, however, that Beard et al. (10) did systematically include ways to incorporate thermodynamic constraints into biochemical models and that Mulquiney et al. (11) and Mulquiney and Kuchel (12,13) included pH effects on kinetics of selected reactions in a detailed model of erythrocyte glycolysis. We test the validity of the resultant pH-dependent model by analysis of glycogenolytic flux and pH time course measured in a reconstituted mixture of enzymes in vitro (14,15). Our starting point is a mathematical model adapted for skeletal muscle glycogenolysis (16), which is augmented by addition of a glycerol-3-phosphate dehydrogenase kinetic model. We dynamically compute proton stoichiometries, pH-dependent apparent equilibrium constants of all biochemical reactions in that model, and pH dependence of enzyme activities of some of the enzymes. Then we incorporate these computations in that kinetic model with simultaneous computation of the pH time course. The effects of ionic strength and temperature on these computations are considered whenever the relevant data are available.
A realistic model and simulation could add to the understanding of the biochemical pathway for glycogenolysis and glycolysis. Research on this pathway has had an extraordinarily rich history in muscle energy metabolism starting at the beginning of the last century when glycogen breakdown to lactic acid was thought to be the primary chemical reaction driving muscle contraction (17,18). A significant problem has been the identification of the reaction(s) that is (are) the source of glycolytic acid production (17,19–21). While it is clear that the lactate dehydrogenase reaction itself does not generate acid, the mechanistic question of the source(s) of acid production in the glycolytic network has not been rigorously answered in any cell despite considerable progress in mammalian erythrocytes (11–13) and much earlier in several mammalian cell types by the work of Garfinkel and colleagues (22–24). Analysis of glycogenolysis and glycolysis in a metabolically closed cell system (one without exchange of metabolites into or out of the cell) will allow strict accounting of mass balance. Glycogenolysis and glycolysis in a muscle cell is ideal for these purposes and a basic model was recently developed for these purposes (16).
METHODS
pH and Temperature Effects on Equilibria and Kinetics of Biochemical Reactions gives the logic and principles from physical chemistry needed for computing proton stoichiometries and apparent equilibrium constants of biochemical reactions and describes how these are incorporated into computations with kinetic models of metabolic pathways in general. Our work is built on the solid foundation provided by the work of Alberty and colleagues (6), and we use the nomenclature and symbols given therein as defined in Table 1. The differential equation for pH is also derived in this section based on proton mass balance constraint. Application of pH and Kapp and Vmax(pH) Computations to Glycogenolysis applies the principles developed in the first part specifically to an in vitro enzyme mixture reconstituting muscle glycogenolysis from Scopes (14,15). Simulation of Scopes' Experiments and Fitting the Model to Data describes the methods for software implementation and numerical computations, including those for solving ordinary differential equations, parameter adjustment, computing parameter sensitivities, and parameter optimization. At several places we also describe how to apply this analysis to a realistic skeletal muscle, and the additions needed to model an intact muscle cell as an open system compared to a closed system in vitro.
TABLE 1.
Nomenclature
| Symbol | Definition | Units |
|---|---|---|
| I | Ionic strength | M |
| Ka,ℓ | Dissociation constant for the lth protonation reaction | M |
| pKaℓ | −log10 (Ka,ℓ/c0) | Dimensionless |
| KM,m | Dissociation constant for metabolite binding to metal 
|
M |
| pKaM,m | −log10 (KM,m/c0) | Dimensionless |
| c0 | Reference concentration for all species (1 M) | M |
| Pi | Binding polynomial for ith metabolite | Dimensionless |
 |
Average proton binding of ith metabolite at specified T, P, pH, pMg, and I | Dimensionless, noninteger |
| nk | Proton consumption stoichiometry of the kth reference reaction | Dimensionless |
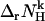 |
Proton consumption stoichiometry of the kth biochemical reaction at specified T, P, pH, pMg, and I | Dimensionless, noninteger |
| NH(j) | Number of H atoms in species j | Dimensionless, integer |
| zj | Charge on species j | Dimensionless, integer |
 (I = 0) (I = 0) |
Gibbs energy of formation at zero ionic strength of species j | KJ/mol |
 |
Stoichiometric coefficient of species j in the kth reference reaction | |
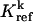 |
Equilibrium constant of kth reference reaction | Dimensionless |
 |
Standard free energy of kth reference reaction | KJ/mol |
 |
Apparent equilibrium constant of kth biochemical reaction | Dimensionless |
 |
Standard transformed free energy of kth biochemical reaction | KJ/mol |
 |
Standard enthalpy of jth proton dissociation reaction at a given ionic strength | KJ/mol |
 |
Flux through reaction kth biochemical reaction | M/min |
| Ci | Concentration of metabolite i | M |
| β | Buffer capacity for pH | M |
pH and temperature effects on equilibria and kinetics of biochemical reactions
The pH and temperature of the milieu affects biochemical reactions in two ways: reaction equilibria and enzyme activity.
Reaction stoichiometry, proton balance, and calculation of intracellular pH
Most intracellular metabolites are anions at the pH of the cellular milieu and exist in various forms bound to protons and metal cations such as K+ and Mg2+. The binding reactions occur simultaneously and independently, and the equilibria of the multiple species depend on pH and cation concentration as well as on the concentration of all the other ligands. As the reactions proceed, redistribution of ion-bound forms of a metabolite as fractions of the total metabolite concentration governs the proton stoichiometry of reactions in which it participates. Proton stoichiometry of each biochemical reaction is computed by accounting for protons bound to reactants and products, along with the net change in number of protons in the chemical structures of reactants and products in its reference reaction (6). These principles are derived from thermodynamics of electrolyte solutions (25).
Muscle cells have a finite buffer capacity that is itself a function of the physiological status of the cell, which results in variation of pH due to metabolic proton loads. This situation contrasts to most calculations and experiments in vitro, which are implemented with essentially infinite buffer capacity. For example, Alberty (6) elaborates proton stoichiometry computation for biochemical reactions when the pH or free Mg2+ concentration is fixed. Na+ binding is similar to K+, and Ca2+ binding is similar to Mg2+, but their concentrations are an order-of-magnitude smaller in the cell. For this reason they were not included in our analyses, but could be with a modest increase in the complexity of the algebraic functions used.
Buffer capacity is defined as the number of strong base equivalents added or protons consumed, divided by the corresponding pH change in the reaction medium (see (26)). In a dynamic situation, the rate of change of pH equals the rate of proton consumption divided by the buffer capacity, since the numerator and denominator are functions of reaction flux and therefore of time. The rate of proton consumption in a reaction is its proton consumption stoichiometry multiplied by the flux through the reaction. Summing this flux for all reactions under consideration gives the total proton consumption flux, which, when divided by the buffer capacity of the medium at that time, gives the rate of change of pH. Solving this differential equation simultaneously with the concentration differential equations of the biochemical system gives the pH time course. Solution of this problem requires accounting for the total proton binding, cation binding, the distribution of these moieties as fractions of the total metabolite present, and finally, for the change in total buffer capacity with the dynamic metabolic flux. Accounting for proton and other cation stoichiometries in biochemical reactions also makes them balanced with respect to charge. The details of these calculations are given below.
Calculation of proton binding fraction for each metabolite using multiple cation equilibria
Let [L] represent the concentration of unbound anionic form of metabolite L and [Ltotal] be its total concentration in all forms,
 |
(1) |
where H is a proton and Mm is the mth metal ion. The second term on the right refers to the sum of the protonated moieties and the third term refers to the sum of the metal-bound moieties. Here we consider only K+ and Mg2+ but any number of cations can be included if their dissociation constants are known. We assume that, at most, only one metal ion of each type binds to L. Additionally, we do not consider proton binding to any metal-bound species, because their contribution is small, although these forms could also be added as done previously (8). Note that the net charge on each of the species is not stated here for simplicity of notation, but this information can be derived from the information given in Tables 2 and 3. The concentration [LHp] is given by the equilibrium relation
 |
(2) |
where Ka,l is the dissociation constant for the reaction,  and
and  at a given temperature and ionic strength.
at a given temperature and ionic strength.
TABLE 2.
pKa (I = 0.1), free energy of formation, and enthalpy of dissociation (I = 0) values used in model at T = 298 K
| Reference species |  |
NH(j) | pKa1 | ΔH(I = 0) | pKa2 | ΔH(I = 0) | pKaMg | ΔH(I = 0) | pKaK | ΔH(I = 0) |
|---|---|---|---|---|---|---|---|---|---|---|
| G1P2− | −1756.87 | 11 | 6.09 | −1.7 | — | — | 2.48 | −12 | — | — |
| G6P2− | −1763.94 | 11 | 6.11* | — | — | — | — | — | — | — |
| F6P2− | −1760.80 | 11 | 5.89 | — | — | — | — | — | — | — |
| FDP4− | −2601.40 | 10 | 6.4 | — | 5.92 | — | 2.7 | — | — | — |
| G3P2− | −1339.25† | 5 | 6.22 | −3.1 | — | — | 1.63 | — | — | — |
| DHAP2− | −1296.26 | 5 | 5.9 | — | — | — | 1.57 | — | — | — |
| GAP2− | −1288.60 | 5 | 6.45* | — | — | — | — | — | — | |
| 13DPG4− | −2356.14 | 4 | 7.5* | — | — | — | — | — | — | |
| 3PG3− | −1502.54 | 4 | 6.21‡ | — | — | — | — | — | — | |
| 2PG3− | −1496.38 | 4 | 7.0 | — | — | — | 2.45 | — | 1.18 | — |
| PEP3− | −1263.65 | 2 | 6.35 | — | — | — | 2.26 | — | 1.08 | — |
| PYR− | −472.27 | 3 | 2.49* | — | — | — | — | — | — | |
| LAC− | −516.72 | 5 | 3.67 | −0.33 | — | — | 0.98 | — | — | — |
| HPi2− | −1096.10 | 1 | 6.75 | 3 | — | — | 1.65 | −2.9 | 0.5 | — |
| IMP2− | Not avail. | 11 | 6.34 | −2 | — | — | 1.67 | — | — | — |
| AMP2− | −1040.45 | 12 | 6.29 | −3 | — | — | 1.92 | −7.5 | — | — |
| ADP3− | −1906.13 | 12 | 6.38 | −3 | — | — | 3.25 | −15 | 1.0 | — |
| ATP4− | −2768.10 | 12 | 6.48 | −5 | — | — | 4.19 | −18 | 1.17 | −1 |
| HPCr2− | Not avail. | 9 | 4.5¶ | 2.66§ | — | — | 1.6 | 8.19§ | 0.31¶ | — |
| HCr0 | Not avail. | 8 | 2.63 | — | — | — | — | — | — | — |
| Glycogen(n)‖ | 0 | 10n+1‖ | — | — | — | — | — | — | — | — |
| Glycogen(n–1)‖ | 679.1‖ | 10(n−1)+‖ | — | — | — | — | — | — | — | — |
 |
0 | 26 | — | — | — | — | — | — | — | — |
 |
22.65** | 27 | — | — | — | — | — | — | — | — |
| H+ | 0 | 1 | — | — | — | — | — | — | — | — |
References are from the NIST Database 46: Critical Stability Constants (29), unless noted otherwise.
Dawson et al. (26).
See Thermodynamic Parameters (in text).
Robitaille and Robitaille (53).
Kushmerick (8).
Teague et al. (30).
Beard and Qian (54); note also that n indicates number of glycosyl units.
Alberty (6).
TABLE 3.
Definition of reference reactions
| Enzyme (EC No.) | Reference reaction | n | ΔrNH(pH = 7.4)* |
 * *
|
|---|---|---|---|---|
| Glycogen phosphorylase (2.4.1.1) | Glycogen(n) + HPi2− = G1P2− + Glycogen(n−1) | 0 | −0.101 | 1.54 |
| Phosphogluco mutase (2.7.1.41) | G1P2− = G6P2− | 0 | 0.009 | −6.67 |
| Phosphogluco isomerase (5.3.1.9) | G6P2− = F6P2− | 0 | −0.019 | 3.19 |
| Phosphofructokinase (2.7.1.11) | F6P2− + ATP4− = FDP4− + ADP3− + H+ | −1 | −0.929 | −25.4 |
| Aldolase (4.1.2.13) | FDP4− = DHAP2− + GAP2− | 0 | 0.053 | 20.5 |
| Triosephosphate isomerase (5.3.1.1) | GAP2− = DHAP2− | 0 | −0.071 | −7.51 |
| Glyceraldehyde-3-phosphate dehydrogenase (1.2.1.12) |  |
−1 | −0.685 | −0.8781 |
| Glycerol-3-phosphate dehydrogenase (1.1.1.8) |  |
−1 | −1.03 | 19.34 |
| Phosphoglycerate kinase (2.7.2.3) | DPG4− + ATP3− = 3PG3− + ATP4− | 0 | −0.519 | −10.02 |
| Phosphoglycerate mutase (5.4.2.1) | 3PG3− = 2PG3− | 0 | 0.084 | 3.71 |
| Enolase (4.2.1.11) | 2PG3− = PEP3− + H2O | 0 | −0.103 | −3.82 |
| Pyruvate kinase (2.7.1.40) | PEP3− + ADP3− + H+ = PYR− + ATP4− | 1 | 0.94 | −27.05 |
| Lactate dehydrogenase (1.1.1.27) |  |
1 | 1 | −21.73 |
| ATP hydrolysis | ATP4− + H2O = ADP3− + HPi2− + H+ | −1 | −0.84 | −37.08 |
| Creatine kinase (2.7.3.2) | HPCr2− + ADP3− + H+ HCr0 + ATP4− | 1 | 0.98 | −7.16 |
| Adenylate kinase (2.7.4.3) | ATP4− + AMP2− = 2ADP3− | 0 | −0.016 | −1.41 |
| AMP deaminase (3.5.4.6) |  |
1 | 1.01† | — |
T = 303.15 K, I = 0.1 M, K+ = 0.08 M, Mg2+ = 5.1e–4 M.
T = 303.15 K, I = 0.14 M, K+ = 0.12 M, Mg2+ = 7.3e–4 M.
The pKa values listed in Table 2 were reported at 298.15 K and ionic strength 0.1 M whereas the conditions to be simulated and experiments to be modeled were performed at a range of temperatures and ionic strengths. The pKa value at the given experimental temperature was calculated as shown below, assuming that the standard enthalpy ΔH0 of the dissociation reaction is constant over that temperature range. This relation can be obtained by integrating the van 't Hoff equation  from T1 to T2:
from T1 to T2:
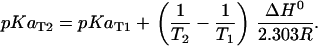 |
(3) |
T2 is the given experimental temperature, and T1 = 298.15 K and R = 8.314 J/K/mol is the universal gas constant.
The enthalpy of dissociation reactions is a function of ionic strength. Table 2 lists the available enthalpies at zero ionic strength whereas experimental ionic strength is nonzero. The following empirical equation (see Eqs. 3.6–5 in (6)) gives ionic strength dependence of enthalpy at 298.15 K,
 |
(4) |
The pK values which are reported at ionic strength I1 = 0.1 M, were corrected to ionic strength I of experimental or physiological medium using the empirical equation (6) of
 |
(5) |
The numerical constants in Eqs. 4 and 5 are derived from Debye-Hückel theory in sections 3.6 and 3.7 of Alberty (6), based on the analysis of Clarke and Glew (27). In summary, the pK value at 298.15 K and 0.1 M ionic strength is first corrected for the experimental ionic strength using Eq. 5. The ionic-strength-corrected pK value at 298.15 K is then temperature-corrected using Eq. 3, in which the dissociation enthalpy is first corrected for ionic strength using Eq. 4.
The concentration of each of the mth metal bound species [LMm] is given by
 |
(6) |
We assume in Eq. 6 that each species binds, at most, one metal ion per molecule.
From the above equations the average proton binding for metabolite L is given by
 |
(7) |
which is the average number of dissociable protons covalently bound to L. The value p in Eqs. 2 and 7 denotes the number of dissociable protons bound in each proton-bound form of the metabolite L.
The average number of protons bound for each metabolite calculated by Eq. 7 considers all ligands and is the important quantity needed to proceed with our analysis.
Proton consumption stoichiometry of a biochemical reaction
The first step in computing the stoichiometry for proton consumption of a biochemical reaction advancing to a unit extent is to define the reference reaction, which is the reaction defined in terms of the reference species of the metabolites. The reference species are defined as the most deprotonated species in the pH range 5.5–8.5, which are used to calculate the average proton binding  for each metabolite L. We use the most deprotonated form of metabolites over the range of pH 5.5–8.5 as the reference species, noting that other conventions are possible but we found them less tractable.
for each metabolite L. We use the most deprotonated form of metabolites over the range of pH 5.5–8.5 as the reference species, noting that other conventions are possible but we found them less tractable.
Protons are included on the reactant or product side to balance the reference reaction with respect to both mass and charge (Table 3). Proton consumption stoichiometry for the reference reaction is denoted by n, which is the difference in protons between the reactants and the products in their reference states. The creatine kinase reaction is shown below in terms of its reference species to illustrate this point:
 |
A biochemical reaction is defined in terms of sums of species constituting each of the reactants and products. The creatine kinase reaction is shown below in terms of sums of species for each metabolite:
 |
The proton imbalance in a biochemical reaction is the proton consumption stoichiometry, ΔrNH, which is the difference between the average proton binding of the reactants and the products plus the proton consumption stoichiometry, n, of the reference reaction. Defined in this way, n is an integer but ΔrNH is not because the first two terms on the RHS of Eq. 8, which are sums of average proton binding of the reactants and the products from Eq. 7, are nonintegers. Note that the first two terms on the right-hand side of Eq. 8 go to zero at highly alkaline pH values,
 |
(8) |
