

Abstract
SPARC, a 32-kDa glycoprotein, participates in the regulation of morphogenesis and cellular differentiation through its modulation of cell-matrix interactions. Major functions defined for SPARC in vitro are de-adhesion and antiproliferation. In vivo, SPARC is restricted in its expression to remodeling tissues, including pathologies such as cancer. However, the function of endogenous SPARC in tumor growth and progression is not known. Here, we report that implanted tumors grew more rapidly in mice lacking SPARC. We observed that tumors grown in SPARC null mice showed alterations in the production and organization of ECM components and a decrease in the infiltration of macrophages. However, there was no change in the levels of angiogenic growth factors in comparison to tumors grown in wild-type mice, although there was a statistically significant difference in total vascular area. Whereas SPARC did inhibit the growth of tumor cells in vitro, it did not have a demonstrable effect on the proliferation or apoptosis of tumor cells in vivo. These data indicate that host-derived SPARC is important for the appropriate organization of the ECM in response to implanted tumors and highlight the importance of the ECM in regulating tumor growth.
Introduction
The development, growth, and spread of cancer occur in the context of the ECM and are influenced by its constituents (1, 2). The ECM contains a mixture of glycoproteins, proteoglycans, growth factors, and structural proteins that provide a context for cellular function and act to maintain homeostasis in tissues by influencing cell communication and adhesion. Tumor cells must respond and adapt to the local microenvironment to progress to malignant disease, a process involving not only deregulated proliferation of tumor cells but also modification of the immediate environment to favor cell survival, the recruitment of new blood vessels, and the metastatic spread of the tumor (3, 4). It is therefore important to identify the proteins that regulate cell-matrix interactions in malignant disease.
SPARC (secreted protein acidic and rich in cysteine), also called osteonectin or BM-40, is a matricellular glycoprotein that modulates cellular interaction with the ECM and is expressed in tissues undergoing remodeling (5). It functions as a de-adhesive protein, as a modulator of growth factor activity, and as a cell-cycle inhibitor. SPARC can bind many proteins that reside in the ECM — for example, the fibril-forming collagens (types I, III, and V), collagen IV (basement membranes), vitronectin (6), and laminin-1 (LN1; our unpublished data). Mice with a homozygous null mutation in the SPARC gene (SP–/–) (7) undergo progressive early-onset cataractogenesis (8) and exhibit altered dermal connective tissue characterized by accelerated closure of cutaneous wounds (9) and enhanced fibrovascular invasion of subcutaneous sponge implants (10).
The context of the expression of SPARC is critical for an understanding of its functions in a given microenvironment. SPARC is generally considered an antiangiogenic protein because it blocks VEGF- and FGF-2–induced proliferation of endothelial cells. It can also inhibit PDGF activity on stromal cells (6). Expression of SPARC by melanoma and glioma cells has been linked to an invasive phenotype in vivo (11, 12). In contrast, overexpression of SPARC by ovarian carcinoma cells led to increased tumor cell apoptosis, and the levels of SPARC were inversely correlated with tumor progression in vivo (13). Using genetically deficient mice, we have tested the contribution of host SPARC to the progression of syngeneic tumors. Here we report that (1) tumor growth is enhanced in mice lacking endogenous SPARC, (2) the increased tumor size in the SP–/– mice is not due to an increase in tumor angiogenesis, and (3) the increased tumor growth results from changes in the ECM of the SP–/– mice that create a more permissive environment for tumor progression.
Methods
Cell cultures.
Lewis lung carcinoma (LLC) cells were obtained from Judah Folkman (Children’s Hospital, Boston, Massachusetts, USA), bovine aortic endothelial (BAE) cells were isolated as previously described (14), bEnd.3 cells were provided by the late Werner Risau (Bad Nauheim, Germany), and EL4 cells were provided by Brad Nelson (Benaroya Research Institute, Seattle, Washington, USA). All of the cell lines were grown in DMEM supplemented with L-glutamine (2 mM), penicillin (100 U/ml), streptomycin sulfate (100 μg/ml), and 5–10% fetal bovine serum (Life Technologies Inc., Grand Island, New York, USA).
Tumor growth in vivo.
C57Bl/6 × 129SvJ SPARC null (SP–/–) and wild-type (SP+/+) mice were generated as described (9). The mice were backcrossed against wild-type C57Bl/6 for at least four generations before use in the tumor studies. For subcutaneous tumor growth, LLC cells were removed from tissue culture flasks with trypsin, or EL4 cells were pelleted from suspension; cells were washed two times with HBSS, counted, and resuspended at 1 × 107 (LLC) or 5 × 106 (EL4) cells per milliliter. Cells (200 μl in HBSS) were injected subcutaneously into mice. For intravenous injection, LLC cells were resuspended at 1 × 106 cells per milliliter, and 100 μl was injected into the tail vein. For intraperitoneal injection, EL4 cells (200 μl at 5 × 106 cells per milliliter) were injected into the peritoneal cavity. The mice were housed in a pathogen-free facility, and experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of the Hope Heart Institute and Fred Hutchinson Cancer Research Center (Seattle, Washington, USA). Subcutaneous tumors were excised, weighed, and divided such that one third of each tumor was frozen, one third was immersed in methyl Carnoy’s fixative, and the remaining third was fixed in 10% neutral buffered formalin. The lungs of the mice injected intravenously with LLC cells were removed, weighed, and immersed in Bouin’s solution (0.9% picric acid, 9% formaldehyde, and 5% acetic acid). The lungs were examined under a dissecting microscope, and the surface tumor colonies were counted. Mice injected intraperitoneally with EL4 cells were dissected and evaluated for ascites production and tumor growth in the peritoneal cavity. All organs were monitored for disseminated disease.
Histology and immunohistochemical analysis.
