

Abstract
Biogenesis of a superfamily of surface structures by gram-negative bacteria requires the chaperone/usher pathway, a terminal branch of the general secretory pathway. In this pathway a periplasmic chaperone works together with an outer membrane usher to direct substrate folding, assembly, and secretion to the cell surface. We analyzed the structure and function of the PapC usher required for P pilus biogenesis by uropathogenic Escherichia coli. Structural analysis indicated PapC folds as a β-barrel with short extracellular loops and extensive periplasmic domains. Several periplasmic regions were localized, including two domains containing conserved cysteine pairs. Functional analysis of deletion mutants revealed that the PapC C terminus was not required for insertion of the usher into the outer membrane or for proper folding. The usher C terminus was not necessary for interaction with chaperone-subunit complexes in vitro but was required for pilus biogenesis in vivo. Interestingly, coexpression of PapC C-terminal truncation mutants with the chromosomal fim gene cluster coding for type 1 pili allowed P pilus biogenesis in vivo. These studies suggest that chaperone-subunit complexes target an N-terminal domain of the usher and that subunit assembly into pili depends on a subsequent function provided by the usher C terminus.
Gram-negative bacteria have evolved a number of pathways for extracellular protein secretion (39). Proteins targeted for secretion in gram-negative bacteria must cross the periplasm and outer membrane (OM) in addition to the cytoplasmic or inner membrane (IM). One of the best-understood gram-negative bacterial secretion pathways is the chaperone/usher pathway responsible for biogenesis of a superfamily of surface structures associated with pathogenesis (40). The prototype members of the chaperone/usher pathway are the pap and fim gene clusters of uropathogenic Escherichia coli that code for P and type 1 pili (fimbriae), respectively. P pili consist of six structural proteins which interact to form a fiber composed of two distinct subassemblies: a 6.8-nm-thick helical rod comprised mainly of PapA and a 2-nm-diameter linear tip fibrillum comprised mainly of PapE (7, 20). The PapG adhesin is located at the distal end of the tip fibrillum and binds to Galα(1-4)Gal moieties present in kidney glycolipids (6, 11). PapD and PapC are the chaperone and usher for P pili, respectively (10, 21, 26). Type 1 pili have a short tip fibrillum, containing the FimH adhesin (23), joined to the distal end of the FimA pilus rod (17). The FimH adhesin binds to mannosylated glycoproteins present in the bladder epithelium (1). FimC is the type 1 pilus chaperone, and FimD is the OM usher (16, 18).
The chaperone/usher pathway is a terminal branch of the general secretory pathway (27, 37). Following translocation across the IM via the Sec system, pilus subunits must interact with the periplasmic chaperone. The periplasmic chaperone consists of two immunoglobulin-like (Ig) domains (13) and has three main functions: it facilitates the folding of pilus subunits, caps their interactive surfaces, and maintains the subunits in stable conformations. The three functions of the chaperone are all part of the same process, as revealed by crystal structures of chaperone-subunit complexes (8, 29). Pilus subunits are comprised of a single Ig domain, except that they are missing the seventh β-strand present in canonical Ig folds. The absence of this strand produces a deep groove on the surface of the folded subunit, exposing the subunit's hydrophobic core. The chaperone functions by donating its G1 β-strand to fill this groove, facilitating subunit folding in a mechanism termed donor strand complementation (4, 8, 29). The chaperone remains bound to the folded subunit, thus stabilizing it. The subunit groove also comprises an interactive surface involved in subunit-subunit interactions. Thus, donor strand complementation couples the folding of the subunit with the simultaneous capping of its interactive surface.
Periplasmic chaperone-subunit complexes must next target the OM usher. In the absence of the usher, complexes accumulate in the periplasm, but no pili are assembled or secreted (18, 26, 42). Pilus assembly is thought to occur at the periplasmic face of the usher, concomitant with secretion of the pilus fiber through the usher to the cell surface (40). This process appears to be self-energized and does not require the transduction of energy from the IM (14). Pilus subunits have a highly conserved N-terminal extension that is exposed in the chaperone-subunit complex. At the usher, the G1 β-strand of the chaperone is thought to be exchanged for the N-terminal extension of an incoming subunit in a process termed donor strand exchange, which couples chaperone dissociation with pilus assembly (4). Pili are built from the top down; the adhesin is incorporated first, followed by assembly of the tip fibrillum and finally the rod. The usher facilitates this organization by differentially recognizing chaperone-subunit complexes according to their final position in the pilus (10, 31). Thus, chaperone-adhesin complexes from both P and type 1 pili bind with highest affinity to their respective ushers. The P pilus usher PapC and the type 1 pilus usher FimD assemble into ring-shaped oligomeric complexes containing central pores 2 to 3 nm in diameter (30, 41). This is large enough to allow secretion of folded pilus subunits.
We analyzed the structure and function of the P pilus usher PapC to gain insights into the molecular mechanisms governing pilus biogenesis across the OM. The usher is thought to fold as a β-barrel in the OM, and circular dichroism (CD) spectroscopy confirmed a largely β-sheet secondary structure for PapC. Topology modeling predicted that PapC contains 24 transmembrane (TM) β-strands, with extensive regions exposed to the periplasm. We were able to experimentally localize N- and C-terminal periplasmic regions of PapC, including two domains containing conserved cysteine pairs. Functional analysis of the usher was carried out using C-terminal truncation mutants of PapC. The results suggest a model in which the usher possesses distinct N- and C-terminal domains for chaperone-subunit targeting and subunit assembly into pili, respectively.
MATERIALS AND METHODS
Strains and plasmids.