The reference reaction is also balanced with respect to charge via the n protons consumed. The biochemical reaction, defined in terms of sums of species for each metabolite, is also balanced with respect to charge due to the proton stoichiometry defined in Eq. 8, which includes the binding change in addition to n. Although the reference reactions defined here have zero magnesium and potassium stoichiometries, the biochemical reactions do have a binding change for these ions for some of the metabolites and accounting for them results in the total charge balance for the biochemical reactions. For the kth biochemical reaction in the system, the proton stoichiometry is defined as  and that of its corresponding reference reaction is defined as nk.
and that of its corresponding reference reaction is defined as nk.
pH change due to proton consumption flux through a set of reactions
The proton consumption flux through a reaction is given by the product of the proton consumption stoichiometry for that reaction and the flux through the reaction. For a set of reactions defining a metabolic network, the total proton consumption flux is given by a summation of the individual proton consumption fluxes for all biochemical reactions,
 |
(9) |
where  is the flux through the kth biochemical reaction.
is the flux through the kth biochemical reaction.
Proton consumption flux divided by pH buffer capacity gives the rate of pH change.
Buffer capacity and the differential equation for pH
Buffer capacity β is defined as the derivative of base equivalents added (protons consumed) to the reaction medium with respect to pH. The rates of pH change and proton consumption due to a biochemical reaction network are related through the buffer capacity β as shown below:
 |
(10) |
Equation 10 holds for a constant free magnesium ion concentration, which will be shown through the complete derivation of the pH differential equation in the remainder of this section.
The differential equation for pH and the expression for buffer capacity are derived by starting with a mass balance expression for protons,
 |
(11) |
where [H+] is the free proton concentration,  is the metabolite bound dissociable proton concentration, and
is the metabolite bound dissociable proton concentration, and  is the concentration of protons in the reference species of all metabolites.
is the concentration of protons in the reference species of all metabolites.
Differentiating Eq. 11 with respect to time, we get
 |
(12) |
 is the sum of average proton binding of a metabolite
is the sum of average proton binding of a metabolite  multiplied by the total metabolite concentration (Ci), for all metabolites in the system. Differentiating this summation with respect to time and applying the chain rule, we obtain
multiplied by the total metabolite concentration (Ci), for all metabolites in the system. Differentiating this summation with respect to time and applying the chain rule, we obtain 
 |
(13) |
where  is a function of free proton and magnesium concentration, which are functions of time. This leads to
is a function of free proton and magnesium concentration, which are functions of time. This leads to
 |
(14) |
Note that free potassium concentration is considered a constant and is therefore a parameter in the above equation, whereas change in free Mg2+ with time is included.
The value ( ) is obtained from summation of the product of proton consumption stoichiometry of each of the reference reactions with the flux through the reaction:
) is obtained from summation of the product of proton consumption stoichiometry of each of the reference reactions with the flux through the reaction:
 |
(15) |
The total proton load due to the biochemical reactions is the sum of  and term 3 of the right-hand side of Eq. 14 for
and term 3 of the right-hand side of Eq. 14 for  This proton load is defined by the following equation in terms of the reaction proton consumption stoichiometry and the flux through the reaction
This proton load is defined by the following equation in terms of the reaction proton consumption stoichiometry and the flux through the reaction
 |
(16) |
Using Eqs. 13–16 in Eq. 12 and transposing terms, we obtain the following equation for d[H+]/dt:
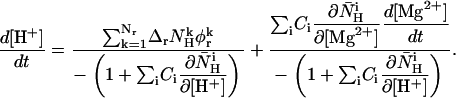 |
(17) |
The value pH is defined as −log10([H+]), which leads to
 |
(18) |
Using Eq. 17 in Eq. 18, we obtain
 |
(19) |
The numerator of the right-hand side of Eq. 19 reduces to  for a constant free Mg2+, since d[Mg2+]/dt = 0 for that case.
for a constant free Mg2+, since d[Mg2+]/dt = 0 for that case.
Comparing Eqs. 10 and 19, we obtain the expression for the buffer capacity β,
 |
(20) |
Equation 20 remains the same for constant or variable free magnesium, since the denominator of Eq. 19 is the same for both cases.
From Eq. 20, buffer capacity (see (26)) of a monoprotic acid HX and its conjugate base X− with a proton binding defined by  is given by
is given by
 |
(21) |
where [X]T = [HX] + [X−]. The buffers in the cytoplasm are not all monoprotic. However, we only consider the proton dissociation reaction with the pKa value close to the physiological pH (±2 log units) in each case. The practical result is that the buffering capacity term for single proton dissociation is used for each metabolite, and each metabolite contributes to the total buffer capacity in the metabolic system. Equation 20 shows that all metabolites (Ci) contribute to proton balance and to buffer capacity.
In muscle, the buffers that still need to be accounted for are the proteins in the cell, the dipeptides carnosine and anserine (together called “fixed buffers”), and the  buffer pair. Note that inorganic phosphate, a well-known component of intracellular buffering in muscle (28), is accounted for in the set of metabolites in Eq. 20. In the Scopes' experiment, inorganic phosphate concentration decreases as creatine is phosphorylated whereas in muscle it increases during periods of increased contractile activity contributing to buffer capacity significantly.
buffer pair. Note that inorganic phosphate, a well-known component of intracellular buffering in muscle (28), is accounted for in the set of metabolites in Eq. 20. In the Scopes' experiment, inorganic phosphate concentration decreases as creatine is phosphorylated whereas in muscle it increases during periods of increased contractile activity contributing to buffer capacity significantly.
We analyzed the biochemical experiments of Scopes (14). Note that Scopes used carnosine and tris buffers for his reconstituted glycolytic system at concentrations given in Table 5. The buffer capacity for his system is calculated by applying Eqs. 20 and 21, as shown:
 |
(22) |
TABLE 5.
Initial solution composition at time zero for Experiment 29
| Metabolite/buffer | Experiment 29: concentration (mM) | Experiment 45: concentration (mM) | Postmortem glycolysis: concentration (mM) |
|---|---|---|---|
| Mg2+ (total) | 5 | 5 | 5 |
| Mg2+ (free)* | 0.513 | 0.491 | 0.728 |
| K+ | 80 | 80 | 120 |
| Pi | 30 | 35 | 6 |
| NAD | 0.5 | 0.5 | 0.5 |
| Cr | 30 | 30 | 6 |
| PCr | 0 | 0 | 24 |
| ATP | 5 | 5 | 5 |
| Glycogen | 40 (in glucose equivalents) | 40 (in glucose equivalents) | 65 |
| Tris | 15 | 15 | 15 |
| Carnosine | 25 | 25 | 25 |
| Acetate | 10 | 10 | 50 |
| Histidine | 0 | 0 | 30 |
Note that, in Experiment 29, pH is allowed to vary freely for the first minute, after which it is kept constant at 7.4 by a pH-stat (pH = 7.8; T = 303 K) in Scopes (14); in Experiment 45, pH is fixed at 7.3 throughout the experiment (pH = 7.3; T = 310 K) in Scopes (14) and postmortem glycolysis simulation (pH = 7.25; T = 303 K) in Scopes (15).
Free Mg2+ at time zero is calculated by solving Eq. 41 for Mg2+ mass balance wherein the total Mg2+ equals sum of free Mg2+ and Mg2+ bound to ATP and Pi, and PCr also in case of the postmortem glycolysis simulation.
Here, 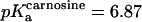 and
and  (T = 298.15 K, see (26)). In the experiment simulating postmortem glycolysis (15), additional buffers Histidine (pKa = 6.3) and Acetate (pKa = 4.8) (29) were added and included in Eq. 22 when that experiment was simulated by our model. Note for situations other than those studied here, additional buffers used can be included in the last term of Eq. 22.
(T = 298.15 K, see (26)). In the experiment simulating postmortem glycolysis (15), additional buffers Histidine (pKa = 6.3) and Acetate (pKa = 4.8) (29) were added and included in Eq. 22 when that experiment was simulated by our model. Note for situations other than those studied here, additional buffers used can be included in the last term of Eq. 22.
Computation of apparent equilibrium constant as a function of pH
Because the biochemical reaction network is defined as fully reversible, changes in the apparent equilibrium constant due to pH will impact the reverse Vmax as defined in the model by the Haldane relationship.
The standard free energy of the kth reference reaction is the difference between the free energies of formation of the products and the reactants in that reaction,
 |
(23) |
The free energy of formation of each reference species is a function of pH and ionic strength, which is an empirical expression taken from Alberty (6) at 298.15 K,
 |
(24) |
Since the reference reaction is balanced with respect to protons, the free energy of the reference reaction,  is independent of pH and therefore a constant at a given ionic strength and temperature.
is independent of pH and therefore a constant at a given ionic strength and temperature.
The equilibrium constant of the reference reaction is given by
 |
(25) |
Apparent equilibrium constant of a biochemical reaction is defined in terms of the metabolite concentrations at equilibrium (indicated by the subscript eq in Eq. 26), which are sums of their constituent species,
 |
(26) |
where n is the proton stoichiometry of the reference reaction, and P is the binding polynomial for a metabolite given by
 |
(27) |
The standard transformed free energy for a biochemical reaction is given by
 |
(28) |
The apparent equilibrium constant of creatine kinase reaction was calculated starting from the Kref value reported by Teague et al. (30) at 298.15 K and 0 M ionic strength with appropriate temperature and ionic-strength corrections, since the free energies of formation of HCr and PCr2− were unavailable. The Kref value reported was 2.58e8 and the standard enthalpy of the reference reaction was 17.55 KJ/mol.
In summary, the following steps and calculations are needed to account for H+ and metal cation binding to metabolites, buffer capacity, and pH change, and the effects of these parameters on reaction equilibria, to calculate:
The binding polynomial for each metabolite that describes the distribution of all forms of the metabolite as mole fractions of the total and the average number of protons in each metabolite as a function of pH.
The stoichiometry of net proton consumption (or uptake) for each biochemical reaction by tallying the number of protons in the products minus the number in the reactants.
The buffer capacity.
The pH change due to net proton change in the reaction network.
The apparent equilibrium constant as a function of pH and the relevant ionic strength.
The model analyses and simulations in this article include these calculations in the progress curves at the specified pH, temperature, and ionic strength.
Enzyme activity as a function of pH: pH effects on kinetics
Enzyme activities depend on pH. These mechanisms influence only the rate of a reaction in its dynamic response to a perturbation from one steady or equilibrium state to another and include pH effects on the binding of substrate or product and on catalytic rate due to altered structure and charge of the enzyme. These effects are complex and discussed in detail in textbooks on enzymology (e.g., (31,32)). We used an empirical approach to adjust enzyme activity as a function of pH in our simulations. The dependence of enzyme activity on pH over the physiological range has been reported for most of the glycolytic enzymes (33–41). These data were fit to an algebraic function for each enzyme and normalized such that at optimal pH reported, the algebraic function is equal to 100% activity. The enzyme activity at a specified pH is a product of this algebraic function and the activity at optimal pH, i.e., the maximal activities reported in Table 4. Thus, changes from the optimal pH in the simulation or in the fitting of data will result in a fraction of the specific activity given in Table 4 according to the value of the algebraic function. In this way Vmax is a function of pH in the model. The following algebraic functions were obtained from data reported in the literature in the pH range listed for each function:
 |
(29) |
 |
(30) |
 |
(31) |
 |
(32) |
 |
(33) |
 |
(34) |
 |
(35) |
 |
(36) |
 |
(37) |
 |
(38) |
TABLE 4.
Enzyme activities
| Enzyme (EC No.) | Enzyme abbreviation | Activity (U/mg)* | Concentration (mg/ml) | Species | Vmax†(M/min) |
|---|---|---|---|---|---|
| Glycogen phosphorylase (2.4.1.1) | GPa + GPb | 25 | 2 | Rabbit | 0.05 |
| Phosphogluco mutase (2.7.1.41) | PGLM | 800 | 0.6 | Pig | 0.48 |
| Phosphogluco isomerase (5.3.1.9) | PGI | 1100 | 0.8 | Pig | 0.88 |
| Phosphofructokinase (2.7.1.11) | PFK | 160 | 0.35 | Rabbit | 0.056 |
| Aldolase (4.1.2.13) | ALD | 16 | 6.5 | Rabbit | 0.104 |
| Triosephosphate isomerase (5.3.1.1) | TPI | 6000 | 2 | Rabbit | 12.0 |
| Glyceraldehyde-3-phosphate dehydrogenase (1.2.1.12) | GAPDH | 115 | 11 | Rabbit | 1.265 |
| Glycerol-3-phosphate dehydrogenase (1.1.1.8) | G3PDH | 275 | 0.3 | Rabbit | 0.0825 |
| Phosphoglycerate kinase (2.7.2.3) | PGK | 800 | 1.4 | Rabbit | 1.12 |
| Phosphoglycerate mutase (5.4.2.1) | PGM | 1400 | 0.8 | Pig | 1.12 |
| Enolase (4.2.1.11) | EN | 80 | 2.4 | Rabbit | 0.192 |
| Pyruvate kinase (2.7.1.40) | PK | 450 | 3.2 | Rabbit | 1.44 |
| Lactate dehydrogenase (1.1.1.27) | LDH | 600 | 3.2 | Pig | 1.92 |
| Adenylate kinase (2.7.4.3) | ADK | 2200 | 0.4 | Pig | 0.88 |
| Creatine kinase (2.7.3.2) | CK | 100 | 5 | Pig | 0.5 |
The above equations are valid for the pH range specified in the data from which they are derived. Equations of the form  which are bell-shaped functions with a maximum at the pH optimum, are based on a very simplified picture of the enzyme as a dibasic acid (31,32) and therefore have some mechanistic interpretation. Extrapolations of these functions outside of the pH range specified may therefore not be totally incorrect as opposed to simple polynomial functions, which are more likely to be incorrect outside of the pH range from which they are obtained. The above expressions apply for simulation of Experiments 29 and 45 in which the pH range is 7.2–7.8. For the postmortem glycolysis simulation, the pH range is 7.25–5.5, which requires extrapolation of some of the equations described above. The pH dependence of LDH activity described by a straight line covers the entire pH range encountered in both studies. We did not find data for the pH-dependence of activity in the physiological range for the enzymes G3PDH, TPI, PGK, and EN; therefore, these reactions' kinetics have no dependence on pH in the simulation but do have an accounting for pH dependence on reaction equilibria. When correction for temperature was required, a rate coefficient Q10 = 1.9 was used. We did not include the pH dependence of creatine kinase (CK) and adenylate kinase (ADK) activities because their activities in this experiment are very high and the reactions are very close to equilibrium. The sensitivity analysis portrayed in Fig. 14 also shows that a 10% change in the activities of these enzymes has negligible effect on the measured concentration variables. Note that the dependence of reaction fluxes in these simulations follows standard experimental practice. Each flux is modeled as a function of concentrations of metabolites involved in the corresponding biochemical reaction as described in Lambeth and Kushmerick (16).
which are bell-shaped functions with a maximum at the pH optimum, are based on a very simplified picture of the enzyme as a dibasic acid (31,32) and therefore have some mechanistic interpretation. Extrapolations of these functions outside of the pH range specified may therefore not be totally incorrect as opposed to simple polynomial functions, which are more likely to be incorrect outside of the pH range from which they are obtained. The above expressions apply for simulation of Experiments 29 and 45 in which the pH range is 7.2–7.8. For the postmortem glycolysis simulation, the pH range is 7.25–5.5, which requires extrapolation of some of the equations described above. The pH dependence of LDH activity described by a straight line covers the entire pH range encountered in both studies. We did not find data for the pH-dependence of activity in the physiological range for the enzymes G3PDH, TPI, PGK, and EN; therefore, these reactions' kinetics have no dependence on pH in the simulation but do have an accounting for pH dependence on reaction equilibria. When correction for temperature was required, a rate coefficient Q10 = 1.9 was used. We did not include the pH dependence of creatine kinase (CK) and adenylate kinase (ADK) activities because their activities in this experiment are very high and the reactions are very close to equilibrium. The sensitivity analysis portrayed in Fig. 14 also shows that a 10% change in the activities of these enzymes has negligible effect on the measured concentration variables. Note that the dependence of reaction fluxes in these simulations follows standard experimental practice. Each flux is modeled as a function of concentrations of metabolites involved in the corresponding biochemical reaction as described in Lambeth and Kushmerick (16).
FIGURE 14.