Formalin- and methyl Carnoy’s–fixed tissues embedded in paraffin were sectioned by the Histopathology Laboratory at the University of Washington. Formalin-fixed sections were stained with hematoxylin and eosin, Masson’s trichrome, and picrosirius red according to standard protocols (15, 16). Methyl Carnoy’s–fixed sections were deparaffinized under standard conditions. If necessary, endogenous peroxidases were blocked in methanol with 1.0% H2O2 for 30 minutes. Some of the sections were subsequently treated with AutoZyme (10 μl of enzyme concentrate/1 ml of buffer for 6 minutes at room temperature; BioMeda Corp., Foster City, California, USA). Antibodies requiring AutoZyme treatment were goat anti-mouse SPARC (R&D Systems Inc., Minneapolis, Minnesota, USA), rabbit anti-LN1 (BD Biosciences, San Diego, California, USA), rabbit anti-LN1 (Sigma-Aldrich, St. Louis, Missouri, USA), rat anti-LNα1 chain (Chemicon, Temecula, California, USA), rat anti-F4/80 (Serotec, Raleigh, North Carolina, USA), rat anti–Mac-3 (BD Biosciences), and rabbit anti-VEGFR2 (T014) (17). The antibodies that did not require antigen retrieval were rabbit anti-mouse collagen type I (Biodesign International, Saco, Maine, USA), rabbit anti-collagen type IV (BD Biosciences), rabbit anti–phospho-histone H3 (Upstate Biotechnology Inc., Lake Placid, New York, USA), rabbit anti–active caspase-3 (R&D Systems Inc.), and rat anti-mouse endothelial cell MECA32 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, Iowa, USA). The sections were incubated with primary antibody for 1 hour, washed with PBS containing 0.2% Tween-20, incubated for 1 hour with the appropriate peroxidase-labeled or FITC-labeled secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA), developed with Stable DAB (ResGen, Huntsville, Alabama, USA), counterstained with hematoxylin, and coverslipped in Permount (Fisher, Fair Lawn, New Jersey, USA). Slides treated with FITC were coverslipped in ProLong antifade mounting media (Molecular Probes, Eugene, Oregon, USA). Sections were examined on a Leica DMR microscope, and images were captured digitally with an RT-Spot camera (Diagnostic Instruments, Sterling Heights, Michigan, USA).
RT-PCR.
RNA was isolated with Tri Reagent (Molecular Research Center Inc., Cincinnati, Ohio, USA) according to the manufacturer’s instructions. PCR was performed as described (18) with the following primer sequences: SPARC sense (5′-GTCCCACACTGAGCTGGC-3′) and anti-sense (5′-AAGCACAGAGTCTGGGTGAGTG-3′), collagen 1A1 sense (5′-GAACTTGGGGCAAGACAGTCA-3′) and anti-sense (5′-GTCACGTTCAGTTGGTCAA-AGG-3′), LNα1 sense (5′-GATGCCATTGGCCTAGAGATTG-3′) and anti-sense (5′-GGATGGGAATGGGAGCTGA-3′), TGF-β1 sense (5′-ACCATCCATGACATGAACCG-3′) and antisense (5′-GGCTTGCGACCCACGTA-3′), VEGF sense (5′-AGGCTGCTGTAACGATGAAGC-3′) and antisense (5′-CCGCCTTGGCT-TGTCAC-3′), VEGFR1 sense (5′-GTGTGCTTAGGTCGTGCACAC-3′) and anti-sense (5′-CTG-GATGAGAAACCTC-AGGCT-3′), VEGFR2 sense (5′-CGAGTCTGTCTACCTTGGAGGC-3′) and anti-sense (5′-CAGCCTGAGCCTTTACCGC-3′), neuropilin-1 (Npn-1) sense (5′-ACCACTGAT-AACTCGATTTGTCCG-3′) and anti-sense (5′-CGATCTTG-AACTTCCTCATGAACAC-3′), SC1 sense (5′-TGGTTCTTGCACGAACTTCC-3′) and anti-sense (5′-GAGAAGTTCAATGGGATGGTCTC-3′), and rpS6 sense (5′-AAGCTCCGCACCTTCTATGAGA-3′) and anti-sense (5′-TGACTGGACTCAGACTTAGAAGTAGAAGC-3′).
Western blotting and ELISA.
Pieces of frozen tumor were homogenized in a modified RIPA buffer (19), and protein concentration was determined by a bicinchoninic acid assay (Pierce Chemical Co., Rockford, Illinois, USA). Lysate (25 μg) from each tumor was loaded onto duplicate 12% SDS-polyacrylamide gels, one of which was stained with Coomassie blue while the other was used for electrophoretic transfer to a PVDF membrane. The membrane was blocked with Aquablock (East Coast Biologics Inc., Berwick, Maine, USA) for 1 hour at room temperature. The membrane was probed overnight with an anti-SPARC antibody. Proteins secreted by LLC cells in vitro were probed with 303, a mouse anti-SPARC monoclonal antibody that binds to mouse but not bovine SPARC (our unpublished data). The blots were developed as described (19). For ELISA, 100 μg of tumor lysate was assayed for VEGF (R&D Systems Inc.) or 20 μg was assayed for TGF-β1 (Promega, Madison, Wisconsin, USA), according to the manufacturers’ instructions.
Determination of vascular area.
Using Adobe Photoshop, we analyzed the green channel of images of FITC-labeled MECA32-positive blood vessels. The “magic wand” tool was used to detect regions of dissimilar color; the threshold for detection (0–255) was set by eye and ranged between 4 and 35, and the background was measured at multiple sites within each image. Images were measured at different threshold levels and with different background regions; for each image, we found insignificant quantitative differences in vascular area among these parameters.
In vitro proliferation assay.
Quiescent cells were stimulated with serum in the presence or absence of recombinant human SPARC, and incorporation of [3H]thymidine (PerkinElmer, Torrance, California, USA) into DNA was measured as described (20).