The E. coli strains and plasmids used in this study are described in Table 1. All strains were grown in Luria-Bertani broth containing appropriate antibiotics at 37°C with aeration (shaking broth), unless indicated otherwise. Plasmid genes under the control of Para were induced at an optical density at 600 nm of 0.6 for 1 h by addition of 0.1% l-arabinose. Plasmid genes under the control of Ptac or Ptrc were induced by addition of 10 μM isopropyl-β-d-thiogalactoside (IPTG). Strain MM294 was purchased from the American Type Culture Collection (Manassas, Va.). C-terminal deletion mutants of PapC were created using a procedure based on that in reference 3. Briefly, plasmid pFJ20 (14) was treated with DNase in the presence of ethidium bromide to randomly nick the plasmid circle. Subsequent treatment with ExoIII, Bal 31, and Klenow created a linearized blunt DNA. This was then cut with HindIII, which cuts 3′ of the papC gene. The DNA was then ligated with a DNA fragment containing a kanamycin resistance cassette with a blunt BamHI site on one end and a cohesive HindIII site on the other. Transformants were screened for PapC expression by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of whole-cell lysates and immunoblot analysis. Plasmids containing genes for stable PapC truncation mutants were sequenced to verify the site of truncation. To aid purification, hexahistidine tags were appended to the truncated papC genes by digesting with KpnI and HindIII to remove the kan cassette and ligating with a DNA fragment made by annealing oligonucleotides encoding six histidines in the proper reading frame for the particular truncation mutant.
TABLE 1.
E. coli strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)c | Residues added with His tag | Reference |
|---|---|---|---|
| Strainsa | |||
| W3110II5 | Derivative of W3110 [F−mcrA mcrB IN(rrnD-rrnE)1 λ−] | 14 | |
| SF100 | ΔOmpT [F− Δ(ompT−entF) Δ(lacIPOZY)X74 galE galK thi rpsL ΔphoA] | 2 | |
| MM294 | fim+ (F−endA1 thi-1 hsdR17 supE44 λ−) | 5 | |
| AAEC185 | Δfim (MM294 Δfim recA) | 5 | |
| Plasmids | |||
| pPAP5 | pap gene cluster in pBR322, Ampr | 26 | |
| pMJ2 | ΔpapC pap operon under Ptrc in pACYC185, Tetr | 41 | |
| pMON6235Δcat | pBR322-based vector with Para, Ampr | 15 | |
| pMJ3 | PapC(809)-Hisb | (H)6 | 41 |
| pPAPC747Δ | PapC(Δ748-809)-His | RSPGT(H)6 | This study |
| pPAPC640Δ | PapC(Δ641-809)-His | GSPGT(H)6 | This study |
| pPAPC596Δ | PapC(Δ597-809)-His | RSPGY(H)6 | This study |
| pPAPC562Δ | PapC(Δ563-809)-His | IPGY(H)6 | This study |
| pPAPC450Δ | PapC(Δ451-809)-His | DPRVP(H)6 | This study |
| pPAPC366Δ | PapC(Δ367-809)-His | IPGY(H)6 | This study |
| pPAPC292Δ | PapC(Δ293-809)-His | YPGY(H)6 | This study |
All strains are K-12.
The papC genes each have a C-terminal His tag and are under the control of Para in pMON6235Δcat.
Abbreviations: Ampr, ampicillin resistance; Tetr, tetracycline resistance; Para, arabinose-inducible promoter; Ptrc, IPTG-inducible promoter.
OM isolation and PapC purification.
OM was isolated by Sarkosyl extraction (Sigma, St. Louis, Mo.) (25). Following growth and induction as described above, bacteria were chilled on ice, washed with 20 mM Tris-HCl (pH 8), and resuspended to a volume of 3 ml per 100 ml of starting culture in the same buffer containing complete protease inhibitor cocktail (Roche, Indianapolis, Ind.). Bacteria were broken by one passage through a French pressure cell (SLM Instruments, Rochester, N.Y.) at 14,000 lb/in2. Unbroken cells were removed by centrifugation (15 min, 3,000 × g, 4°C). Sarkosyl was added to the supernatant fraction to a final concentration 0.5%, and the mixture was rocked for 5 min at 25°C. OM was harvested by centrifugation (1 h, 100,000 × g, 4°C) and resuspended in 20 mM HEPES (pH 7.5)-0.3 M NaCl. Purification of His-tagged PapC from isolated OM was carried out as described previously (41), except PapC was solubilized by rocking for 1 h at 25°C in 20 mM HEPES (pH 7.5)-0.3 M NaCl-0.1% n-dodecyl-β-d-maltoside (Anatrace, Maumee, Ohio) and purified using a Pharmacia fast-performance liquid chromatography apparatus (Amersham Pharmacia Biotech, Piscataway, N.J.).
CD.
CD spectra were obtained with an Olis RSM CD spectrophotometer at room temperature using a 1-mm-path-length cell. PapC purified as described above was dialyzed into 5 mM K phosphate (pH 7.6)-150 mM NaCl-0.05% n-dodecyl-β-d-maltoside and used at a final concentration of 0.2 mg/ml. Eighty scans were taken at a rate of 1 s per datum, with the contribution of the buffer-detergent mixture alone subtracted. Estimation of secondary structure from the CD spectrum was performed with the DICROPROT software package (9).
Topology modeling.
Putative TM β-strands of PapC were identified by the method developed by Schirmer and Cowan (33). The secondary structure of PapC was analyzed by the PHD program (28), available by e-mail (predictprotein@embl-heidelberg.de) from the EMBL database server.
Epitope mapping.
Polycolonal anti-PapC antibody was generated by Covance Research Products (Richmond, Calif.), by injecting New Zealand White rabbits with His-tagged PapC purified as described above. The anti-PapC98-111 polyclonal antibody, raised against the PapC peptide ADFHGLPGVDIRPD, was kindly provided by MedImmune (Gaithersburg, Md.). Enzyme-linked immunosorbent assays (ELISAs) of whole bacteria (SF100/pMON6235Δcat or SF100/pMJ3) and OM (prepared from W3110II5/pMON6235Δcat or W3110II5/pMJ3) were carried out as described previously (22).