Row and column normalized grayscale image maps of mean-square error sensitivity maps of the nine variables with respect to all kinetic parameters in Experiment 29: GPb 99.8% are shown respectively in the left- and right-hand panels. Columns 1–9 represent the nine measurements: 1, PCr; 2, lactate; 3, ATP; 4, ADP; 5, AMP; 6, hexose monophosphates; 7, fructose diphosphates; 8, glycerol-3-phosphate; and 9, phosphoglycerates. The rows correspond to enzyme kinetic parameters and are labeled with the abbreviated enzyme name at the start of each enzyme's parameter group, which is the Vmax of that enzyme. The rest of the kinetic and allosteric parameters are all included in the Supplementary Material and follow their order of presentation in a detailed tabular form defining all of the parameters and their values. After the enzyme kinetic parameters, sensitivities with respect to ionic-strength I followed by the buffer concentrations are shown in the figure.
In this section we have adjusted enzyme activity as a function of pH empirically. This empirical adjustment would account for observed variation of enzyme activity, which could be due to variation of enzyme affinity to the substrate with pH and/or a variation in distribution of the substrate ionic forms. In the subsection above, Reaction Stoichiometry, Proton Balance, and Calculation of Intracellular pH, we computed the distribution of all moieties of metabolites, used in the computation of proton stoichiometries of biochemical reactions and the apparent equilibrium constants. However, the fluxes are simulated as functions of metabolite concentrations (sums of species), as observed experimentally.
Application of pH and Kapp and Vmax(pH) computations to glycogenolysis
Experiments by Scopes (14,15) used a cell-free reconstituted glycolytic system in a defined buffer solution. In the 1973 study (14), Scopes asked whether the reconstituted in vitro system of glycogenolysis and glycolysis could synthesize lactate and phosphocreatine. In the 1974 study (15), Scopes let the same reconstituted glycogenolytic system go toward equilibrium in the presence of ATPase and AMP deaminase, experimentally simulating postmortem glycolysis. The enzymes were purified from porcine or rabbit muscle and reconstituted in appropriate activities measured in extracts of mixed muscle; the reported values for enzyme activity (Table 4) were used in the enzyme flux equations of the model. The composition of the buffer solution and initial metabolite concentrations were the same as reported (Table 5). Scopes' experimental results were simulated by the model described above to test the validity of the model with the dynamic biochemical results.
The reconstituted system used by Scopes (14) consisted of rabbit muscle glycerol-3-phophate dehydrogenase (G3PDH; EC No. 1.1.1.8) in addition to the glycogenolytic enzymes modeled in Lambeth and Kushmerick (16). We used the following kinetic equation (42,43) and Haldane constraint for the flux through G3PDH with parameters from rabbit muscle:
 |
(39) |
The value KG3P = 0.18 mM; KNAD = 0.012 mM; KDHAP = 0.22 mM; and KNADH = 0.008 mM (44). The value Vmaxr is defined in Table 5 and the Haldane constraint was used to define Vmaxf:
 |
(40) |
The value  was computed as a function of pH using methods described in Reaction Stoichiometry, Proton Balance, and Calculation of Intracellular pH.
was computed as a function of pH using methods described in Reaction Stoichiometry, Proton Balance, and Calculation of Intracellular pH.
For experiments with added potato ATPase, the following irreversible Michaelis-Menten flux expression was used:
 |
(41) |
Scopes added AMP deaminase in the experiment simulating postmortem glycolysis, for which we use the flux expression
 |
(42) |
The experiment of Scopes (14) was a closed system for all metabolites and had a fixed total Mg2+ concentration:
 |
(43) |
Differentiating Eq. 42 with respect to time, we obtain
 |
(44) |
 |
(45) |
Equations 41 and 42 lead to
 |
(46) |
We consider free K+ as a constant parameter at its given concentration here. Therefore, average magnesium binding  is a function of dynamic variables pH and free magnesium, which results in the following development on expanding the RHS of Eq. 46 using the chain rule of differentiation:
is a function of dynamic variables pH and free magnesium, which results in the following development on expanding the RHS of Eq. 46 using the chain rule of differentiation:
 |
(47) |
 |
(48) |
Rearranging Eq. 48 to obtain the derivative of free magnesium ion concentration, we get
 |
(49) |
The denominator of Eq. 49 is analogous to the expression for pH buffer capacity in Eq. 20.
The following set of differential equations including Eq. 49 comprised the model used for analysis of the experiments in Scopes (14):
 |
(50) |
 |
(51) |
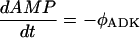 |
(52) |
 |
(53) |
 |
(54) |
 |
(55) |
 |
(56) |
 |
(57) |
 |
(58) |
 |
(59) |
 |
(60) |
 |
(61) |
 |
(62) |
 |
(63) |
 |
(64) |
 |
(65) |
 |
(66) |
 |
(67) |
 |
(68) |
 |
(69) |
 |
(70) |
 |
Fluxes φCK, φADK, φGP, φPGLM, φPGI, φPFK, φALD, φTPI, φGAPDH, φPGK, φPGM, φENOL, φPK, and φLDH are defined in Lambeth and Kushmerick (16). The value φG3PDH is defined by Eqs. 39 and 40. The complete computer code for this model and a table of the parameter values are given in the Supplementary Material.
For simulating the postmortem glycolysis study, the differential equation for AMP was modified to include AMP deaminase flux in the following manner:
 |
(71) |
The differential equation portion of Eq. 70 for pH also holds for the entire duration of this experiment, because pH is allowed to vary freely without any pH-stat.
Simulation of Scopes' experiments and fitting the model to data
The augmented glycogenolysis model with pH computation was fitted to experimental data found in Scopes (14,15); the data shown in plots were translated into numerical tables and thus were subject to rounding and approximation. Four of the experiments in the 1973 study and one from the 1974 study were analyzed in detail with initial conditions given in Table 5:
The time course of PCr resynthesis at a low fraction of 0.2% of the active, a, form of glycogen phosphorylase, Experiment 29: Glycogen Phosphorylase Almost Entirely in b-Form and Figs. 3–6 for Experiment 29 of Scopes (14).
The time course of PCr resynthesis at a higher fraction, 40%, of the a-form of glycogen phosphorylase, Incomplete Predictions If pH Effects Are Ignored and Figs. 8 and 9 for the second part of Experiment 29 of Scopes (14).
PCr synthesis with progressive increases in the fraction of the a-form of glycogen phosphorylase from 0.1% to 4% by adding enzyme during the course of the experiment discussed in Experiment 29: Fraction of Glycogen Phosphorylase-a is 40%, and Fig. 15 for Experiment 45 of Scopes (14).
PCr concentrations measured after adding varying amounts of ATPase; see Table 7. The model structure and parameter values were identical for each experiment with the exception of the defined fractions of glycogen phosphorylases-a and -b and duration of the experiment; see Table 6.
Scopes (1974) postmortem glycolysis study: pH time course with 1 unit/ml of ATPase and 10 units/ml of AMP deaminase added to the same enzyme mixture used in Experiment 29 with 0.2% GPa, and a starting solution with more glycogen and PCr defined in Table 5: Figs. 12 and 13.
FIGURE 3.