Results
Host-derived SPARC influences tumor growth.
Given that there are alterations in the ECM of mice lacking different matricellular proteins (21, 22), we investigated the importance of host-derived SPARC on the growth of implanted tumors. SP+/+ and SP–/– mice were injected subcutaneously with murine LLC or T-cell lymphoma (EL4) cells. At the time of sacrifice, the SP–/– mice had significantly larger LLC tumors than the SP+/+ mice (Figure 1a), with mean tumor volumes of 1930 and 405 mm3 and mean tumor weights of 1.5 and 0.46 g, respectively. The rate of tumor take for both genotypes was 100%. A three- to fivefold difference in LLC tumor size between SP–/– and SP+/+ mice, 3–4 weeks after tumor cell injection, was consistent in separate experiments in three different litters of mice. We also challenged the mice by injecting LLC cells intravenously and found a significant difference in the number of detectable tumor colonies on the surface of the lungs between the SP+/+ and SP–/– mice, with a mean value (± SE) of 6 ± 2.3 and 22 ± 6 colonies, respectively (Figure 1b).
Figure 1.

Tumor growth is enhanced in SPARC null mice. SP+/+ and SP–/– mice were injected subcutaneously with 2 × 106 LLC or 1 × 106 EL4 cells (a and c, respectively). (a) Comparison of the mean final volume and weight of LLC tumors grown subcutaneously in SP+/+ and SP–/– mice. The results shown are the combination of two independent experiments, which lasted 24–29 days after tumor cell injection. (b) Comparison of the number of lung surface tumor colonies in age-matched SP+/+ and SP–/– mice 14–19 days after intravenous injection of 1 × 105 LLC cells. The results shown are the combination of two independent experiments. Surface lung tumor colonies were visualized under a dissecting microscope after fixation in Bouin’s fixative. (c) Comparison of the mean final volume and weight of EL4 tumors grown subcutaneously in SP+/+ and SP–/– mice. The results shown are the combination of two independent experiments, which lasted 14–16 days after tumor cell injection. (d) Comparison of the animals’ weights after intraperitoneal injection of 1 × 106 EL4 cells, and the weight of the GI tract at sacrifice. P values were determined by Student’s t test.
EL4 cells also grew more rapidly in the SP–/– mice after subcutaneous injection (Figure 1c). However, in contrast to the LLC tumors, only 50% of the SP+/+ mice showed evidence of established subcutaneous EL4 tumors at the time of sacrifice, whereas all of the SP–/– mice had large, well-established subcutaneous tumors. After intraperitoneal injection, we found that 100% of the SP–/– mice produced a large of amount of ascitic fluid and had substantial tumor growth that involved the entire gastrointestinal (GI) tract. Although 50% of the SP+/+ mice also produced copious ascitic fluid and exhibited substantial tumor growth, 25% of these mice produced only a small amount of ascitic fluid, and 25% produced none. The levels of ascitic fluid were correlated with the weight of the animals and of the GI tract (Figure 1d). The latter was significantly different between SP+/+ and SP–/– mice, with mean values (± SD) of 4.4 ± 1.1 g and 6.3 ± 0.6 g, respectively.
SPARC influences matrix deposition.
We hypothesized that the lack of host-derived SPARC might influence the growth of the tumor cells in vivo by alteration of (1) the deposition of ECM in and around the tumor mass, (2) angiogenesis within the tumor, and/or (3) the survival or proliferation of the tumor cells. The gross morphology of tumors grown in both genotypes appeared similar (Figures 2a and 2b). However, differences between the tumors became apparent after staining with Masson’s trichrome and picrosirius red, both of which provide information on extracellular collagen. Collagen fibers (mainly collagens I and III), stained bright blue by Masson’s trichrome (15), were clearly evident in the cap-sules surrounding the tumors. Representative sections (Figure 2a) show a reduction in collagen in the capsules of tumors grown in SP–/– mice, as well as a reduction in the birefringence of the collagen fibers stained with picrosirius red. This reduction indicates that the collagen fibers (yellow-green color) surrounding the tumors in SP–/– mice are of a smaller diameter and have fewer cross-links than the more mature collagen fibers (red) in the capsules surrounding tumors grown in SP+/+ mice (16, 23).
Figure 2.

Tumor growth in SPARC null mice is associated with changes in collagen organization. Representative serial sections of LLC (a) and EL4 (b) tumors grown in SP+/+ or SP–/– mice and stained with hematoxylin and eosin, Masson’s trichrome, and picrosirius red. The bottom panel of (a) represents an internal highway of connective tissue in an LLC tumor from SP+/+ and SP–/– mice. Scale bars: 50 μm. (c) Immunostaining for collagen I of LLC tumors grown in SP+/+ or SP–/– mice. The top panel represents the capsule area, and the lower panel represents the interior of the tumor. Arrows denote capsular fibroblasts and arrowheads denote tumor cells.
EL4 tumors revealed a similar pattern of reactivity (Figure 2b). Immunostaining for collagen I (Figure 2c) was consistent with the data shown in Figure 2a. The majority of reactivity was in or around the capsule of the tumors and appears to be largely the result of host cell production of collagen. There was only light and sporadic staining in tumor cell nests, as seen in both SP+/+ and SP–/– animals (Figure 2c, lower panels). There were also differences in the interiors of large LLC tumors stained with picrosirius red. Figure 2a (bottom panel) shows a “highway” of connective tissue that was laid down in the tumor. In the SP+/+ mouse, this stromal component exhibits a red fluorescence, whereas a comparable tract of connective tissue in the tumor grown in the SP–/– mouse exhibits a yellow and green fluorescence, typical of less mature collagen fibers. In summary, the collagen that is laid down in response to the tumor does not mature at the same rate in the absence of host-derived SPARC.
SPARC is expressed in LLC tumors.