Fluorescence-activated cell sorter (FACS) analysis of whole bacteria (W3110II5/pMJ3) was done as follows. Cultures were harvested at mid-exponential phase, washed, and resuspended in phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA). The cells were blocked by incubation in PBS-BSA for 1 h at 4°C. Primary antibodies were added to the blocked cells (anti-PapC98-111 at a 1:25 dilution, anti-OmpA at a 1:3,000 dilution, anti-β-lactamase at a 1:2,000 dilution), and the samples were incubated for 2 h at 4°C. Cells were washed once with PBS, resuspended in PBS-BSA solution, and reacted with fluorescein isothiocyanate-conjugated secondary antibodies at a 1:50 dilution for 1 h at 4°C. The cells were washed twice, resuspended in PBS, and scanned by a FACSort fluorescence-activated cell sorter (Becton Dickinson, San Jose, Calif.).
Proteolysis.
Protease digests of whole bacteria were done using a 50-μg/ml concentration of trypsin, chymotrypsin, or proteinase K (Sigma). Following growth and induction as described above, whole bacteria were washed and resuspended to one-fourth of the original culture volume in 20 mM HEPES (pH 7.5). Aliquots of 50 to 100 μl were incubated with protease for 5 min at 4°C or 1 h at 37°C. Digestion was terminated by addition of Pefabloc SC (0.5 mg/ml; Roche, Indianapolis, Ind.) and treatment at 95°C in SDS sample buffer. Samples were subjected to SDS-PAGE and analyzed by Coomassie blue staining or immunoblotting with anti-PapC antibody. For OM digests, approximately 0.1 mg of isolated OM (as determined by bicinchoninic assay; Pierce, Rockford, Ill.) was treated as above. N-terminal sequencing was performed by Midwest Analytical (St. Louis, Mo.).
Carboxypeptidase A (CPA) digestion of PapC and the PapC truncation mutants for localization of the His tag was performed as follows. Following growth and induction, bacteria were washed and resuspended to 1/30 of the original culture volume in PBS. Aliquots of 10 ml were treated with or without CPA (250 μg/ml; Sigma) for 1 h at 25°C. Cysteine was added to 1 mM to inhibit CPA, and the bacteria were lysed by sonication (5 cycles of 30 s on and 30 s off on ice). OMs were isolated by Sarkosyl extraction as described above, resuspended in 10 ml of PBS, and treated with or without CPA (250 μg/ml) for 1 h at 25°C. Cysteine was added to 1 mM, and the membranes were solubilized for nickel-affinity chromatography as described previously (41).
Analysis of PapC deletion constructs.
To determine expression of the various PapC C-terminal truncation mutants in whole bacteria, strain SF100 harboring pMJ3 or one of the PapC truncation mutants was grown and induced as described above. Equal amounts of whole bacteria were lysed in SDS-PAGE sample buffer (95°C, 5 min), subjected to SDS-PAGE, and transferred to nitrocellulose membrane for immunoblotting. The membrane was probed with anti-PapC polyclonal antibody, followed by a horseradish peroxidase-conjugated secondary antibody. The blot was developed using SuperSignal West Dura chemiluminescent substrate (Pierce) and imaged on a Fluor-S MultiImager System (Bio-Rad, Hercules, Calif.). To determine targeting and folding of the various PapC C-terminal truncation mutants, OM was isolated from SF100 harboring pMJ3 or one of the PapC truncation mutants as described above. The OM samples were treated in SDS-PAGE sample buffer at 25 or 95°C for 5 min and then analyzed by immunoblotting as for whole bacteria.
Binding of PapDG to the PapC C-terminal truncation mutants was performed by an overlay assay (10). Equal amounts of OM isolated as described above from SF100 harboring pMON6235Δcat, pMJ3, or one of the PapC truncation mutants were treated in SDS sample buffer (95°C, 5 min), subjected to SDS-PAGE, and transferred to polyvinylidene difluoride membrane. The membrane was blocked overnight with 2% BSA in buffer A (10 mM Tris [pH 8.2]-0.5 M NaCl-0.5% Tween 20) and then incubated for 1 h with purified PapDG (2 μg/lane) or PapD only (0.9 μg/lane) in buffer A-BSA. The membrane was washed with buffer A and then immunoblotted with a polyclonal anti-PapDK antibody, followed by an alkaline phosphatase-conjugated secondary antibody and visualization with BCIP (5-bromo-4-chloro-3-indolyl phosphate)-nitro blue tetrazolium substrate. The C-terminal His tag appended to the PapC constructs is not required for binding of PapDG to PapC (10) and does not allow nonspecific binding of PapDG to PapC (data not shown).
Analysis of pilus biogenesis.
Hemagglutination assays were performed by serial dilution in microtiter plates as described previously (36). P pili were tested for agglutination of human red blood cells (donated by laboratory volunteers), and type 1 pili were tested for agglutination of guinea pig red blood cells (Colorado Serum Company, Denver, Colo.). Bacteria were grown and induced as described above or grown in static Luria-Bertani broth at 37°C for 48 h to induce type 1 pili. Specificity of type 1 pilus agglutination was demonstrated by inhibition with 0.5% d-mannose (Sigma). Purification of P pili from the bacterial surface was done as follows. For strains grown with aeration (shaking broth), 100-ml cultures were induced for 1 h with 0.1% arabinose and 10 μM IPTG, washed, and resuspended into 1 ml of 5 mM Tris (pH 8.0)-75 mM NaCl. Samples were heated to 65°C for 30 min to release P pili and then let cool to 25°C. Samples were centrifuged (6,000 × g, 5 min, 4°C), and the supernatant fractions were transferred to fresh tubes and centrifuged again (16,000 × g, 20 min, 4°C). The supernatant fractions were transferred to fresh tubes, and pili were precipitated by sequential addition of 0.25 ml of 1 M NaCl and 0.35 ml of 0.5 M MgCl2. The tubes were rocked for 2 h at 25°C, and pili were collected by centrifugation (16,000 × g, 30 min, 4°C). The pellets were resuspended in 0.1 ml of 1 mM Tris (pH 8.0) by rocking overnight at 4°C. Finally, any insoluble material was removed by centrifugation (16,000 × g, 30 min, 4°C). For strains grown in static broth, 200-ml cultures were grown for 48 h and then induced for 2 h with 0.1% arabinose and 10 μM IPTG. Cultures were washed and resuspended into 5 mM Tris (pH 8.0)-75 mM NaCl as above, except the resuspension volume was adjusted according to the wet weight of the cell pellets to give equal concentrations of bacteria. Pili were then harvested from 1 ml of the resuspended cultures as described for the shaking broth cultures. Purified pili were incubated (95°C, 5 min) with SDS sample buffer containing 4 M urea to dissociate the pili. The samples were then subjected to SDS-PAGE and visualized by Coomassie blue staining for PapA detection or by immunoblotting with anti-P pilus tip antisera for detection of PapG and PapE.