ATP (○), ADP (▴), and AMP (□) time course data and model simulations from Scopes (14) for Experiment 29: 99.8% GPb.
FIGURE 4.

PCr (○) and lactate (▴) time course data and model simulations from Scopes (14) for Experiment 29: 99.8% GPb.
FIGURE 5.

Hexose monophosphates (○) and fructose diphosphate (▴) time course data and model simulations from Scopes (14) for Experiment 29: 99.8% GPb.
FIGURE 6.

Glycerol-3-phosphate (○) and phosphoglycerates (▴) time course data and model simulations from Scopes (14) for Experiment 29: 99.8% GPb.
FIGURE 8.

PCr (○), lactate (□), and Pi (▴) time course data and model simulations from Scopes (14) for Experiment 29: 60% GPb.
FIGURE 9.

Hexose monophosphates (○) and fructose diphosphate (▴) time course data and model simulations from Scopes (14) for Experiment 29: 60% GPb.
FIGURE 15.

PCr (○) and Pi (▴) time course data and model simulations from Scopes (14) for Experiment 45 with increasing GPa fractions and added ATPase at 110 min.
TABLE 7.
Effect of adding ATPase on the PCr concentration after initial accumulation of PCr, with GPa fraction 0.1% at pH 7.2 and 303.15 K
| Time of ATPase addition (min) | ATPase activity(units/mL) | Time of measurement(min) | PCr measured(mM) | PCr, simulated(mM) | Lactate, simulated(mM) |
|---|---|---|---|---|---|
| — | 0 | 30 | 13.1 | 13.1 | 8.5 |
| 30 | 0.5 | 60 | 10.2 | 8.69 | 15.15 |
| 30 | 1 | 60 | 6.5 | 7.39 | 23.93 |
| 30 | 2 | 60 | <4.4 | 6.26 | 42.5 |
TABLE 6.
Parameter values adjusted and optimized to match model simulations to the data
| Experiment No. | GPb fraction | Parameter | Definition | Reported value | Adjusted value | Optimized value |
|---|---|---|---|---|---|---|
| 29 | 99.8% | K′AMP | Intrinsic affinity of GPb for AMP | 9.7e–5 M | 3e–3 M | 2.66e–3 M |
| 29 | 99.8% | nH | Hill coefficient for AMP activation of GPb | 1.75 | 1.75 | 1.7505 |
| 29 | 99.8% |  |
Vmax of aldolase | 0.104 M/min | 9e–3 M/min | 1.07e–2 M/min |
| 29 | 60% | KiGLY* | Inhibition constant of glycogen for GPa | 2e–3 M | 1.33e–3 M | — |
| 45 | 99.9–96% | K′AMP† | Intrinsic affinity of GPb for AMP | 9.7e–5 M | 2e–3 M | 3.3e–3 M |
| 45 | 99.9–96% | nH† | Hill coefficient for AMP activation of GPb | 1.75 | 2.5 | 2.66 |
| Postmortem glycolysis | 99.8% | KATP | Km of ATP for ATPase | 0.1 mM | 0.5 mM | 0.358 mM |
| Postmortem glycolysis | 99.8% | KAMP | Km of AMP for AMP deaminase | 1–3 mM | 2 mM | 5.91 mM |
Appropriate temperature and initial conditions as given in Table 5 were applied to this experiment.
This parameter was adjusted keeping the adjusted values of K′AMP and  from Experiment 29 with GPb fraction 99.8%.
from Experiment 29 with GPb fraction 99.8%.
These parameters were optimized, keeping the adjusted value of KiGLY = 1.33e–3 M.
FIGURE 12.

Model-predicted and experimentally measured (○) time courses in the postmortem glycolysis simulation experiment.
FIGURE 13.

Model-predicted proton consumption flux time courses in the postmortem glycolysis simulation experiment.
Sums of metabolites in groups are defined as in the original article of Scopes (14), with hexose monophosphates = G1P + G6P + F6P, fructose diphosphate = 1, 6 fructose bis-phosphate + (GAP + DHAP)/2, and phosphoglycerates = 13DPG + 3PG + 2PG + PEP. During Experiment 29 pH dropped from an initial value of 7.8 to a lower value, not measured, in the first minute of the experiments and subsequently maintained at pH 7.4 with a pH-stat for the remainder of the time courses. In the model, the pH variation during the first minute was computed by solving for dpH/dt in Eq. 70, and the operation of the pH-stat was simulated by setting pH to 7.4 after the first minute. For Experiment 45 pH was held constant at 7.3 throughout the experiment, and for the experiment with varying amounts of ATPase added, 7.2. In the postmortem glycolysis study, pH was allowed to vary freely until the end of the experiment from a starting pH of 7.25.
The system of differential equations in the model was solved for the various experiments (14) using initial conditions listed in Tables 5 and 6. Numerical solution of the differential equations was obtained in MatLab (The MathWorks, Natick, MA) computational platform, using MatLab's ode15s solver, and the stiff differential equation solver RADAU (45). This system was also simulated with the JSim 1.6 system (http://nsr.bioeng.washington.edu) using the RADAU solver. The most sensitive parameters initially identified by inspection were adjusted to match the model predictions to the data. Later a mean-square error sensitivity analysis was performed to obtain the sensitivities of all experimental variables to all parameters and initial conditions. The parameters optimized for fitting were chosen based on this sensitivity analysis and are given in Table 6, which lists the literature-reported value of the parameters, adjusted values, and optimized values in their respective units. Parameter optimization was performed using the MatLab Optimization Toolbox function “fmincon” (constrained function minimization to minimize the cost function) for Experiment 29. Unconstrained minimization function “fminsearch” was used for minimizing the cost function for Experiment 45. Cost function was defined in each case as the sum of squares of residuals.
RESULTS
Proton stoichiometry of biochemical reactions
Fig. 1 shows proton consumption stoichiometry of selected reactions of the glycogenolysis pathway and the creatine kinase reaction. The four reactions were selected to illustrate the main results obtained. The proton stoichiometric coefficient has a positive value if the reaction consumes protons progressing in the direction written for the reference reaction in Table 3; this is the convention used throughout this article. The calculation of proton stoichiometries was made as a function of pH at fixed free Mg2+ (0.513 mM) and K+ (80 mM) concentrations at 303 K and an ionic strength = 0.1 M, by applying Eqs. 1–7 for the distribution of metabolite forms, binding polynomial and average number of protons for each reactant and product.
FIGURE 1.

Variation in the net proton stoichiometry with pH for four selected biochemical reactions. Units of the ordinate scale is mole of H+ per mole of advancement of the reaction to the right as defined in Table 3; positive means protons are consumed. Stoichiometries are computed using Eq. 7.
Every biochemical reaction in the network, derived from reference reactions given in Table 3, has an associated proton stoichiometry that is computed from the binding polynomials and the number of protons in the reactants and products as defined by Eq. 8 in Methods. Due to changes in metabolite-proton binding, all reactions, even those biochemical reactions whose reference reactions do not show any net proton change, have a proton stoichiometry defined by Eq. 8. This stoichiometry is influenced by a change in pH through variation of average proton binding ( ) differences between reactants and products as shown in Eq. 8. At a highly alkaline pH, the proton consumption stoichiometry of a biochemical reaction converges to that of its reference reaction as each
) differences between reactants and products as shown in Eq. 8. At a highly alkaline pH, the proton consumption stoichiometry of a biochemical reaction converges to that of its reference reaction as each  term approaches zero, i.e., all reactants and products are almost entirely in their deprotonated forms. The results show specific examples of the general rules that each biochemical reaction has a proton stoichiometry associated with it as defined by the binding polynomials, that the proton stoichiometry is dependent on pH, and that the stoichiometric coefficients per unit advancement of the reaction are usually not integers.
term approaches zero, i.e., all reactants and products are almost entirely in their deprotonated forms. The results show specific examples of the general rules that each biochemical reaction has a proton stoichiometry associated with it as defined by the binding polynomials, that the proton stoichiometry is dependent on pH, and that the stoichiometric coefficients per unit advancement of the reaction are usually not integers.
The four reactions selected to illustrate these results are GAPDH, PGK, LDH, and CK. Results for all biochemical reactions in the model are given in the Supplementary Material.
Glyceraldehyde phosphate dehydrogenase (EC No. 1.2.1.12) includes one proton and one molecule of nadred as a product in the reference reaction (Table 3):
 |
(72) |
The corresponding biochemical reaction consists of summations for each participating metabolite, which results in a stoichiometry for the number of protons released per unit advancement of reaction (defined in Eqs. 7 and 8) that depends on pH in a complex fashion as shown in the lowest curve in Fig. 1. The proton consumption stoichiometry varies from −1.57 at pH 6 to peak at −0.682 at pH 7.32 to approach −1 at very alkaline pH. This complex dependence of proton stoichiometry is simply due to the algebraic summation of the average proton binding for the five reactants and products.
Phosphoglycerate kinase (EC No. 2.7.2.3) does not have a proton in its reference reaction (Table 3),
 |
(73) |
but it does contribute to proton balance because of the rule that biochemical reactions require representation as sums of bound and free forms as discussed in pH and Temperature Effects on Equilibria and Kinetics of Biochemical Reactions; the results are shown in Fig. 1. Its proton stoichiometry is −0.6 at pH 6, −0.707 at pH 6.8 and tends to zero at alkaline pH values.
Lactate dehydrogenase (EC No. 1.1.1.27) includes one molecule of nadred as a reactant and has one proton in its reference reaction:
 |
(74) |
LDH reaction per se takes up a proton (alkalinizes the cytoplasm). In the pH range 5.5–8.5, the reactants in this reaction are almost entirely in the deprotonated state due to their low pKa values. Therefore, the proton stoichiometry of this reaction deviates negligibly from that of its reference reaction, i.e., 1.
Creatine kinase (EC No. 2.7.3.2) in its reference reaction consumes a proton in the direction of PCr breakdown:
 |
(75) |
The proton stoichiometry of its biochemical reaction varies from 0.654 at pH 5.5 and converges to 1 at high pH. This deviation from reference reaction stoichiometry at lower pH is due to the proton-binding changes in ATP and ADP mostly since the pKa of H2PCr− is 4.5, whereas the pKa values of HADP2− and HATP3− are 6.38 and 6.48, respectively. The calculated results for CK agree with Kushmerick (8) at comparable free magnesium and potassium concentrations.
For a system of reactions at constant pH and at steady state, it is a simple matter to sum the proton stoichiometric coefficients for the net reactions. For net lactate production from glycolysis producing PCr (through the coupling of creatine kinase with glycolytic ATP synthesis) this has been done (8,46). However, the realistic situation in the experiments simulated in this study, and in muscle metabolism during activity, is dynamic as the system progresses from one steady state (e.g., rest) to another (e.g., submaximal exercise) with respect to net flux, the distribution of mass within the network, and pH change. Thus for all non-steady-state situations, the analysis described in this section is necessary, requiring solution of the coupled differential equations.
Apparent equilibrium constants and reference free energies
The necessary consequence of the proton stoichiometric coefficients of the reactions is that the apparent equilibrium constants of all biochemical reactions are functions of pH. The reason is every biochemical reaction in the cell has an associated proton stoichiometry; see Eq. 26. In contrast, the equilibrium constant of a reference reaction is independent of pH as shown by Eqs. 23–25. To see this dependence of a biochemical reaction in more detail, take the logarithm to the base 10 on both sides of Eq. 28 to get
 |
(76) |
The first term on the right of Eq. 73 shows the equilibrium constant of the reference reaction is a component of the apparent equilibrium constant of the biochemical reaction in the cell. The second term shows a linear dependency on pH for reactions with a nonzero n. The nonlinear pH dependency of Kapp is due to the third term that contains the binding polynomials.
The important meaning of Eq. 76 is that apparent equilibrium constants can change by n orders of magnitude for each unit of pH change, in addition to the positive or negative contribution by the nonlinear term (given in the third term on the right-hand side of Eq. 76). Fig. 2 displays the apparent equilibrium constants as a function of pH for the same example-reactions shown in Fig. 1; these results were computed with Eq. 76. Over the easily achievable physiological range of pH 6.5 to 7, there are substantial ranges of the apparent equilibrium constants. The apparent equilibrium constants decrease by 2–3 orders-of-magnitude from pH 5.5 to 8.5 for the LDH and CK reactions and increase by the same magnitude for PGK and GAPDH reactions. A graph of the results for all reactions is given in the Supplementary Material. These results show the magnitude of changes in Kapp as a function of pH. This dependence means there is a change in mass distribution in all reactions, including those near equilibrium, in the reaction network. This effect will propagate through the network and could influence the kinetics of all reactions. These changes must be taken into account in the computational model to obtain correct thermodynamics compatible with a real system.
FIGURE 2.