Cultured LLC but not EL4 cells contained detectable amounts of SPARC mRNA (Figure 3a) and protein (data not shown). SPARC was consistently produced at a higher level in LLC tumors grown in SP+/+ mice, as shown by RT-PCR (Figure 3b), Western blotting (Figure 3c), and immunohistochemistry (Figure 3d). That the LLC tumors grown in SP–/– mice did show SPARC mRNA and protein indicated that the tumor cells themselves produce SPARC in vivo. We found no significant differences in the profile of proteins resolved by SDS-PAGE (Figure 3c). There was a difference, however, in the distribution of SPARC between the LLC tumors grown in SP+/+ and SP–/– mice (Figure 3d). In the former, SPARC was located in the outer one third of the tumor and capsule, with staining largely in the cytoplasm of host cells (possibly endothelial cells and fibroblasts) as well as some tumor cells. In contrast, in the tumors from the SP–/– mice, SPARC appeared less frequently and was confined to the cytoplasm of tumor cells and to some necrotic areas. There was considerably less staining for SPARC in the outer one third of the tumors grown in SP–/– relative to SP+/+ hosts. Expression of SPARC was restricted to host cells in the EL4 tumors grown in SP+/+ mice and was not present in tumors grown in SP–/– hosts (see supplemental Figure 1, http://www.jci.org/cgi/content/full/111/4/497/DC1).
Figure 3.

Expression of SPARC mRNA and protein by LLC cells and tumors. (a) RT-PCR analysis of LLC, EL4, and bEnd.3 cells grown in culture. (b) RT-PCR analysis of LLC tumors grown in SP+/+ or SP–/– mice (n = 6). RNA from bEnd.3 cells grown in vitro was used as a positive control. A representative tumor from the SP+/+ and SP–/– groups is shown. (c) SDS-PAGE of tumor lysates and demonstration of SPARC expression by Western blot analyses. Two identically prepared gels were separated by SDS-PAGE; one was transferred to PVDF for Western blot analysis (left), and the other was stained with Coomassie blue (right). The latter demonstrates a similar protein profile in the lysates from tumors removed from both wild-type and SPARC null mice, and it shows equivalent protein loading among the lanes on the gel. Bars represent molecular weight standards in kDa. (d) Immunohistochemical analysis of SPARC in LLC tumors grown in SP+/+ and SP–/– mice. The rectangular insets are enlarged in the bottom panel. Arrows indicate positive tumor cells; the arrowhead indicates a positive host cell. Scale bars: 50 μm. Col I, collagen type I; rpS6, ribosomal protein S6.
Host SPARC influences protein expression in LLC tumors.
By RT-PCR, LLC and EL4 cells in vitro transcribed VEGF and VEGFR1 but not VEGFR2, Npn-1, or the SPARC homolog SC1/hevin (Figure 3a). bEnd.3, a transformed mouse endothelial cell line, expressed all three VEGFRs as well as SPARC and SC1.
Six tumors from the SP+/+ and SP–/– mice were analyzed by RT-PCR for the level of the indicated proteins; a representative panel from each set is shown (Figure 3b). Steady-state levels of collagen I and TGF-β1 appeared similar in the tumors from the SP+/+ and SP–/– mice. Levels of VEGF mRNA were also similar between LLC tumors grown in SP+/+ and SP–/– mice, but perhaps surprisingly, VEGFR1, VEGFR2, and Npn-1 mRNAs were reduced in tumors grown in SP–/– mice. The absence of host-derived SPARC did not influence the level of SC1 mRNA found in the tumor samples.
We anticipated that there might be changes in the distribution or levels of several major basement membrane proteins. LN1 was in fact reduced in tumors from SP–/– relative to those from SP+/+ mice (Figure 4). Differences in LN1 were most evident on tissues stained with polyclonal antibodies specific for Engelbreth-Holm-Swarm (EHS) tumor-derived laminin (Figure 4, top panel). Monoclonal antibodies specific for either the α1 (Figure 4, middle panels) or β1 chains (not shown) of LN1 also indicated reduced amounts of laminin in SP–/– tumors. The differences between the tumors were most apparent in the outer regions near the capsule, where both laminin and SPARC were preferentially produced in SP+/+ mice. Interestingly, the LNα1 chain was located more centrally in tumors from SP–/– mice, often near necrotic areas consistent with the location of SPARC in these tumors. The distribution of collagen IV was similar in both genotypes (Figure 4, bottom panel). Fibronectin, a major structural component of the ECM, as well as MMP 9, was also distributed in a similar fashion in both sets of tumors (data not shown).
Figure 4.