DNA sequencing.
Both strands of papC were sequenced using primers binding ∼100 nucleotides upstream (primer, 5′-AGAGATGTATACCGTTACG-3′) and downstream (primer, 5′-ATTACTCATACTGCCACTG-3′) of the putative cysteine residue at position 512 of the mature PapC amino acid sequence. The papC gene was sequenced from plasmid pMJ3 as well as from pPAP5. DNA was prepared using the Qiagen (Valencia, Calif.) plasmid midi kit. Sequencing reactions were prepared using the ABI Prism BigDye Terminator cycle sequencing ready reaction kit (PE Biosystems, Foster City, Calif.) and analyzed by the Washington University Nucleic Acid Facility.
Nucleotide sequence accession number.
The corrected papC sequence has been deposited in GenBank under accession number AF481883.
RESULTS
Topology modeling and analysis of the PapC usher.
The ushers are large, integral OM proteins generally 80 to 90 kDa in size. Mature PapC contains 809 amino acids and has a molecular mass of 88.2 kDa. Bacterial OM proteins typically span the membrane via a series of TM β-strands arranged to form a β-barrel (35). OM β-barrel proteins are often stable to SDS unless incubated at high temperatures and thus exhibit a characteristic heat-modifiable mobility on SDS-PAGE (34, 38). Both the PapC and FimD ushers exhibit this heat-modifiable mobility, suggesting they fold as β-barrels (41). We modeled the topology of PapC using an algorithm designed specifically for β-sheet OM proteins (33). Computer analysis predicted that PapC contains 24 TM β-strands, with the strands spread fairly evenly throughout the protein sequence (Fig. 1). To provide support for this prediction, the secondary structure of PapC was experimentally investigated using CD analysis. The spectrum obtained (Fig. 2) is characteristic of a largely β-sheet protein, with a minimum at 214 nm. The spectrum lacks a minimum at 209 nm, indicating an absence of significant α-helical structure. Analysis of the CD spectrum for secondary structure using the DICROPROT software package (9) indicated PapC contains approximately 10% α-helix, 40% β-sheet, 20% turns, and 30% other structure. This analysis is compatible with the 24 TM β-strands predicted by topology modeling, which represent 36% of the mature PapC sequence.
FIG. 1.

Sequence of mature PapC. Boldface lettering indicates TM β-strands predicted by computer modeling. Italic lettering indicates predicted loop regions experimentally localized to the periplasm. The conserved cysteine pairs are labeled with a star, the periplasmic epitope recognized by the αPapC98-111 antibody is underlined, and the periplasmic trypsin cleavage site is indicated by an inverted open triangle. The positions of the His-tagged C-terminal truncations are indicated, as well as the His tag appended to full-length PapC (both shown by inverted filled triangles). The residues boxed in gray (490 to 516) represent the corrected PapC sequence.
FIG. 2.

CD spectrum of PapC. Purified PapC (0.2 mg/ml) was measured in 5 mM K phosphate (pH 7.6)-150 mM NaCl-0.05% dodecyl maltoside. The average of 80 buffer-corrected scans is shown in values of mean residue ellipticity (degrees · square centimeter · decimoles−1).
Members of the usher superfamily contain conserved N- and C-terminal cysteine pairs (Fig. 1). These cysteine pairs form intramolecular disulfide bonds that appear to stabilize usher domains (S. Shu Kin So and D. G. Thanasssi, unpublished data). The published sequence of PapC (26) is unique among the ushers in that it contains a single additional cysteine at position 512 of the mature sequence. To verify the presence of this extra cysteine, which could influence disulfide bond formation and thus the structure of the usher, we resequenced this region of PapC. Sequencing in fact revealed an error encompassing mature amino acids 490 to 516 (Fig. 1). The corrected sequence of this region contains no cysteines (Fig. 1) (GenBank accession number AF481883). Therefore, in agreement with other members of the usher superfamily, PapC contains only the conserved N- and C-terminal cysteine pairs.
Topology modeling predicted that PapC contains relatively short surface-exposed loops, with larger regions extending into the periplasm. This is unusual for a bacterial OM protein (35) but consistent with findings for other members of the usher superfamily (12, 32). Incubation of intact bacteria with trypsin or chymotrypsin produced little cleavage of PapC. In contrast, PapC was readily degraded in isolated OM, confirming exposure of larger domains to the periplasm (data not shown). PapC has some surface-exposed regions, as proteinase K was able to degrade the usher in whole bacteria. In addition, polyclonal antibody raised against full-length PapC was able to bind intact bacteria expressing the usher (Fig. 3A). We were particularly interested in defining periplasmic regions of the usher, as these are likely to function in essential interactions with chaperone-subunit complexes. The PapC topology model predicts a large loop from residue 53 to 109 (Fig. 1). This loop is of interest because it contains the conserved N-terminal cysteine pair. The polyclonal antibody anti-PapC98-111, raised against a peptide in this region (Fig. 1), was used to experimentally localize this predicted loop. Isolated OM containing PapC reacted with the anti-PapC98-111 antibody, indicating exposure of the epitope to solution, consistent with it residing in a loop region (Fig. 3B). In contrast, the anti-PapC98-111 antibody bound only weakly or not at all to whole bacteria expressing PapC, whereas a control antibody recognizing surface epitopes of the OmpA protein reacted strongly (Fig. 3C). Therefore, the loop from residue 53 to 109 is exposed to the periplasm.