Apparent equilibrium constants of CK, LDH, PGK, and GAPDH reactions plotted on logarithmic scale as a function of pH at 0.51 mM free Mg2+, 80 mM free K+, 303.15 K, and I = 0.1 M. The apparent equilibrium constants computed using Eq. 28 are dimensionless, assuming a 1 M reference concentration for all species.
Simulation and model fits of data from cell free reconstituted glycogenolysis experiments by Scopes (14,15)
The kinetic model augmented with appropriate computations for proton stoichiometry and for Kapp was employed to simulate experiments by Scopes (14,15) as described in Application of pH and Kapp and Vmax(pH) Computations to Glycogenolysis. In brief, those experiments measured the rate and extent of PCr synthesis in a reconstituted enzyme mixture for glycogenolysis and glycolysis and pH time course, when the same mixture with high starting PCr, ATPase, and AMP deaminase is allowed to reach equilibrium. The objective of these simulations was to determine to what extent a model assembled with parameters from the literature, including all of the appropriate principles from physical chemistry, could account for the experimental observations. After these simulations we determined the minimal set of parameters to be optimized to obtain simulations reproducing the observed data; Table 6 contains the parameters optimized with their original literature values.
Experiment 29: glycogen phosphorylase almost entirely in b-form
The first experiment reported by Scopes (14) was Experiment 29, in which the lactate production and ATP synthesis by glycolytic flux was coupled to the creatine kinase flux so that the concentrations of PCr and lactate increased and Pi decreased in the experimental mixture in vitro as a function of time. The goal of this experiment was to study the reaction network at low fraction of glycogen phosphorylase in the active (GPa) form; the original article states the activity was negligible at ∼0.2% of total GP activity in the a form. The experimental result was that the reconstituted mixture of enzymes and substrates functioned as the network does in the muscle cell by reducing Pi concentration and synthesizing PCr and lactate. We simulated these results with our model described in Application of pH and Kapp and Vmax(pH) Computations to Glycogenolysis, above. Figs. 3–6 show model predictions for Experiment 29 with GPb 99.8% and parameters adjusted as shown in Table 6. Parameters adjusted for this model were those of AMP activation factors of GPb, which were found to be the most sensitive parameters on inspection. Table 4 of Scopes' article gives the measured PCr and Pi concentrations after 30 min for this experiment as 16.5 and 15.3 mM, respectively. Our model predicts a PCr concentration of 16.9 mM and a Pi concentration of 13.1 mM, which are in reasonable agreement with the measured values.
The kinetics of this experiment were also simulated well by the model. Fig. 3 displays the data and the model simulated curves for ATP, ADP, and AMP concentrations. Fig. 4 shows the early burst of lactate and PCr synthesis followed by a slower increase. The simulation of time courses (using the metabolite sets defined in the original article) for FDP (fructose diphosphate = 1, 6 fructose bis-phosphate + (GAP + DHAP)/2) and to HMP (hexose mono-phosphates = G1P + G6P + F6P) correctly shows the rapid rise in metabolite concentrations observed in the data (Fig. 5). The simulation of FDP correctly displays its rapid decrease, but that of HMP showed a slower time course than the data indicate. The simulation of time courses for glycerol-3-phosphate and phosphoglycerates (phosphoglycerates = 13DPG + 3PG + 2PG + PEP) does not match the transient in the data during the first minute but qualitatively matches the data from the third minute until the end of the experiment (Fig. 6). In the absence of detailed information on each metabolite, further analysis and refinement of model predictions is not possible.
To obtain the simulations described, it was necessary to decrease the intrinsic AMP binding affinity to GPb, i.e., increase K′AMP, compared with the reported affinity given in Table 6. This shift in affinity is realistic since the reported affinity was at pH 6.8, whereas the Scopes experiment starts at pH 7.8 and is maintained at pH 7.4 after the first minute. Optimization of AMP activation of GPb matched the PCr and lactate time courses but did not match the hexose monophosphate and fructose diphosphate time courses even though the behavior of phosphofructokinase flux included and followed regulation by AMP and ATP in a manner compatible with the experiment. Adjustment of Vmax of aldolase by an ∼10-fold reduction enabled the matching of measured and simulated time courses of hexose phosphates. This enzyme was found to be allosterically inhibited by ATP with an I50 of 2.1 mM (47). The ATP concentration in Scopes' experiments is nearly constant at 5 mM so the enzyme was significantly inhibited, which could explain the 10-fold reduction in its activity required to match the data on hexose monophosphates and fructose diphosphates.
Incomplete predictions if pH effects are ignored
The analyses of the kinetic model of glycogenolysis and glycolysis presented so far demonstrate a good matching of model predictions with Scopes' experimental data showing the extent and time course of PCr and lactate synthesis in a reconstituted glycolytic system. Now we show that the model simulates the Scopes' data much less well if pH is fixed at a constant value of 7. This effectively demonstrated a situation wherein the variation of pH is ignored and the simulation of the enzyme kinetics is performed with fixed equilibrium constants taken at this pH instead of the actual experimental pH. Fig. 7 displays the same data as in Fig. 4, with the model prediction for pH fixed at 7.0 throughout the simulation. Qualitatively the model follows the trend of the experimental results, but quantitatively the model predictions for both the kinetics and approach to equilibrium do not describe the data unless proton stoichiometry and effects of pH are properly included. It is necessary to account correctly for H+ consumption, pH changes, and pH-dependence of reaction equilibria in model simulations of metabolic networks.
FIGURE 7.

PCr (○) and lactate (▴) time course data from Scopes (14) for Experiment 29: 99.8% GPb and model predictions when pH is fixed at 7.0 throughout the simulation with the same parameter set as in the Fig. 4. The significant deviation of model predictions from the data emphasizes the importance of including pH regulation of enzyme activities and biochemical reaction thermodynamics in the model.
Experiment 29: fraction of glycogen phosphorylase-a is 40%
Scopes (14) also studied the properties of the reaction mixture at higher activity of glycogen phosphorylase in the active a-form (from Fig. 2 in (14)). The time courses of these data were also simulated by the model. Figs. 8 and 9 display the model simulation for the kinetics of PCr, Pi, lactate, hexose monophosphates (HMP), and fructose diphosphates (FDP) at a GPa fraction of 40% of the total phosphorylase activity. Starting with the parameter set for GPb from the previous experiment, we reduced the inhibition constant of glycogen for GPa KiGLY by twofold in the flux expression for GPa flux to match the measured concentrations.
Fig. 8 shows the time experimental data and model predictions of PCr, lactate, and Pi time courses for a high GPa fraction (40%).The model prediction for PCr and lactate from 2 min until the end of the experiment at the 10th minute is closer to the measured concentrations than to the predictions in the first two minutes of the experiment. Scopes also made a measurement of PCr and lactate concentrations at the 30th minute, which are higher than the model predictions at this time point. After the first minute, the experiment is essentially a trajectory of the system toward equilibrium at pH 7.4 whereas during the first minute, the trajectory toward equilibrium starts at pH 7.8 and pH rapidly decreases. Equilibrium concentrations of metabolites were obtained by running the model for a long time until the derivatives of the state variables were ∼1e–15 M/min or less in magnitude. The predicted equilibrium concentrations of PCr and lactate and Pi are 7.8 mM, 7.35 mM, and 5.9e–8 mM, respectively. The same concentrations are obtained for GPa 0.2% but it takes longer for this system to reach equilibrium compared to the case with higher GPa fraction. The important conclusion from this simulation to equilibrium is that Scopes' experiment captured only the rising part of the PCr and lactate transients, which are only an apparent steady state at ∼10–15 min; however, in reality they are an overshoot toward equilibrium, as the very long simulations show.
The measured and model-predicted time courses of HMP and FDP are shown in Fig. 9. The model prediction for FDP qualitatively shows the fast rise and fall during the first minute as shown by the data, but a rise to a level significantly higher than the measured values toward the end of the experiment. For HMP, the model prediction does not show the transient during the first minute as seen in the measurements, but it does show qualitatively the transient after the first minute until the end of the experiment.
Predicted time courses of pH, proton fluxes, and metabolic proton load and lactate production
We now consider how the model accounts for the time course of the pH change in the experiments of Scopes (14,15). This is a useful test of the model to account for H+ uptake in biochemical reactions, changes in buffer capacity, and pH as the reaction progresses. Unfortunately in the original Experiment 29, no pH was measured during the first minute and thereafter pH was clamped at pH = 7.4 by a pH-stat. Apparently it was realized that starting the reactions in the network produced acid at a high rate, and this is the likely reason the pH at the start of the experiment was set high, to pH = 7.8. For Scopes' Experiment 29, already simulated in Experiment 29: Glycogen Phosphorylase Almost Entirely in b-Form (Figs. 3–6), the model predicts a large and rapid decrease of pH in the reaction medium during the first minute (Fig. 10). The simulation shows a greater decrease in pH with the higher fraction of GPa (40%) and therefore greater flux during the first minute.
FIGURE 10.

Predicted pH time courses for Experiment 29 at both 99.8% and 60% GPb and for CK knockout. A pH-stat is operated at 7.4 starting at the end of the first minute. Note the greater fall in pH for 60% GPb owing to higher creatine phosphorylation flux. The pH transient goes in alkaline direction for CK knockout since the fluxes other than creatine phosphorylation are alkalinizing.
The task of identifying which reaction(s) contributes to these changes in pH is simplified because the creatine kinase reaction so clearly dominates proton uptake. Fig. 11 shows the proton consumption fluxes of the creatine kinase reaction itself, compared to the sum of all the other reactions in the system during the first minute of simulation and to the total proton consumption by the network of reactions as the reaction proceeds. Note that the vertical scale of the graph is greatly expanded compared to the inset. The inset shows a very rapid proton consumption dominated by the creatine kinase reaction. At ∼0.2 min there is a minor transient in proton consumption stoichiometry by the other reactions and creatine kinase. The rapid decrease in pH is due to the creatine kinase reaction going in the direction of creatine phosphorylation, and this reaction leads to the sharp drop in pH by ∼0.15 units during the first minute of the experiment. This result demonstrates that the reaction responsible for acidification by glycolysis in this reconstituted and closed system is the phosphorylation of creatine by the creatine kinase reaction, because the sum of proton fluxes due to all the other reactions contributes negligibly to the observed change in pH. If pH is allowed to vary freely throughout the simulation from the initial pH, the system converges to an equilibrium pH of 7.04 for both cases. The higher GPa fraction experiment results in a greater undershoot in pH before converging to the equilibrium value. For this case, the equilibrium concentrations of metabolites are different compared to when the pH is fixed at 7.4.
FIGURE 11.