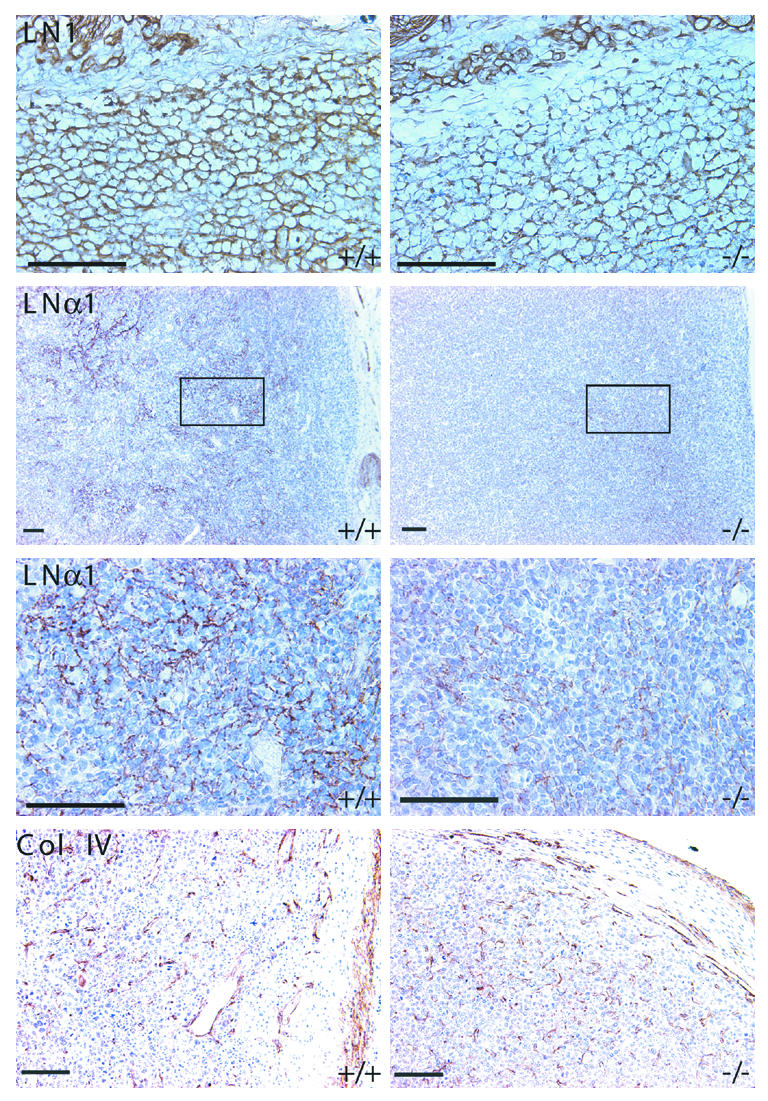
Changes in the expression of LN1 are associated with increased tumor growth in SPARC null mice. Immunohistochemical analysis of LN1, LNα1, and collagen type IV in LLC tumors grown in SP+/+ or SP–/– mice. Scale bars: 50 μm. Col IV, collagen type IV.
Angiogenesis in LLC tumors from SP+/+ and SP–/– mice.
We examined the number and distribution of blood vessels in the tumors by immunohistochemical identification of endothelial cells with two antibodies, one specific for VEGFR2 (17) and MECA32, a pan-endothelial cell marker (24) (Figure 5a). Counting the number of immunoreactive vessels in high-power fields yielded no significant differences in the number of vessels per unit area (data not shown). However, analysis of vascular area, as determined by MECA32-positive structures, showed a statistically significant decrease in vascular area in the tumors grown in SP–/– mice (Table 1). We also examined the levels of VEGF and TGF-β1 protein by ELISA of lysates from the LLC tumors and found no significant differences in either growth factor, irrespective of genotype; VEGF was present at 5.0 ± 2.9 ng and 3.9 ± 1.9 ng per 100 μg of LLC lysate from SP+/+ and SP–/– mice, respectively. Corresponding values for TGF-β1 were 70.4 ± 12.8 ng and 71.4 ± 12.2 ng per 20 μg of lysate from SP+/+ and SP–/– mice, respectively, with approximately 30% in the active form in each case.
Figure 5.

Analysis and quantification of markers of angiogenesis in LLC tumors grown in SP+/+ and SP–/– mice. (a) Blood vessels of tumors grown in SP+/+ versus SP–/– mice stained with either anti-VEGFR2 or MECA32. (b) Representative immunohistochemical staining with F4/80, a marker of mature macrophages, showing the extent of infiltration into the LLC tumors grown in SP+/+ or SP–/– mice. Scale bars: 50 μm.
Table 1.
Percent vascular area of LLC tumors

Host SPARC supports macrophage infiltration.
We have seen no evidence of rejection or spontaneous regression of LLC tumors grown in either SP+/+ or SP–/– mice. However, there were differences in the number and distribution of macrophages. The antigens F4/80 (Figure 5b) and Mac-3 (see supplemental Figure 2, http://www.jci.org/cgi/content/full/111/4/497/DC1), both expressed on mature murine macrophages (25, 26), were more apparent in tumors from SP+/+ mice. F4/80-positive cells were found in both tumor sets near the capsular region of the tumor; however, the intensity of staining diminished rapidly in the interior of tumors from SP–/– mice, whereas it was sustained in tumors from SP+/+ mice.
Tumor cell proliferation and apoptosis in LLC tumors from SP+/+ and SP–/– mice are equivalent.
Since SPARC is known to block proliferation of some cell types in G1 (20), we evaluated whether SPARC could inhibit serum-induced proliferation of LLC cells in vitro. Using a 3H-thymidine incorporation assay, we found that purified recombinant human (rh) SPARC (27) inhibited serum-induced proliferation of LLC and BAE cells in a concentration-dependent manner (Table 2).
Table 2.
SPARC inhibits the proliferation of LLC tumor cells in vitro

We evaluated the proliferation of the tumor cells in vivo with an antibody against phosphorylated histone H3 (p-H3) (28) (Table 3 and supplemental Figure 3, http://www.jci.org/cgi/content/full/111/4/497/DC1). Quantification of the number of nuclei positive for p-H3 in tumors grown in SP+/+ and SP–/– mice showed no significant difference in the proliferation of the tumor cells, despite the fact that the tumors were, on average, three- to fourfold larger in the SP–/– mice. Importantly, the number of nuclei per unit area was similar in LLC tumors grown in SP+/+ and SP–/– mice, as determined by quantification of high-power fields of non-necrotic areas with DAPI (Table 3). We also examined the extent of apoptosis in the LLC tumors by determining the levels of active caspase-3 by immunohistochemistry and immunoblotting of tumor lysates (data not shown). There was no consistent or significant difference in the number of tumor cells positive for active caspase-3 between the tumors grown in SP+/+ and those grown in SP–/– mice (Table 3). The tumors examined for proliferation and apoptosis were excised from SP+/+ and SP–/– mice 3 weeks after tumor cell injection and reflected the difference in tumor size shown in Figure 1a. These results indicate that there were no substantial differences in the rate of cell proliferation or programmed cell death in the LLC tumors grown in SP+/+ and SP–/– mice.