FIG. 3.

Epitope mapping of PapC. (A) ELISA performed with polyclonal anti-PapC antibody versus BSA as a background control, or whole bacteria expressing either vector alone (pMON6235Δcat) or PapC (pMJ3). Error bars, standard deviations. (B) ELISA performed with the epitope-specific antibody anti-PapC98-111 versus BSA, or OM isolated from bacteria expressing either vector alone or PapC. Error bars, standard deviations. (C) FACS analysis of whole bacteria. Bacteria were unstained (no reaction with primary antibody), or stained with either anti-OmpA (αOmpA) as a control for surface-exposed epitopes, anti-β-lactamase (αBla) as a control for periplasmic epitopes, or the anti-PapC98-111 antibody (αPapC98-111). All samples were reacted with fluorescein isothiocyanate-conjugated secondary antibodies. Each panel indicates the percent of the input bacteria with fluorescence intensities greater than the threshold indicated by the left end of the horizontal bar.
Limited trypsin digestion of OM containing PapC enabled two cleavage products to be excised for N-terminal sequencing. One fragment, running at 66 kDa, contained the intact mature N terminus (sequence, VEFNTD). The second fragment, running at 20 kDa, contained the sequence LLVDTD, indicating cleavage between residues Arg652 and Leu653 (Fig. 1). This likely represents the C-terminal portion of the 66-kDa fragment. This cleavage site resides in a large C-terminal loop predicted by the topology model (Fig. 1). The 20-kDa fragment was not visible when intact bacteria were treated with trypsin, indicating a periplasmic location for the loop from residue 638 to 674 (data not shown). Limited digestion of OM with chymotrypsin allowed sequencing of a 65-kDa band, which also contained an intact N terminus. Thus, the C-terminal region of PapC is preferentially cleaved during limited proteolysis.
The PapC C terminus was localized using a C-terminal hexahistidine tag (His tag) as a marker. Intact bacteria or isolated OM was incubated with CPA, which cleaves proteins from the C terminus, and the samples were processed for nickel-affinity chromatography. Loss of the His tag and subsequent loss of binding to the nickel column only occurred when isolated OM was incubated with CPA (Fig. 4). Thus, the usher C terminus, which contains the C-terminal conserved cysteine pair (Fig. 1), resides in the periplasm. The same CPA approach was used to map the termini of three His-tagged PapC C-terminal truncation mutants: PapC747Δ, PapC640Δ, and PapC596Δ (Fig. 1 and see below). For each of the PapC truncation mutants, loss of the His tag and subsequent loss of binding to the nickel column only occurred when isolated OM was incubated with CPA (Fig. 4). These results localize PapC residues 747, 640 and 596 to the periplasm. Residue 640 resides in the same large loop identified by the trypsin mapping experiments described above, thus confirming a periplasmic location for this loop.
FIG. 4.

Localization of the C termini of full-length PapC and PapC C-terminal truncation mutants. No protease was added (−), or CPA was added to whole bacteria (WB) or to isolated OM expressing full-length His-tagged PapC (PapC809) or the indicated His-tagged PapC truncation mutant. The samples were then processed for nickel-affinity chromatography. Proteins were eluted with imidazole, subjected to SDS-PAGE, and visualized by Coomassie blue staining. Note that the different amounts of protein recovered from the nickel column reflect differences in truncation mutant stability.
Functional analysis of PapC.
A set of seven C-terminal PapC truncation mutants (each modified with a C-terminal His tag) was used to probe the function of the usher C terminus (Fig. 1). Analysis of expression in whole bacteria indicated that each of the truncation mutants, except the two largest deletion mutants, PapC366Δ and PapC292Δ, was stably expressed (data not shown). The PapC366Δ and PapC292Δ truncation mutants were not analyzed further. Each of the remaining PapC truncation mutants was able to localize to the OM as judged by immunoblots of OM preparations (Fig. 5). This indicates that the C terminus is not required for targeting of the usher to the OM. The expression level of the truncation mutants varied, and degradation products were apparent for each of the truncation mutants, as well as for full-length PapC. This may partly be due to overexpression of the ushers from plasmids. Note also that the blots shown in Fig. 5 were overexposed to better visualize the more weakly expressed truncation mutants. To analyze folding of the PapC truncation mutants in the OM, we exploited the heat-modifiable mobility of the usher on SDS-PAGE. Full-length PapC, PapC747Δ, and PapC640Δ each shifted to faster-migrating species when treated at 25°C in sample buffer (Fig. 5B). Therefore, the PapC747Δ and PapC640Δ truncation mutants are able to adopt the wild-type SDS-resistant fold (41). Truncation mutants larger than PapC640Δ did not undergo this mobility shift, indicating a loss of SDS resistance (Fig. 5B). In addition, full-length PapC, PapC747Δ, and PapC640Δ each produced prominent high-molecular-weight bands when treated at 25°C (Fig. 5B), indicative of the native oligomeric form of the usher (41). The amount of this high-molecular-weight species decreased significantly in truncation mutants larger than PapC640Δ. Thus, the C-terminal 169 residues of PapC, including the conserved C-terminal cysteine pair, do not appear to be required for stable protein folding or for usher oligomerization.
FIG. 5.