Predicted H+ consumption flux time courses for Experiment 29 at 99.8% GPb. Inset shows the full range of CK proton consumption flux transient in the first 0.05 min. Note ∼100-fold difference in scale between main graph and inset.
The pH time course was measured in the subsequent study simulating postmortem glycogenolysis, which we use to validate the pH prediction portion of our model. Figs. 12 and 13 show the pH time course and the proton consumption fluxes in Scopes' (14) experimental simulation of postmortem glycogenolysis, respectively. The reaction system, which now has 1 unit/ml activity of ATPase and 10 units/ml of AMP deaminase, starts in a solution with high initial PCr concentration of 24 mM and an initial pH of 7.25. pH declines as ATP is hydrolyzed with a slight alkalinization at the very beginning due to breakdown of PCr. The pH declines almost linearly to a pH of 5.64 at 190 min after which the rate of pH decreases more slowly to 5.48 at 300 min. The model describes the measured pH data very closely, validating the pH prediction of the model, while the previous experiments validated the glycolytic measurements and creatine phosphorylation described by the model. Proton fluxes in Fig. 13 show that proton consumption flux due to creatine kinase is initially alkalinizing after which it declines. Glycolytic proton flux, which is initially alkalinizing, turns acidifying below a pH of 6.7, and proton consumption flux increases almost linearly in the negative direction after this point. At the same time, ATPase proton consumption flux, which is initially negative, i.e., acidifying and flat during the first few minutes due to creatine kinase buffering of ATP concentration, starts to decrease in magnitude almost linearly toward zero. Due to these transients, the sum total, which shows an alkaline transient initially, subsequently becomes acidifying and remains acidifying due to the contribution of proton fluxes from both glycolytic reactions and the ATPase reaction. Glycolytic rate increases sharply initially, then plateaus until the end of the linear pH decline period, after which it declines sharply due to reduction in glycogen and degradation of the adenine nucleotide pool. These simulations are consistent with Scopes' 1974 observations (15).
Sensitivity
In a metabolic network with so many parameters, it is important to test the model solutions to variations in parameter values. This gives an impression of the robustness of the model to variations in parameters and identifies those parameters that have the greatest influence in the simulations. Whereas conventional definitions of sensitivity are useful for steady states, the definition of sensitivity for dynamic stimulations is more difficult to define and somewhat arbitrary. We opted for a sensitivity measured over the entire time course of the simulation. Mean-square error sensitivity of jth variable with respect to a 10% change in ith parameter taken from the adjusted parameter set is defined as
 |
(77) |
The sensitivity was calculated for the nine measured variables in Experiment 29: GPb 99.8% with respect to all kinetic parameters and initial conditions in the model. The resulting sensitivity values were stored in a matrix with each column corresponding to a measured variable and each row to a parameter. A qualitative picture of relative column magnitudes and relative row magnitudes computed by appropriate normalization schemes is shown in Fig. 14. Fig. 14 shows the results represented in grayscale images of the matrix with two normalization schemes. The normalization of this matrix was done in three ways:
By normalizing each element of a column to the maximum element in that column: This gives a measure of which parameter sensitizes a particular variable relative to other parameters in terms of the shade of gray, white corresponds to a value of 1 (highest), and black to 0 (none).
By normalizing each element of a row to the maximum element in that row: This gives a measure of which variable is sensitized by a particular parameter relative to other variables in terms of the shade of gray, white corresponds to a value of 1 (highest), and black to 0 (none).
By normalizing all elements of the matrix with respect to the maximum element (not shown in Fig. 14): This gives the relative magnitude of all sensitivities in terms of the shade of gray, white corresponds to a value of 1 (highest), and black to 0 (none).
The left-hand panel in Fig. 14 shows the column normalized sensitivity map. We see that PCr (column 1), lactate (column 2), ATP (column 3), ADP (column 4), hexose monophosphates (column 6), glycerol-3-phosphate (column 8), and phosphoglycerates (column 9) are most sensitive to the Hill coefficient nH for AMP activation of glycogen phosphorylase-b. Note that GPb being in the exponent of the activation function, amplifies any sensitivity due to the interaction coefficient and intrinsic AMP binding affinity Kgp_amp (Table 6). Fructose diphosphates (column 7) and AMP (column 5) time courses are most sensitive to the Vmax of aldolase (Vffald). Also, note the significant sensitivity of most variables to Vmax of glycogen phosphorylase (Vfgly). This parameter was considered fixed for modeling purposes but the sensitivity result indicates that this parameter must be measured carefully. There is small but visible sensitivity of ATP, ADP, and AMP to concentrations of the fixed buffers carnosine and tris. AMP, hexose monophosphates and fructose diphosphates are mildly sensitive to PFK activity. AMP, glycerol-3-phosphate, and phosphoglycerates are also sensitive, though to a lesser extent, to activities of enolase and triose phosphate isomerase.
The right-hand panel in Fig. 14 shows the row-normalized sensitivity map, which gives groups of parameters that are important for each variable. PCr is sensitive to creatine kinase, glycogen phosphorylase, glycerol-3-phosphate dehydrogenase, and phosphoglycerate kinase. The lactate time course is sensitive to creatine kinase, adenylate kinase, glycogen phosphorylase, and all of the glycolytic enzymes from aldolase to lactate dehydrogenase. This shows the strong coupling of creatine kinase and adenylate kinase to the glycolytic pathway. Phosphofructokinase parameters sensitize hexose monophosphates more than other metabolites that are downstream to these metabolites, and glycogen phosphorylase, phosphoglucomutase, and phosphoglucoisomerase parameters sensitize fructose diphosphates, which are upstream of these measurements. Note also the significant sensitivity of all measured variables to the ionic strength (Fig. 14 I). This sensitivity underlines the importance of accurate knowledge of this parameter.
Experiment 45: fraction of GPa is progressively increased from 0.1% to 4%, and addition of increasing amounts of ATPase
A third experiment (Experiment 45 and Fig. 4 of (14)) showed the effects of increasing the percentage of glycogen phosphorylase in the active (GPa) form at a higher temperature (37°C or 310.15 K) than the other experiments (Table 5). In this experiment GPa activity is increased in nonuniform time increments: initially 0.1% to 0.4% at 40 min, then to 1% at 80 min and to 4% at 100 min. Simulation of this experiment and adjusting the model parameters to reproduce the original data is a stringent test of the model calculation, because it uses the built-in temperature corrections to enzyme kinetics, pKa values, and free energies of formation of reference species. Our simulations were able to predict the time courses of PCr and Pi in the experiment wherein the fraction of GPa was increased from 0.1% to 4%, as Fig. 15 shows. A second component of Experiment 45 was the addition of an enzyme catalyzing ATP hydrolysis to the glycolytic system as it approached a steady state of PCr synthesis at 110 min. This addition caused a rapid decrease in PCr and increase in Pi because the activity of the ATPase exceeded the glycolytic flux synthesizing ATP and hence PCr (Fig. 15).
Another set of experiments was conducted at pH 7.2 and a temperature of 303.15 K with various amounts of ATPase added, described in Table 5 of Scopes (14), and initial metabolite concentrations as in Experiment 29. PCr was measured at 30 min after addition of ATPase, before which the reaction mixture was allowed to synthesize PCr for 30 min without any added ATPase. This feature of the original experiments is also simulated by the model as shown in Table 7, which compares the observed data on PCr concentrations at four levels of ATPase activity 30 min after the addition of ATPase with the calculated results. The model prediction of lactate (not measured in the experiments) shows the proportionality of glycolytic response to ATPase activity.
Coupling of creatine kinase and adenylate kinase to biochemical pathway for glycolysis
The basic model for muscle glycogenolysis and glycolysis coupled the pathway to creatine kinase, adenylate kinase, and ATPase, because that is the functional coupling that occurs in muscle (16) and is obvious from the experiments of Scopes. One of the important conclusions of that analysis was that the ATPase flux had the major control strength for glycolytic flux; that is when the biochemical network was coupled to the reactions external to it, the flux control was transferred from enzymes within the pathway to the ATPase. The same reactions were coupled in the present work because this was the design of experimental data analyses here (14,15). The analysis presented so far highlighted the role of H+ balance and pH in the function of the coupled pathway and identified the creatine kinase reaction as the dominant mechanism governing proton uptake. The reaction mixture acidified because creatine was phosphorylated and that releases protons; this reaction alone accounted quantitatively for the proton release and pH change in Experiment 29: Fraction of Glycogen Phosphorylase-a is 40% and Figs. 10 and 11.
Inspection of the ATPase reference reaction (Table 3) and analysis of the proton-binding change show that this reaction releases protons as it proceeds, or if ATP is synthesized the reaction takes up protons. If only ATPase and ATP synthesis were coupled to the glycolytic network, instead of the creatine kinase, the effect would be an alkalinization for glycolytic lactate synthesis coupled to ATP synthesis. In other words, without creatine and creatine kinase, the pH in Scopes' experiments would go alkaline. Simulations showed these effects. When the enzyme activity of the creatine kinase reaction was set to zero, initial ADP to 5 mM, and initial ATP to 10−9 M, the glycolytic flux rapidly synthesized ATP and lactate and raised pH to >7.8 during the first minute of reaction, before the pH was clamped to 7.4, as shown in Fig. 10. The simulation results in this section show that the initial conditions of this glycolytic network have a large influence on the results obtained. It should be clear that the pH goes acidic when PCr is synthesized from Cr and coupled to lactate production in the network but the pH goes alkaline when CK is inactive and ATP is synthesized from ADP. We have not yet analyzed the relevance of these results as relevant to the interpretation of creatine kinase and adenylate kinase knockout models in cardiac and skeletal muscle.
Another prediction of the model is that coupling of ATP or PCr synthesis is necessary for glycolytic flux, because the network generates ATP from ADP as required by pathway stoichiometry (Fig. 16 A). Therefore, in the absence of creatine kinase and ATPase fluxes, there should be no glycolytic flux, no lactate production, and no change in pH. Simulation also showed these effects (data not shown); when there was no flux in creatine kinase or ATPase, the concentrations of PCr, ATP, Pi, and lactate did not change here as opposed to the results shown in Figs. 4, 7, 8, and 15.
FIGURE 16.