Table 3.
Analysis of tumor cell proliferation and apoptosis in vivo

Discussion
The major findings to emerge from this study are that SPARC influences the response of host tissue to implanted tumor cells and that a lack of endogenous SPARC results in (1) a diminished capacity to encapsulate the growing tumor; (2) an alteration in the deposition of the ECM constituents collagen and LN1, and (3) decreased invasion of the tumor by mature macrophages.
We found that tumor burden was increased in SP–/– as compared with SP+/+ mice after tumor cells were injected subcutaneously, intravenously, or intraperitoneally. Both tumor and stromal cells within tumors have been shown to produce SPARC (29, 30), and other studies have implicated tumor-derived SPARC as an important modulator of tumor progression and metastasis (11, 13, 31). However, our results with three different murine tumor cell lines — LLC, EL4, and PAN02, a pancreatic adenocarcinoma cell line (our unpublished data) — show an inverse correlation between the production of SPARC by the tumor cells and the rate of tumor growth in vivo. It is unclear whether tumor-derived SPARC is functionally different from endogenous SPARC produced by host stromal cells. It is possible that host-derived SPARC preferentially affects the stromal cells in the tumor and therefore has a greater influence on the host response to the tumor, which includes the formation and maturation of a capsule rich in collagen (32, 33).
SPARC binds to several collagen types, including collagen I and III (6, 34), the major structural proteins of the ECM produced by host cells in response to cutaneous wounds (35), implanted biomaterials (36), and certain injected tumor cells (37). Recent studies have revealed that the skin from SP–/– mice contains less hydroxyproline (and therefore less collagen) than that from SP+/+ counterparts (9). Furthermore, electron microscopy has shown that the mean diameter of collagen fibrils is reduced in SP–/– relative to SP+/+ dermis (38, 39). These results are consistent with our findings that there is a reduced amount of collagen in the capsules around the tumors grown in SP–/– mice, and that the collagen fibrils that are present contain fewer cross-links and are of smaller diameter. Together, these results highlight a function for SPARC in the regulation of the production and organization of the ECM in response to tumor growth.
It is clear that tumors are angiogenesis-dependent for growth and progression to metastatic disease (2, 40, 41). Therefore, we were surprised that there appeared to be no difference in the microvessel density of LLC tumors grown in SP–/– and SP+/+ mice. That there was actually a larger percent vascular area in tumors grown in SP+/+ mice was also unexpected but consistent with the PCR analysis of the tumors, which showed a reduced level of VEGFRs in tumors from SPARC null mice (Figure 3b). In support of this finding, the foreign body response to biomaterials showed a reduced number of blood vessels in the capsule surrounding the implant in SP–/– relative to SP+/+ mice (39). SPARC can be a proangiogenic factor when it is cleaved by proteases such as plasmin (42, 43), which are present in the tumor microenvironment (44). This property, coupled with an abundant ECM that is less permissive for tumor growth, could lead to an increased vascular area but a smaller overall size of the tumor in wild-type animals.
Macrophages influence the response of host tissue to tumor growth and can directly alter the progression of tumors (45). Our results indicate that host-SPARC influences infiltration of tumors by host macrophages, although it is unclear whether this is due to a direct interaction between SPARC and macrophages. Mice deficient in colony-stimulating factor (CSF-1) also show reduced macrophage infiltration of subcutaneous LLC tumors (46) and primary mammary tumors (47). However, in these mice tumor growth is reduced as compared with that in wild-type mice. Although these results differ from ours with respect to tumor progression, the tumors formed in CSF-1–deficient mice also exhibit reduced amounts of stroma. In fact, modulation of the ECM might point to a common mechanism in the regulation of tumor growth in CSF-1– and SPARC-deficient mice.
Although SPARC inhibited serum-stimulated growth of LLC cells in vitro, we were unable to show an increase in the number of proliferating tumor cells in vivo. We examined the expression of proliferating cell nuclear antigen and Ki-67 (data not shown), as well as p-H3, by immunohistochemistry; in each case, there were no apparent differences in the number of cycling cells in tumors grown in either genotype. SPARC can block cells in the G1 phase of the cell cycle but has no effect on cells in S phase (20, 48). It is possible that the effect of SPARC on tumor cell proliferation in vivo is slight, such that the cell cycle is lengthened but not inhibited. This effect could lead to a significantly greater number of tumor cells in the SP–/– mice over extended periods of time. We also found no difference in the number of cells positive for active caspase-3 or cleaved poly ADP-ribose polymerase (PARP) (data not shown) in the tumors grown in either genotype; hence, there was no substantial difference in the number of cells undergoing apoptosis in the tumors. Our results are consistent with the report from Yiu et al. (13), who found an inverse correlation between the levels of SPARC and the clinical grade of ovarian carcinoma samples from patients. This situation is comparable to the phenomenon we have observed in SP+/+ and SP–/– mice: tumor growth is slower or more controlled in the tissue that is expressing endogenous SPARC.
The ECM can influence tumor progression directly. For example, solid stress, which is comparable to the stress the ECM puts on the tumor mass, inhibits the growth of tumor spheroids in culture (49). These results therefore indicate that a less restrictive ECM allows greater growth. This concept is supported by a recent mathematical study on the mechanics of capsule formation, which predicts that a more robust ECM and capsule result in slower growth of the tumor (50). Additionally, a number of reports demonstrate the importance of the tumor microenvironment, particularly the ECM, as a determinant of drug delivery, gene expression, and angiogenesis (51, 52). These studies show that an increase in the collagen content of the ECM increases the mechanical stiffness and transport resistance of tumors. Increased collagen content and mechanical stiffness are properties that would also increase solid stress and therefore slow the growth of the tumor.