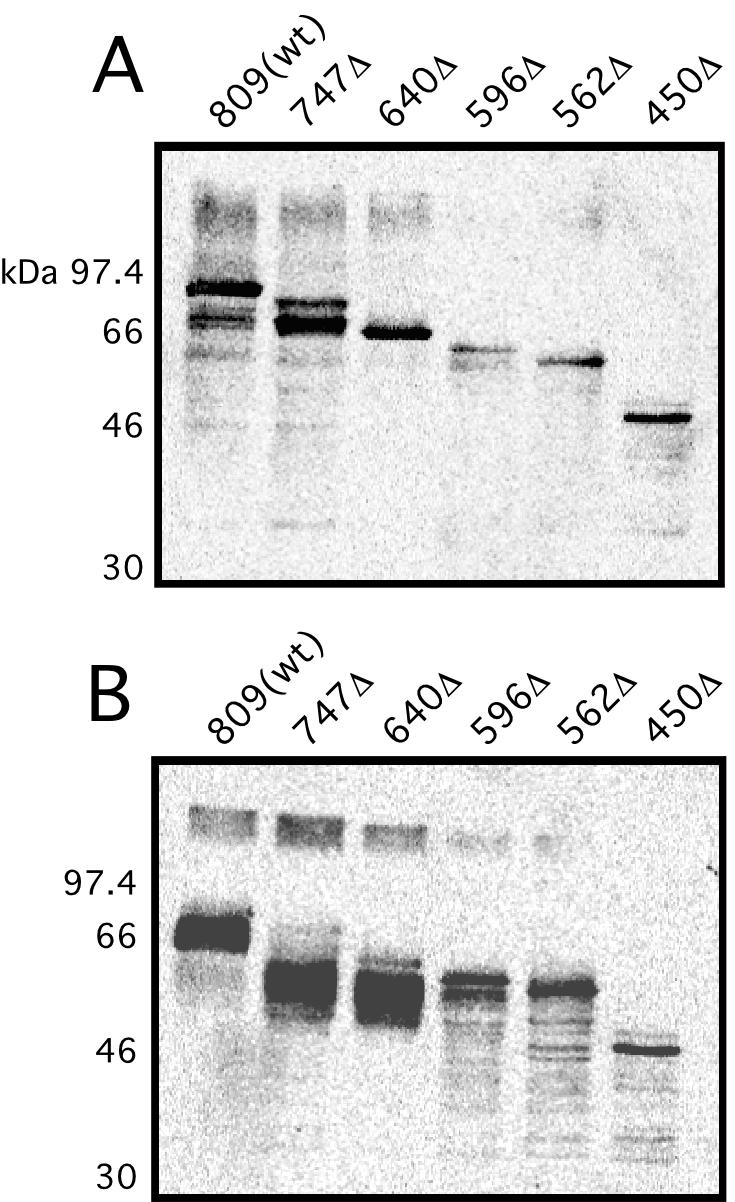
Targeting and folding of the PapC C-terminal truncation mutants in the OM. OM was isolated from bacteria expressing full-length PapC [809(wt)] or the indicated truncation mutant and incubated in SDS sample buffer at 95°C (A) or 25°C (B). The proteins were detected by immunoblotting with anti-PapC antibody and visualized by chemiluminescence.
The PapC C-terminal truncation mutants bind chaperone-adhesin complexes in vitro but are unable to assemble pili in vivo.
An important measure of usher function is the ability to bind chaperone-subunit complexes. Chaperone-adhesin complexes target with highest affinity to the usher (10, 31), and stable chaperone-adhesin-usher complexes appear to initiate pilus biogenesis in vivo (31). Purified PapDG chaperone-adhesin complexes were tested for binding to the PapC C-terminal truncation mutants using an overlay assay (10). Surprisingly, PapDG bound to each of the PapC truncation mutants (Fig. 6), indicating that the C-terminal half of the usher is not required for this crucial initial step of pilus biogenesis.
FIG. 6.

Overlay assay for chaperone-adhesin binding to the PapC C-terminal truncation mutants. OM was isolated from bacteria expressing either full-length PapC [809(wt)], the indicated PapC truncation mutant, or vector only (pMON6235Δcat). The OM was incubated in sample buffer at 95°C, separated by SDS-PAGE, and transferred to polyvinylidene difluoride membrane. The membrane was incubated with purified PapDG chaperone-adhesin complexes (A), or PapD only as a negative control (B). PapD alone does not bind to the usher. Binding of PapDG or PapD was determined by immunoblotting with anti-PapD antibody and visualized by alkaline phosphatase development. The bands present in panel B and the vector-alone lane in panel A represent background binding to OM proteins.
The in vivo functionality of the PapC C-terminal truncation mutants was tested by assaying their ability to complement a ΔpapC pap operon (plasmid pMJ2) to assemble adhesive pili on the surface of E. coli strain AAEC185. Pilus biogenesis was monitored using hemagglutination assays (HA). The His-tagged full-length PapC (PapC809) was fully functional for pilus biogenesis, as previously demonstrated (41), giving an HA titer of 256. The HA titer is the highest dilution of bacteria able to agglutinate human red blood cells. In contrast, and despite their ability to bind chaperone-adhesin complexes in vitro, none of the PapC truncation mutants was able to produce an HA-positive phenotype in vivo, giving HA titers of zero. To confirm that the PapC truncation mutants were defective for pilus biogenesis, pili were purified from the bacterial surface by heat extraction and magnesium precipitation. No pilus fibers were recovered from AAEC185/pMJ2 bacteria complemented with the PapC truncation mutants or vector only, whereas pili were readily recovered from bacteria complemented with full-length PapC (Fig. 7A). Thus, the usher C terminus appears to be required for an assembly step subsequent to the initial targeting of chaperone-subunit complexes to the usher.
FIG. 7.

Pilus biogenesis by the PapC C-terminal truncation mutants. (A and B) AAEC185 (Δfim)/pMJ2 (ΔpapC pap operon) was transformed with vector alone (pMON6235Δcat), full-length PapC (pMJ3) [809(wt)], or the indicated PapC truncation mutant. Bacteria were grown at 37°C in shaking broth (A) or static broth (B). P pili assembled on the cell surface were harvested by heat extraction and magnesium precipitation, followed by separation on SDS-PAGE. The PapA major rod subunit was visualized by Coomassie blue staining (upper panel in each figure). The PapG adhesin and PapE major tip subunit were visualized by immunoblotting with anti-P pilus tip antibody (lower panel in each figure). The positions of the PapA, PapG, and PapE subunits are indicated. (C and D) P Pili were purified from strain MM294 (fim+)/pMJ2 transformed with the indicated PapC construct as described above. Bacteria were grown in shaking broth (C) or static broth (D). Growth in shaking or static broth represses or induces type 1 pilus expression, respectively.