(A) Model predictions of PCr and lactate time courses with no CK activity, starting with 5 mM ADP and 1 nM ATP at conditions of Experiment 29: GPb 99.8%. Note the rapid rise of lactate and no PCr synthesis. (B) Model predictions of ATP, ADP, and AMP time courses with no CK activity, starting with 5 mM ADP and 1 nM ATP at conditions of Experiment 29: GPb 99.8%. Note the rapid synthesis of ATP, and the consequent drop in ADP and AMP. Glycolytic flux is generating ATP, without creatine phosphorylation.
DISCUSSION
Scopes' reconstituted system simulated here is different from an intact muscle in several ways. As opposed to Scopes' reconstituted system, intact muscle always has various ATPases working at different rates, oxidative synthesis of ATP by the mitochondria, a multitude of proton buffering systems, and proton and metabolite transport systems on the sarcolemma. The Scopes' system is a closed system that can only go toward equilibrium whereas a normally perfused intact muscle is an open system capable of attaining steady states. Full simulations including the additional biochemical reactions and transport and buffering components for proton regulation in intact muscle cells must be performed before extending the results presented in this study to an intact muscle. The main objective of this study was to establish and validate with experimental data the method for computing the pH time course due to a biochemical network using a proton mass balance-based approach and its influence on biochemical reaction kinetics and thermodynamics simultaneously. The feasibility of extending the present analysis to a living muscle was shown by preliminary results for perfused mouse skeletal muscles. This required integration of the Lambeth and Kushmerick glycogenolysis model with a phenomenological oxidative phosphorylation model and monocarboxylate transporter isoforms 1 and 4 models. This integrated model simulated PCr, Pi, and pH time courses from transient anoxia experiments (48).
pH and temperature effects on enzyme kinetics are well known and studied (31,32), as are the dependencies of reaction equilibria on pH, temperature, [Mg2+], and other cations in the milieu that bind significantly to the metabolites (6). The contribution of our work is a synthesis of this information to compute pH time course dynamically through proton mass balance and its application to a particular pathway relevant to muscle physiology, glycogenolysis, and glycolysis. Here we matched our simulations, whose parameters are taken from in vitro enzyme literature, to an experimental study of a reconstituted enzyme system. When full consideration is made of the multiple equilibria of the ionic species of the reactants and products, it is possible to account quantitatively for H+ uptake and release by the constituent biochemical reactions into a medium of finite buffer capacity and to account for pH dependence of reaction free energy. We incorporated this synthesis into a mathematical model of the enzyme kinetics in that pathway. The physical chemical and thermodynamic effects are straightforward in principle, and, for steady states, are relatively easy to calculate. However, the detailed kinetics analysis we made is necessary when the evolution of dynamic states is considered. This necessity arises because the time-dependent quantities of proton reaction stoichiometry and pH are functions of each other and because the buffer capacity changes. The resultant ordinary differential equations can be solved for particular cases as was done here by numerical methods. Solution of the interactions of proton uptake and release by the network allows explicit calculation of apparent equilibrium constants for each reaction from first principles. Definition of pH in the reaction network also allows explicit but empirical adjustment of enzyme activities to account for their variation with pH. All of this detail was then incorporated into a kinetic model of glycogenolysis and glycolysis in this closed system.
The combined thermodynamic and kinetic effects of the accounting for H+ consumption and changes in buffer capacity and pH not only influence the behavior of the model studied, as expected, but also provide quantitative constraints on the system behavior. These constraints serve as an integrating mechanism because the pH effects affect all the reactions in complex ways that can be fully understood in basic physical chemical mechanisms and principles. Considering the biochemical pathways as networks, pH is the central, most commonly connected node.
Physical chemistry
The first principles we used are those built upon the extensive work of Alberty and colleagues, summarized in his article (6), explaining how to account for proton consumption or release by biochemical reactions and reaction equilibria in pH and Mg2+ buffered reaction media. We also built upon the work of George and colleagues (2) explaining the mechanisms by which H+ is involved in accounting for the large range of values of free energy in biochemical reactions and were guided by the extensive analysis of glycolysis in the red cell (11–13). However, none of these earlier studies considered a system with time variable pH and finite buffer capacity.
Equilibrium calculations and binding change computations in media with clamped pH or magnesium have been developed and applied in detail by Alberty (6) and the references therein. Those methods for proton binding change or stoichiometry of biochemical reactions in infinitely buffered reactions have been well developed and give an indication of proton loads imposed on the reaction medium by each individual biochemical reaction. However, they need to be extended to physiological reaction media, which have a finite buffer capacity. Moreover, the proton load imposed by a biochemical reaction is a product of the proton stoichiometry of the reaction and the flux through the reaction. Alberty (6) shows proton stoichiometries of individual reactions as a function of pH and sums up the individual reaction stoichiometries to show the net pathway proton stoichiometry, which is valid only for steady-state linear pathways with no branching. Full kinetics should be taken into account for a realistic description of proton dynamics in a reaction network as shown by Eqs. 9–14 in pH and Temperature Effects on Equilibria and Kinetics of Biochemical Reactions.
Precise mass balance of protons and magnesium in closed systems is possible by our method as shown by Eqs. 11–19 and 43–49, provided the numerical methods used to solve them are appropriate. The equations for rate of change of pH and rate of change of free magnesium form a set of coupled differential equations that were decoupled before solving. To verify the conservation of protons by the numerical methods used, we computed  the total protons bound for a simple system consisting of creatine kinase in a solution, with the starting composition as in Experiment 29 but without any buffers, and found a drift in this quantity which was six orders-of-magnitude less than the total proton concentration. We also observed the total magnesium in the system, which showed a similar change. Upon reducing the tolerance parameters of the solver, we found an improved ability of the system to conserve magnesium and protons. However, this significantly increases the computation time. In addition, a change of variable was tried by solving for free protons rather than the pH as the dynamic variable. However, this variable change increased the magnitude of total proton drift. When hydrolysis reactions are involved, water should be taken into account for verifying this conservation. In a physiological system, free magnesium concentration is heavily buffered and can be regarded as a constant that makes the free and metabolite bound magnesium variables. Total intracellular Mg = 6–10 mM but free Mg ranges from 0.6 to 1 mM (49). If the Mg2+ bound to other buffers than metabolite buffers is also accounted for, then precise mass balance is possible in a closed system without any magnesium transport into or out of the system. K+ binding was also included in the binding polynomial terms but we considered K+ concentration to be fixed. This is not strictly valid physiologically but was justified in our calculations because the K+ concentration at 80 mM was much higher than the possible binding changes. In muscle cells, K+ concentration is on the order of 120 mM, and even a several-millimeter change over the time course of the metabolic fluxes simulated would not change the value of the free potassium significantly.
the total protons bound for a simple system consisting of creatine kinase in a solution, with the starting composition as in Experiment 29 but without any buffers, and found a drift in this quantity which was six orders-of-magnitude less than the total proton concentration. We also observed the total magnesium in the system, which showed a similar change. Upon reducing the tolerance parameters of the solver, we found an improved ability of the system to conserve magnesium and protons. However, this significantly increases the computation time. In addition, a change of variable was tried by solving for free protons rather than the pH as the dynamic variable. However, this variable change increased the magnitude of total proton drift. When hydrolysis reactions are involved, water should be taken into account for verifying this conservation. In a physiological system, free magnesium concentration is heavily buffered and can be regarded as a constant that makes the free and metabolite bound magnesium variables. Total intracellular Mg = 6–10 mM but free Mg ranges from 0.6 to 1 mM (49). If the Mg2+ bound to other buffers than metabolite buffers is also accounted for, then precise mass balance is possible in a closed system without any magnesium transport into or out of the system. K+ binding was also included in the binding polynomial terms but we considered K+ concentration to be fixed. This is not strictly valid physiologically but was justified in our calculations because the K+ concentration at 80 mM was much higher than the possible binding changes. In muscle cells, K+ concentration is on the order of 120 mM, and even a several-millimeter change over the time course of the metabolic fluxes simulated would not change the value of the free potassium significantly.
Our model for glycogenolysis coupled with creatine kinase, adenylate kinase, and ATPase in muscle extends our original model (16) to allow analysis of pH dynamics in conjunction with a detailed kinetic model. This method is applicable to buffered systems, and developed to describe biological systems as they are buffered with respect to pH and Mg. An exact mass balance approach may also be taken as demonstrated for free magnesium computation in Scopes' experiments, which are closed systems in vitro. A closed cellular system is of limited utility for normal physiological conditions but the model can be extended further and applied to an open system by proper accounting of metabolite, cation, and H+ fluxes across the sarcolemma, by inclusion of appropriate transport kinetic modules at the interface and definition of composition of the compartment outside the cell of interest.
The kinetic model of the glycogenolysis pathway due to Lambeth and Kushmerick (16) used here models all reactions as reversible. Irreversible steps introduced within biochemical pathway models can potentially isolate the biochemical reactions upstream and downstream of the irreversible reactions, resulting in a distribution of metabolic control that would be markedly different from what could be observed in a real system as shown by Cornish-Bowden and Cardenas (50). Their broader conclusion is that if some form of inhibitory feedback exists between the reactions downstream and upstream of the irreversible step, then reversibility may not be critical. In this study, we have included thermodynamic reversibility to the possible extent except for some irreversible boundary fluxes like the ATPase reaction.
Thermodynamic parameters
The method developed in this article relies on the availability of equilibrium constants of metabolite binding to H+, Mg2+, and K+, enthalpies of dissociation, and metabolite free energies of formation to account for proton binding change, for the influence of pH on apparent equilibrium constants and for the influence of temperature and ionic strength on the aforementioned parameters and variables. We used binding constants shown in Table 2 mostly taken from the NIST Standard Reference Database 46, Ver. 7.0, or from other sources as cited. Only those proton dissociation constants whose pKa values are within two pH units of the physiological range were used. Magnesium and potassium dissociation constants were used wherever available. These dissociation constants, reported at 298 K and 0.1 M ionic strength, were corrected to the experimental temperature and ionic strength when the dissociation enthalpies were known. This left a certain portion of pKa values uncorrected for these variables and can have some effect on the simulations obtained and the proton stoichiometries and likely account for some of the discrepancies reported between the observed data and simulated curves. An important limitation to detailed kinetic and thermodynamic modeling of cellular systems is the incompleteness of the available database. The metabolite and ion composition in the starting mixture given in Table 5 gives a calculated ionic strength of 0.1 M for Experiments 29 and 45, and 0.14 M for the postmortem glycolysis experiment. These ionic strength values are used in the calculations for correcting enthalpies, free energies of formation, and pK values. The presence of ions in the added enzyme mixture and buffer during the course of the experiments may change the ionic strength of the medium. This is an uncertainty in the experimental information available, which could have been improved upon had the compositions of all solutions used and their quantities been given.
Free energies of formation of metabolites in their deprotonated states were taken from Alberty (6) when available. Reference equilibrium constants for the reactions listed in Table 3 were computed using Eq. 18 as the difference of free energies of formation of products and reactants and Eq. 19 to compute the Kref. This approach was not possible for creatine kinase reaction because the free energies of formation of HPCr2− and HCr are not available. Therefore we started directly from Kref = 2.58e8 and an enthalpy of formation of −17.55 KJ/mol, at I = 0 and 298 K given in Teague et al. (30). Apparent equilibrium constants, computed using free energies of formation and metabolite binding polynomials, were in good agreement with values from reference data in Goldberg and Tewari (51) and Goldberg et al. (52), except that of glycerol-3-phosphate dehydrogenase. Apparent equilibrium constant of glycerol-3-phosphate dehydrogenase was corrected by adjusting the free energy of formation of glycerol-3-phosphate2− from Alberty's value of −1358.96 KJ/mol to −1339.25 KJ/mol. The values for free energies of formation of the biochemical species were given for 298.15 K and 0 ionic strength in Alberty (6). Temperature corrections were not made for these values, because the enthalpies of formation required for the van 't Hoff equation were not available for most of the species. Creatine kinase is the only reaction whose Kref is temperature-corrected. In summary, the available information on free energies of formation is yet to be completely verified against experimental measures of apparent equilibrium constants for all the reactions in the glycolytic network simulated; this made empirical corrections in the data set necessary. In addition to this, availability of enthalpies of formation will enable temperature corrections of free energies of formation and therefore the equilibrium constants.
Kinetic parameters
Kinetic parameters of an enzyme-catalyzed reaction may vary with pH. This can occur due to change in ionizable groups in the enzyme active site or a shift in predominant ionic form of the substrate or both (31,32). We included empirical dependence of Vmax values of some of the enzymes for which pH-dependence information was available. Similarly, temperature effects were accounted for by using a temperature factor of 1.9 for a 10°C change in temperature (14). More complete descriptions of pH and temperature sensitivity of enzymes may be used if the data becomes available, else the simulations will be restricted to kinetic parameters obtained specifically at the temperature and in the expected pH range of the system in question. The effect of pH on reaction free energy is also relevant as discussed in Thermodynamic Parameters.
It is possible that the rapid equilibrium assumption used in all of the enzyme kinetics may need to be replaced by detailed metabolite on and off binding-reaction kinetics. We did not include these details in the present model simulations because the simulated flux compared to the maximal flux predicted for each enzyme is a small fraction.
Goodness of fit of model to experimental data
Our model aims to explain the influence of pH on biochemical reaction kinetics and equilibria by prediction of experimental data from the study of Scopes (14) on in vitro glycogenolysis using a cell free reconstituted enzyme system. We find that a wide range of experimental data, 13 time series out of 15 from three experiments, are explained by adjustment of only three kinetic parameters out of nearly 100 parameters, which leads to a significant conclusion that accurate accounting for the physical chemistry leads to a model with greater predictive power.
Insights for glycogenolysis and glycolysis
The model analysis gives insight to important properties of glycogenolysis regulation. Prior analysis of the glycogenolytic model (16) demonstrated the ATPase control over the pathway flux. This regulatory property is further illustrated by Table 7, in which experimental conditions used in Scopes original work were reproduced in silico by the addition of ATPase to the metabolite mixture at specific times. The model was used to simulate lactate accumulation, a property not measured by Scopes, to demonstrate a linear relationship between lactate accumulation and ATPase activity. Faster formation of lactate due to higher percentage of the active form of glycogen phosphorylase leads to more rapid proton accumulation, as illustrated in Fig. 10 by decreases in pH.
Through the biochemical network analyses described here, the biochemical source of acidification from glycogenolysis can be directly and decisively addressed. Our results show that defining the proton source in muscle acidification depends on the entire network or biochemical system defined. In the closed experimental system used, analysis of the contribution of individual reactions to net proton uptake and release identified the creatine kinase reaction as the dominant source proton release and acidification of the medium. All of the simulations in Simulation and Model Fits of Data from Cell Free Reconstituted Glycogenolysis Experiments by Scopes (14,15) demonstrated the stoichiometric coupling of lactate production to synthesis of PCr. The results displayed in Fig. 11 show that the contribution of all other reactions is negligible in comparison.
During anaerobic glycolysis, in the presence of ATPase, lactate production is often used as a measure of metabolic proton load assuming a stoichiometry of one proton per lactate. We demonstrate that the relation between lactate accumulation and metabolic proton load is variable and depends on the biochemical conditions in Fig. 17, A and B. Fig. 17 A shows the metabolic proton load and lactate accumulation during Experiment 29 at 99.8% GPb due to the biochemical reactions and lactate production. The ratio of derivatives of lactate accumulation and proton load gives the stoichiometric relationship between lactate and proton load. This is a variable quantity throughout the transient. Fig. 17 B shows the same quantities when there is no creatine kinase and the initial conditions are set as in the simulations for Fig. 16, A and B. Here the relation between lactate generation and proton load is totally opposite to that in Experiment 29, i.e., alkalinizing as opposed to acidifying in Experiment 29.
FIGURE 17.