We have presented evidence for a role of host-derived SPARC in the response to tumor cells. Although other studies have shown that tumor-derived SPARC can increase the invasiveness of different tumors, our results show the importance of endogenous SPARC in controlling tumor size through the production and organization of the ECM.
Supplementary Material
Acknowledgments
We thank J. Folkman and B. Nelson for the LLC and EL4 tumor cells, respectively; A. Bradshaw, K. Motamed, A. Francki, and R. Vernon for support and insightful discussions; P. Theiss for advice on growing the EL4 cells; and S. Funk for maintenance of the animals. This work was supported in part by grants from the NIH (F32 HL10352 to R.A. Brekken, and RO1 GM40711 and RO1 HL59574 to E.H. Sage), and from The Seattle Foundation to The Hope Heart Institute.
Footnotes
Rolf A. Brekken’s present address is: Departments of Surgery and Pharmacology and the Hamon Center for Therapeutic Oncology Research, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: laminin-1 (LN1); Lewis lung carcinoma (LLC); bovine aortic endothelial (BAE); neuropilin-1 (Npn-1); gastrointestinal (GI); Engelbreth-Holm-Swarm (EHS); recombinant human (rh); phosphorylated histone H3 (p-H3); colony-stimulating factor (CSF-1); poly ADP-ribose polymerase (PARP).
References
- 1.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Radisky D, Hagios C, Bissell MJ. Tumors are unique organs defined by abnormal signaling and context. Semin. Cancer Biol. 2001;11:87–95. doi: 10.1006/scbi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 4.Yeo TK, Brown L, Dvorak HF. Alterations in proteoglycan synthesis common to healing wounds and tumors. Am. J. Pathol. 1991;138:1437–1450. [PMC free article] [PubMed] [Google Scholar]
- 5.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002;14:608–614. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 6.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 7.Gilmour DT, et al. Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. EMBO J. 1998;17:1860–1870. doi: 10.1093/emboj/17.7.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan Q, Sage EH. SPARC, a matricellular glycoprotein with important biological functions. J. Histochem. Cytochem. 1999;47:1495–1506. doi: 10.1177/002215549904701201. [DOI] [PubMed] [Google Scholar]
- 9.Bradshaw AD, Reed MJ, Sage EH. SPARC null mice exhibit accelerated cutaneous wound closure. J. Histochem. Cytochem. 2002;50:1–10. doi: 10.1177/002215540205000101. [DOI] [PubMed] [Google Scholar]
- 10.Bradshaw AD, et al. Increased fibrovascular invasion of subcutaneous polyvinyl alcohol sponges in SPARC null mice. Wound Repair Regen. 2001;9:522–530. doi: 10.1046/j.1524-475x.2001.00522.x. [DOI] [PubMed] [Google Scholar]
- 11.Ledda MF, et al. Suppression of SPARC expression by antisense RNA abrogates the tumorigenicity of human melanoma cells. Nat. Med. 1997;3:171–176. doi: 10.1038/nm0297-171. [DOI] [PubMed] [Google Scholar]
- 12.Rempel SA, Ge S, Gutierrez JA. SPARC: a potential diagnostic marker of invasive meningiomas. Clin. Cancer Res. 1999;5:237–241. [PubMed] [Google Scholar]
- 13.Yiu GK, et al. SPARC (secreted protein acidic and rich in cysteine) induces apoptosis in ovarian cancer cells. Am. J. Pathol. 2001;159:609–622. doi: 10.1016/S0002-9440(10)61732-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sage H, Vernon RB, Funk SE, Everitt EA, Angello J. SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca2+-dependent binding to the extracellular matrix. J. Cell Biol. 1989;109:341–356. doi: 10.1083/jcb.109.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carson, F.L. 1997. Histotechnology: a self-instructional text. ASCP Press. Chicago, Illinois, USA. 304 pp.
- 16.Andrade GB, Montes GS, Conceicao GM, Saldiva PH. Use of the picrosirius-polarization method to age fibrotic lesions in the hepatic granulomas produced in experimental murine schistosomiasis. Ann. Trop. Med. Parasitol. 1999;93:265–272. doi: 10.1080/00034989958528. [DOI] [PubMed] [Google Scholar]
- 17.Feng D, et al. Ultrastructural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressing tumors and adenoviral vectors. J. Histochem. Cytochem. 2000;48:545–556. doi: 10.1177/002215540004800412. [DOI] [PubMed] [Google Scholar]
- 18.Graves DC, Yablonka-Reuveni Z. Vascular smooth muscle cells spontaneously adopt a skeletal muscle phenotype: a unique Myf5(–)/MyoD(+) myogenic program. J. Histochem. Cytochem. 2000;48:1173–1193. doi: 10.1177/002215540004800902. [DOI] [PubMed] [Google Scholar]
- 19.Brekken RA, et al. Selective inhibition of vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1) activity by a monoclonal anti-VEGF antibody blocks tumor growth in mice. Cancer Res. 2000;60:5117–5124. [PubMed] [Google Scholar]
- 20.Funk SE, Sage EH. The Ca2+-binding glycoprotein SPARC modulates cell cycle progression in bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA. 1991;88:2648–2652. doi: 10.1073/pnas.88.7.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J. Clin. Invest. 2001;107:1049–1054. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyriakides TR, et al. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J. Cell Biol. 1998;140:419–430. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koren R, et al. Capsular collagen staining of follicular thyroid neoplasms by picrosirius red: role in differential diagnosis. Acta Histochem. 2001;103:151–157. doi: 10.1078/0065-1281-00587. [DOI] [PubMed] [Google Scholar]