Coexpression of the PapC C-terminal truncation mutants with type 1 pili complements a papC deletion for P pilus assembly.
PapDG chaperone-adhesin complexes do not bind to the type 1 pilus usher FimD (31), and FimD is unable to substitute for PapC in the assembly of P pili (L. Anderson and D. G. Thanassi, unpublished data). FimD presumably lacks the specific site(s) needed to target P pilus subunits. The results presented above suggest that the chaperone-subunit targeting site resides in an N-terminal domain of PapC. We therefore investigated whether coexpression of N-terminal domains of PapC with the fim (type 1) gene cluster would allow targeting of Pap chaperone-subunit complexes to FimD for assembly into P pili. Strain AAEC185 used for the pilus assembly experiments described above has a deletion of the chromosomal fim genes and is thus unable to express type 1 pili (5). Therefore, the isogenic fim+ parent of AAEC185, strain MM294, was transformed with plasmid pMJ2 (ΔpapC pap operon) and the various PapC C-terminal deletion constructs. Expression of the chromosomal fim genes is repressed by growth in shaking broth (aeration) and induced by growth in static broth. Growth of the MM294 strains in shaking or static broth resulted in the absence or presence of type 1 pili, respectively, as judged by mannose-sensitive HA of guinea pig red blood cells (data not shown). Expression of P pilus genes did not affect type 1 pilus production by the MM294 strains (data not shown).
Strain MM294/pMJ2 complemented with vector alone, full-length PapC, or one of the PapC truncation mutants was grown in static broth to induce the fim genes, and the pap genes were induced from the plasmids. P pili assembled on the cell surface were purified from the bacteria by heat extraction and magnesium precipitation. This method allows selective purification of P pili, as little or no type 1 pili are released from the bacteria by heat extraction under the conditions used. No P pili were assembled in the strain complemented with vector only (Fig. 7D, first lane), indicating that the fim gene cluster by itself is unable to substitute for PapC in P pilus biogenesis. The strain complemented with full-length PapC assembled P pili as expected (Fig. 7D, second lane). Interestingly, each of the strains complemented with a PapC truncation mutant was also able to assemble P pili (Fig. 7D). P pilus biogenesis by the PapC truncation mutants was less efficient than that for full-length PapC, and the amount of pili assembled was inversely proportionally to the size of the truncation. The relative mobilities of the pilus subunits on the gel, the absence of pili in the vector complemented control, and the reactivity with anti-P pilus antiserum confirms that P pili were isolated from these strains. This result suggests that the PapC truncation mutants were able to interact with FimD, and possibly other Fim components, to target Pap chaperone-subunit complexes to the FimD usher for assembly into pili, presumably facilitated by the C-terminal domain of FimD. Expression of the fim genes was required, because no P pili were assembled by the PapC truncation mutants when the MM294 strains were grown in shaking broth (Fig. 7C). In addition, P pili were never found to be assembled by the PapC truncation mutants under either growth condition in the AAEC185 (Δfim) strains (Fig. 7A and B).
DISCUSSION
We present here an analysis of the structure and function of the PapC usher required for P pilus biogenesis in uropathogenic E. coli. The chaperone/usher pathway provides a model system for probing the molecular mechanisms governing virulence factor biogenesis and protein secretion across the bacterial OM. We modeled PapC as a β-barrel protein, predicting 24 potential TM β-strands. This is consistent with modeling done on other members of the usher superfamily. The FasD usher required for biogenesis of 987P pili in enterotoxigenic E. coli was predicted to have 28 TM β-strands (32). The FaeD usher required for biogenesis of K88 pili in enterotoxigenic E. coli was predicted to have up to 32 TM β-strands, although experimental evidence supported a model with 22 strands (12, 42). The CD analysis of PapC in the present study supports these modeling results. To our knowledge, this is the first such confirmation of the secondary structure of the ushers. An interesting feature revealed in this and in previous studies is that the β-barrel fold of the ushers appears to contain only short extracellular loops, with larger loops exposed to the periplasm (12, 32). This is in contrast to canonical OM proteins such as the porins, which have larger surface-exposed loops and only short periplasmic turns (35). This difference is likely a reflection of the function of OM export proteins versus diffusion channels such as the porins. Thus, periplasmic regions of the usher are presumably required for interactions with chaperone-subunit complexes, leading to pilus assembly and secretion. Side views of FimD obtained by electron microscopy show a scaffold-like projection at one end of the usher complex (30). This projection may represent the usher's periplasmic domains.
Topology predictions of OM proteins are prone to error and must be verified experimentally. Therefore, we do not present here a model for the arrangement of PapC in the membrane but focus instead on experimental evidence localizing particular domains of the usher. We were particularly interested in identifying periplasmic regions of the usher, as these are likely to function in essential interactions with chaperone-subunit complexes. Mapping studies localized five regions of PapC to the periplasm (Fig. 1). Four of these are predicted to be extensive regions, ranging from 28 to 81 residues, and two of these domains were found to contain the conserved cysteine pairs. Modeling and experimental studies of FaeD also localized both of its cysteine pairs to the periplasm (12). The cysteine pairs form intramolecular disulfide bonds that appear to stabilize usher domains for proper interaction with chaperone-subunit complexes (S. Shu Kin So and D. G. Thanassi, unpublished results).
Only limited analysis had been done to define functional domains of the usher. We constructed C-terminal truncation mutants of PapC to analyze the role of N- and C-terminal regions of the usher. PapC truncation mutants containing deletions of up to 359 residues were still able to target and insert into the OM, although the stability of the deletions varied. Studies of alkaline phosphatase fusions of FasD and FaeD also found that the C terminus was not required for targeting to the OM (32, 42). PapC truncation mutants containing deletions of up to 169 residues were still able to fold into SDS-resistant structures and oligomerize, as judged by heat-modifiable mobility on SDS-PAGE. This indicates that the C terminus is not required for correct global folding of the usher. The PapC640Δ truncation mutant, lacking 169 residues, was the most stable of the deletion mutants. Purification of this mutant and analysis by liposome swelling assay (41) indicated that it retained channel-forming activity similar to full-length PapC (D. G. Thanassi, unpublished data). Taken together, the above data suggest it is unlikely that the C-terminal 169 residues of PapC contribute TM β-strands toward formation of the core β-barrel structure. The PapC C terminus was the most-sensitive region of the usher to proteolysis. Thus, this region is exposed and might completely lack TM β-strands. Therefore, the computer prediction for the PapC C terminus (Fig. 1) may be incorrect, and the terminal 169 residues may instead reside in the periplasm. Similarly, experimental analysis of the FaeD usher led Harms and coworkers to conclude that its C-terminal region resides entirely in the periplasm, despite topology predictions otherwise (12).