(A) Metabolic proton load and lactate production during Experiment 29: GPb 99.8%. Metabolic proton load was computed by solving the equation  and lactate production by solving Eq. 67. (B) Metabolic proton load and lactate production with no CK activity, starting with 5 mM ADP and 1 nM ATP at conditions of Experiment 29: GPb 99.8%.
and lactate production by solving Eq. 67. (B) Metabolic proton load and lactate production with no CK activity, starting with 5 mM ADP and 1 nM ATP at conditions of Experiment 29: GPb 99.8%.
The demonstrated goodness of fits means that the model approach taken here can be recommended in general. Temperature and ionic strength correction of the thermodynamic quantities were included in the model as given in Eqs. 3–5. The validity of the model was tested against two fractions of the active form of glycogen phosphorylase and at two temperatures (Figs. 3–9 and 15). We note that the model simulations matched the data with the optimization of very few parameters, given in Table 6; these correspond to the results of the sensitivity analysis. It is likely that optimization over a larger number of other parameters, specifically parameters for ALD and PFK, would improve the simulations, but the goal of the present work is a proof of principle rather than an exhaustive reanalysis of Scopes' data. Interestingly, the constraints of mass conservation built into the model cause the total adenine nucleotide pool (ATP + ADP + AMP) to remain constant throughout all simulations; the original data in Experiment 29: 99.8% GPb, as illustrated in Fig. 3, does not maintain this relation despite being a closed system due to undisclosed experimental error. Therefore, to a certain extent, further parameter optimization will be limited in the ability to improve the matching of model simulations to all experimental time courses. As discussed in Lambeth and Kushmerick (16) and elaborated upon in this article, the use of reversible reaction kinetics, the accounting for the relevant physical chemistry and thermodynamics and for pH, and taking care to define the total biochemical network leads to good description of experimental data. Of course the data considered were all obtained from known mixtures of enzymes, buffers, salts, and metabolites in vitro with kinetic constants also measured in vitro, so consistency was anticipated.
SPECIAL NOTE
The entire source code for model simulations in MatLab (The MathWorks, Natick, MA) is available for download from Muscle Metabolism/Systems Biology group, Department of Radiology, University of Washington at http://depts.washington.edu/mrmuscle.
Model implementation in JSim 1.6 is also provided for download. Modeling software JSim 1.6 is available for download from the National Simulation Resource, Department of Bioengineering, University of Washington at http://nsr.bioeng.washington.edu.
MatLab interface for the RADAU solver is available for download from Wissenschaftliches Rechnen, Zentrum Mathematik, Technische Universität München at http://www-m3.mathematik.tu-muenchen.de/twiki/bin/view/Software/ODEHome.
The model source code for Experiment 29, 99.8% GPb, is included in the Supplementary Material.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The following individuals gave valuable comments and critiques to the development of this work and to the text of the article: Dr. Dan Beard (Medical College of Wisconsin), Dr. Feng Yang (Medical College of Wisconsin), Dr. Ron Meyer (Michigan State University), and Dr. Paolo Vicini (University of Washington).
This work was supported by a predoctoral fellowship award from the American Heart Association to K.V., by a Whitaker Foundation Fellowship awarded to M.L.K., by National Institutes of Health grant No. R01-AR036281 awarded to M.J.K., and National Institutes of Health grant No. P41-EB-001975.
K. Vinnakota and M. L. Kemp contributed equally to this work.
References
- 1.Rutman, R. J., and P. George. 1961. Hydrogen ion effects in high-energy phosphate reactions. Proc. Natl. Acad. Sci. USA. 47:1094–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.George, P., and R. J. Rutan. 1960. The high energy phosphate bond. Concept. Prog. Biophys. Biophys. Chem. 10:2–53. [PubMed] [Google Scholar]
- 3.George, P., R. C. Phillips, and R. J. Rutman. 1963. Estimates of thermodynamic data for the formation of Mg2+ complexes of ATP and ADP at zero ionic strength. Biochemistry. 2:508–512. [DOI] [PubMed] [Google Scholar]
- 4.Smith, R. M., and R. A. Alberty. 1956. The apparent stability constants of ionic complexes of various adenosine phosphates with divalent cations. J. Am. Chem. Soc. 78:2376–2384. [Google Scholar]
- 5.Alberty, R. A. 2004. A short history of the thermodynamics of enzyme-catalyzed reactions. J. Biol. Chem. 279:27831–27836. [DOI] [PubMed] [Google Scholar]
- 6.Alberty, R. A. 2003. Thermodynamics of Biochemical Reactions. John Wiley and Sons, Hoboken, NJ.
- 7.Harkema, S. J., and R. A. Meyer. 1997. Effect of acidosis on control of respiration in skeletal muscle. Am. J. Physiol. Cell Physiol. 41:C491–C500. [DOI] [PubMed] [Google Scholar]
- 8.Kushmerick, M. J. 1997. Multiple equilibria of cations with metabolites in muscle bioenergetics. Am. J. Physiol. 272:C1739–C1747. [DOI] [PubMed] [Google Scholar]
- 9.Sato, K., Y. Kashiwaya, C. Keon, N. Tsuchiya, M. T. King, G. K. Radda, B. Chance, K. Clarke, and R. L. Veech. 1995. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 9:651–658. [DOI] [PubMed] [Google Scholar]
- 10.Beard, D. A., E. Babson, E. Curtis, and H. Qian. 2004. Thermodynamic constraints for biochemical networks. J. Theor. Biol. 228:327–333. [DOI] [PubMed] [Google Scholar]
- 11.Mulquiney, P. J., W. A. Bubb, and P. W. Kuchel. 1999. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: in vivo kinetic characterization of 2,3-bisphosphoglycerate synthase/phosphatase using 13C and 31P NMR. Biochem. J. 342:567–580. [PMC free article] [PubMed] [Google Scholar]
- 12.Mulquiney, P. J., and P. W. Kuchel. 1999. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: computer simulation and metabolic control analysis. Biochem. J. 342:597–604. [PMC free article] [PubMed] [Google Scholar]
- 13.Mulquiney, P. J., and P. W. Kuchel. 1999. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: equations and parameter refinement. Biochem. J. 342:581–596. [PMC free article] [PubMed] [Google Scholar]
- 14.Scopes, R. K. 1973. Studies with a reconstituted muscle glycolytic system. The rate and extent of creatine phosphorylation by anaerobic glycolysis. Biochem. J. 134:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scopes, R. K. 1974. Studies with a reconstituted muscle glycolytic system. The rate and extent of glycolysis in simulated post-mortem conditions. Biochem. J. 142:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambeth, M. J., and M. J. Kushmerick. 2002. A computational model for glycogenolysis in skeletal muscle. Ann. Biomed. Eng. 30:808–827. [DOI] [PubMed] [Google Scholar]
- 17.Robergs, R. A., F. Ghiasvand, and D. Parker. 2004. Biochemistry of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287:R502–R516. [DOI] [PubMed] [Google Scholar]
- 18.Needham, D. M. 1971. Machina Carnis: the Biochemistry of Muscular Contraction in its Historical Development. Cambridge University Press, London, UK.
- 19.Gevers, W. 1977. Generation of protons by metabolic processes in heart cells. J. Mol. Cell. Cardiol. 9:867–874. [DOI] [PubMed] [Google Scholar]
- 20.Portner, H. O., N. Heisler, and M. K. Grieshaber. 1984. Anaerobiosis and acid-base status in marine invertebrates: a theoretical analysis of proton generation by anaerobic metabolism. J. Comp. Physiol. [B]. 155:1–12. [Google Scholar]
- 21.Hochachka, P. W., and T. P. Mommsen. 1983. Protons and anaerobiosis. Science. 219:1391–1397. [DOI] [PubMed] [Google Scholar]
- 22.Achs, M. J., and D. Garfinkel. 1977. Computer simulation of rat heart metabolism after adding glucose to the perfusate. Am. J. Physiol. 232:R175–R184. [DOI] [PubMed] [Google Scholar]
- 23.Achs, M. J., and D. Garfinkel. 1977. Computer simulation of energy metabolism in anoxic perfused rat heart. Am. J. Physiol. 232:R164–R174. [DOI] [PubMed] [Google Scholar]
- 24.Achs, M. J., D. Garfinkel, and L. H. Opie. 1982. Computer simulation of metabolism of glucose-perfused rat heart in a work-jump. Am. J. Physiol. 243:R389–R399. [DOI] [PubMed] [Google Scholar]
- 25.Robinson, R. A., and R. H. Stokes. 1968. Electrolyte Solutions. Butterworths, London, UK.
- 26.Dawson, R. M. C., D. C. Elliott, W. H. Elliott, and K. M. Jones. 1969. Data for Biochemical Research. Oxford University Press, London, UK.
- 27.Clarke, E. C. W., and D. H. Glew. 1980. Evaluation of Debye-Hückel limiting slopes for water between 0 and 150°C. J. Chem. Soc., Faraday Trans. 1. 76:1911–1916. [Google Scholar]
- 28.Adams, G. R., J. M. Foley, and R. A. Meyer. 1990. Muscle buffer capacity estimated from pH changes during rest-to-work transitions. J. Appl. Physiol. 69:968–972. [DOI] [PubMed] [Google Scholar]
- 29.Martell, A. E., and R. M. Smith. 2003. NIST Standard Reference Database 46 Version 7.0: NIST Critically Selected Stability Constants of Metal Complexes. NIST Standard Reference Data, Gaithersburg, MD.
- 30.Teague, W. E., Jr., E. M. Golding, and G. P. Dobson. 1996. Adjustment of K′ for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria to varying temperature and ionic strength. J. Exp. Biol. 199:509–512. [DOI] [PubMed] [Google Scholar]
- 31.Cornish-Bowden, A. 1995. Fundamentals of Enzyme Kinetics. Portland Press, London, UK.
- 32.Segel, I. H. 1975. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. Wiley, New York.
- 33.Dyson, J. E., and E. A. Noltmann. 1968. The effect of pH and temperature on the kinetic parameters of phosphoglucose isomerase. Participation of histidine and lysine in a proposed dual function mechanism. J. Biol. Chem. 243:1401–1414. [PubMed] [Google Scholar]
- 34.Keleti, T., and J. Batke. 1967. The kinetics of the reactions catalyzed by d-glyceraldehyde-3-phosphate dehydrogenase. 3. The pH dependence of apparent Michaelis constants and maximum velocity. Enzymologia. 33:65–79. [PubMed] [Google Scholar]
- 35.Najjar, V. A. 1948. The isolation and properties of phosphoglucomutase. J. Biol. Chem. 175:281–290. [PubMed] [Google Scholar]
- 36.Paetkau, V., and H. A. Lardy. 1967. Phosphofructokinase. Correlation of physical and enzymatic properties. J. Biol. Chem. 242:2035–2042. [PubMed] [Google Scholar]
- 37.Simon, L. M., M. Kotorman, B. Szajani, and L. Boross. 1985. Comparative studies on soluble and immobilized rabbit muscle pyruvate kinase. Appl. Biochem. Biotechnol. 11:195–205. [DOI] [PubMed] [Google Scholar]
- 38.Stankiewicz, P. J., and L. F. Hass. 1986. The catalytic bimodality of mammalian phosphoglycerate mutase. J. Biol. Chem. 261:12715–12721. [PubMed] [Google Scholar]
- 39.Winer, A. D., and G. W. Schwert. 1958. Lactic dehydrogenase. IV. The influence of pH on the kinetics of the reaction. J. Biol. Chem. 231:1065–1083. [PubMed] [Google Scholar]
- 40.Yunis, A. A., E. H. Fischer, and E. G. Krebs. 1960. Crystallization and properties of human muscle phosphorylases a and b. J. Biol. Chem. 235:3163–3168. [PubMed] [Google Scholar]
- 41.Abraham, M., L. Horvath, and B. Szajani. 1985. Isolation and characterization of pig muscle aldolase. A comparative study. Comp. Biochem. Physiol. B. 80:847–852. [DOI] [PubMed] [Google Scholar]
- 42.Bakker, B. M., P. A. Michels, F. R. Opperdoes, and H. V. Westerhoff. 1997. Glycolysis in bloodstream from Trypanosoma brucei can be understood in terms of the kinetics of the glycolytic enzymes. J. Biol. Chem. 272:3207–3215. [DOI] [PubMed] [Google Scholar]
- 43.Teusink, B., J. Passarge, C. A. Reijenga, E. Esgalhado, C. C. van der Weijden, M. Schepper, M. C. Walsh, B. M. Bakker, K. van Dam, H. V. Westerhoff, and J. L. Snoep. 2000. Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 267:5313–5329. [DOI] [PubMed] [Google Scholar]
- 44.Ostro, M. J., and T. P. Fondy. 1977. Isolation and characterization of multiple molecular forms of cytosolic NAD-linked glycerol-3-phosphate dehydrogenase from normal and neoplastic rabbit tissues. J. Biol. Chem. 252:5575–5583. [PubMed] [Google Scholar]
- 45.Hairer, E., and G. Wanner. 2004. Solving Ordinary Differential Equations II: Stiff and Differential-Algebraic Problems. R. Bank, R. L. Graham, J. Stoer, R. Varga, and H. Yserentant, editors. Springer, New York.
- 46.Meyer, R. A., and J. M. Foley. 1996. Cellular processes integrating the metabolic response to exercise. In Handbook of Physiology. L. B. Rowell and J. T. Shepherd, editors. Oxford University Press, New York. 841–869.
- 47.MacDonald, J. A., and K. B. Storey. 2002. Purification and characterization of fructose bisphosphate aldolase from the ground squirrel, Spermophilus lateralis: enzyme role in mammalian hibernation. Arch. Biochem. Biophys. 408:279–285. [DOI] [PubMed] [Google Scholar]
- 48.Vinnakota, K. C., M. L. Kemp, and M. J. Kushmerick. 2005. Dynamics of pH changes and phoshoenergetics in mouse skeletal muscle during transient anoxia are modeled by physical chemical principles. Late Breaking Abstracts, XXXV International Congress of Physiological Sciences/Experimental Biology, San Diego. LB158.
- 49.Cornell, N. W., M. Leadbetter, and R. L. Veech. 1979. Effects of free magnesium concentration and ionic strength on equilibrium constants for the glyceraldehyde phosphate dehydrogenase and phosphoglycerate kinase reactions. J. Biol. Chem. 254:6522–6527. [PubMed] [Google Scholar]
- 50.Cornish-Bowden, A., and M. L. Cardenas. 2000. Irreversible reactions in metabolic simulations: how reversible is irreversible? In BTK2000: Animating the Cellular Map. J. H. S. Hofmeyr, J. H. Rohwer, and J. L. Snoep, editors. Stellenbosch University Press. Stellenbosch, South Africa. 65–71.
- 51.Goldberg, R. N., and Y. B. Tewari. 1994. Thermodynamics of enzyme-catalyzed reactions: part 2. Transferases. J. Phys. Chem. Ref. Data. 23:547–617. [Google Scholar]
- 52.Goldberg, R. N., Y. B. Tewari, D. Bell, K. Fazio, and E. Anderson. 1993. Thermodynamics of enzyme-catalyzed reactions: part 1. Oxidoreductases. J. Phys. Chem. Ref. Data. 22:515–582. [Google Scholar]
- 53.Robitaille, P.-M. L., and P. A. Robitaille. 1969. J. Gordon Brown, G., and G. G. Brown. 1991. An analysis of the pH-dependent chemical-shift behavior of phosphorus-containing metabolites. J. Magn. Reson. 92:73–84. [Google Scholar]
- 54.Beard, D. A., and H. Qian. 2005. Thermodynamic-based computational profiling of cellular regulatory control in hepatocyte metabolism. Am. J. Physiol. Endocrinol. Metab. 288:E633–E644. [DOI] [PubMed] [Google Scholar]