- 24.Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev. Dyn. 1995;202:325–332. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- 25.Gordon S, et al. Antigen markers of macrophage differentiation in murine tissues. Curr. Top. Microbiol. Immunol. 1992;181:1–37. doi: 10.1007/978-3-642-77377-8_1. [DOI] [PubMed] [Google Scholar]
- 26.Ho MK, Springer TA. Tissue distribution, structural characterization, and biosynthesis of Mac-3, a macrophage surface glycoprotein exhibiting molecular weight heterogeneity. J. Biol. Chem. 1983;258:636–642. [PubMed] [Google Scholar]
- 27.Bradshaw AD, Bassuk JA, Francki A, Sage EH. Expression and purification of recombinant human SPARC produced by baculovirus. Mol. Cell. Biol. Res. Commun. 2000;3:345–351. doi: 10.1006/mcbr.2000.0237. [DOI] [PubMed] [Google Scholar]
- 28.Ajiro K, Yoda K, Utsumi K, Nishikawa Y. Alteration of cell cycle-dependent histone phosphorylations by okadaic acid. Induction of mitosis-specific H3 phosphorylation and chromatin condensation in mammalian interphase cells. J. Biol. Chem. 1996;271:13197–13201. doi: 10.1074/jbc.271.22.13197. [DOI] [PubMed] [Google Scholar]
- 29.Porter PL, Sage EH, Lane TF, Funk SE, Gown AM. Distribution of SPARC in normal and neoplastic human tissue. J. Histochem. Cytochem. 1995;43:791–800. doi: 10.1177/43.8.7622842. [DOI] [PubMed] [Google Scholar]
- 30.Menon PM, Gutierrez JA, Rempel SA. A study of SPARC and vitronectin localization and expression in pediatric and adult gliomas: high SPARC secretion correlates with decreased migration on vitronectin. Int. J. Oncol. 2000;17:683–693. doi: 10.3892/ijo.17.4.683. [DOI] [PubMed] [Google Scholar]
- 31.Golembieski WA, Ge S, Nelson K, Mikkelsen T, Rempel SA. Increased SPARC expression promotes U87 glioblastoma invasion in vitro. Int. J. Dev. Neurosci. 1999;17:463–472. doi: 10.1016/s0736-5748(99)00009-x. [DOI] [PubMed] [Google Scholar]
- 32.Form DM, VanDeWater L, Dvorak HF, Smith BD. Pathogenesis of tumor desmoplasia. II. Collagens synthesized by Line 1 and Line 10 guinea pig carcinoma cells and by syngeneic fibroblasts in vitro. J. Natl. Cancer Inst. 1984;73:1207–1214. [PubMed] [Google Scholar]
- 33.Tuxhorn JA, et al. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002;8:2912–2923. [PubMed] [Google Scholar]
- 34.Sasaki T, Miosge N, Timpl R. Immunochemical and tissue analysis of protease generated neoepitopes of BM-40 (osteonectin, SPARC) which are correlated to a higher affinity binding to collagens. Matrix Biol. 1999;18:499–508. doi: 10.1016/s0945-053x(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 35.Welch MP, Odland GF, Clark RA. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J. Cell Biol. 1990;110:133–145. doi: 10.1083/jcb.110.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams DF. A model for biocompatibility and its evaluation. J. Biomed. Eng. 1989;11:185–191. doi: 10.1016/0141-5425(89)90138-6. [DOI] [PubMed] [Google Scholar]
- 37.Nakanishi H, Oguri K, Takenaga K, Hosoda S, Okayama M. Differential fibrotic stromal responses of host tissue to low- and high-metastatic cloned Lewis lung carcinoma cells. Lab. Invest. 1994;70:324–332. [PubMed] [Google Scholar]
- 38.Bradshaw, A.D., et al. 2003. SPARC null mice display phenotypic skin abnormalities characterized by reduced tensile strength and decreased dermal collagen fibril diameter. J. Invest. Dermatol. In press. [DOI] [PubMed]
- 39.Puolakkainen, P., et al. 2003. Compromised production of extracellular matrix in mice lacking SPARC leads to reduced foreign body reaction to implanted materials. Am. J. Pathol. In press. [DOI] [PMC free article] [PubMed]
- 40.Folkman J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 41.Hahnfeldt P, Panigrahy D, Folkman J, Hlatky L. Tumor development under angiogenic signaling: a dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999;59:4770–4775. [PubMed] [Google Scholar]
- 42.Iruela-Arispe ML, et al. Expression of SPARC during development of the chicken chorioallantoic membrane: evidence for regulated proteolysis in vivo. Mol. Biol. Cell. 1995;6:327–343. doi: 10.1091/mbc.6.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lane TF, Iruela-Arispe ML, Johnson RS, Sage EH. SPARC is a source of copper-binding peptides that stimulate angiogenesis. J. Cell Biol. 1994;125:929–943. doi: 10.1083/jcb.125.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 45.Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J. Leukoc. Biol. 1998;64:275–290. doi: 10.1002/jlb.64.3.275. [DOI] [PubMed] [Google Scholar]
- 46.Nowicki A, et al. Impaired tumor growth in colony-stimulating factor 1 (CSF-1)-deficient, macrophage-deficient op/op mouse: evidence for a role of CSF-1-dependent macrophages in formation of tumor stroma. Int. J. Cancer. 1996;65:112–119. doi: 10.1002/(SICI)1097-0215(19960103)65:1<112::AID-IJC19>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 47.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motamed K, Sage EH. SPARC inhibits endothelial cell adhesion but not proliferation through a tyrosine phosphorylation-dependent pathway. J. Cell. Biochem. 1998;70:543–552. doi: 10.1002/(sici)1097-4644(19980915)70:4<543::aid-jcb10>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 49.Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ, Jain RK. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997;15:778–783. doi: 10.1038/nbt0897-778. [DOI] [PubMed] [Google Scholar]
- 50.Lubkin SR, Jackson T. Multiphase mechanics of capsule formation in tumors. J. Biomech. Eng. 2002;124:237–243. doi: 10.1115/1.1427925. [DOI] [PubMed] [Google Scholar]
- 51.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 52.Monsky WL, et al. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin. Cancer Res. 2002;8:1008–1013. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}