Targeting of chaperone-adhesin complexes to the usher is thought to be a key initiating step in pilus biogenesis (31). The usher C terminus was not required for interaction with chaperone-adhesin complexes in vitro, as 359 residues could be truncated without affecting PapDG binding to PapC in an overlay assay. Therefore, the targeting site for chaperone-adhesin complexes resides in an N-terminal domain of the usher. In fact, N-terminal deletion products of PapC obtained by protease digestion are unable to bind chaperone-adhesin complexes by the overlay assay (D. G. Thanassi, unpublished data). In agreement with this, expression of an N-terminal region of the FaeD usher was found to allow targeting of a K88 pilus subunit to the OM (24). Despite the ability of the PapC C-terminal truncation mutants to bind PapDG and to fold correctly in the OM, none of the truncation mutants was competent for pilus biogenesis in vivo (in the absence of type 1 pilus expression). This indicates that the usher C terminus is essential for assembly and secretion events subsequent to chaperone-subunit targeting. A previous study found that FimCH chaperone-adhesin complexes form stable assembly intermediates with the FimD usher in vivo (31). Proteolysis of these stable complexes revealed that FimCH was bound to a C-terminal 40-kDa fragment of FimD (31). This is in contrast to the targeting of chaperone-subunit complexes to the usher N terminus and suggests that following targeting to the usher, chaperone-subunit complexes switch to a stable assembly site involving the usher C terminus. Thus, the ushers appear to have distinct N- and C-terminal domains for chaperone-subunit targeting and subunit assembly into pili, respectively.
Each chaperone/usher pathway exhibits specificity for its own subunits. The source of this specificity is not completely understood. Studies have shown that the FimC chaperone cannot substitute for the PapD chaperone in P pilus biogenesis, although PapD is able to substitute for FimC in type 1 pilus biogenesis (16). PapDG chaperone-adhesin complexes do not bind to the FimD usher and FimCH chaperone-adhesin complexes do not bind to the PapC usher (31). A study of type 1 and F1C pili demonstrated that although neither the chaperone nor the usher alone could substitute for assembly of the homologous pilus, both could be swapped together, indicating the assembly components work in parental pairs (19). We show here that expression of N-terminal domains of PapC is sufficient to drive assembly of P pili by the chromosomal fim (type 1) system. Expression of type 1 pili alone was not able to complement a ΔpapC pap operon to assemble P pili, indicating that the PapC N-terminal fragments were functional in vivo and acted synergistically with the fim system. The PapC truncation mutants presumably provided targeting specificity for the Pap chaperone-subunit complexes, which then were able to utilize the FimD usher and possibly other type 1 components for assembly into P pili. This result implies the formation of mixed FimD-PapC oligomeric complexes. The inverse relationship between the amounts of pili assembled and the size of the PapC truncation (Fig. 7D) could reflect a need for C-terminal regions of PapC for efficient interaction with FimD and/or the fact that the larger truncations are simply less stable.
The results suggest a model for pilus biogenesis by the usher in which the initial targeting of chaperone-subunit complexes occurs at an N-terminal domain of the usher, followed by a switch to a second site located at the usher C terminus for donor strand exchange and assembly into pili (Fig. 8). In this model, at least two chaperone-subunit complexes could simultaneously be bound to the usher (Fig. 8). This might position the complexes to drive uncapping of the chaperone from the subunit bound at the C-terminal usher site, followed by donor strand exchange and incorporation of the subunit bound at the N-terminal site into the growing pilus fiber at the C-terminal site. Repeated cycles of targeting and donor strand exchange would lead to extension of the pilus coupled with pilus secretion to the cell surface.
FIG. 8.

Model for P pilus biogenesis by the PapC usher. The oligomeric usher complex contains N-terminal sites (n) and C-terminal sites (c) for interaction with chaperone-subunit complexes. (A and B) Periplasmic chaperone-subunit complexes target the N-terminal usher site. The PapDG chaperone-adhesin complex has the highest affinity for the usher and targets first. (C) PapDG moves from the N-terminal targeting site to the C-terminal assembly site, forming a stable assembly intermediate. The N-terminal site is now open for targeting of the next chaperone-subunit complex, PapDF, to the usher. (D) The N-terminal extension of the bound PapF subunit undergoes donor strand exchange with the subunit domain of PapG, causing uncapping of the chaperone from PapG. (E) Donor strand exchange between PapF and PapG generates the first link in the pilus tip. This results in a shift of the PapDF complex to the usher C-terminal assembly site, allowing targeting of the next chaperone-subunit complex, PapDE, to the usher. Repeated cycles of targeting and assembly would result in extension and secretion of the pilus tip and rod.
Acknowledgments
We thank J. S. Pinkner for purified PapD and PapDG, M.-J. Lombardo for construction of His-tagged versions of the PapC C-terminal truncation mutants, D. Stevens and the Georgiou laboratory for FACS analysis, S. O. Smith and the Smith laboratory for use of and assistance with the CD spectrophotometer, and N. Misiti for assistance with the pilus purification experiments. We are grateful to R. Curtiss III for providing laboratory resources and supporting this collaborative work.
This work was supported by Public Health Service grants GM62987 (D.G.T.), AI24533 (C.S.), AI07172-20 (C.S.), and AI29549 (S.J.H.) from the National Institutes of Health.
REFERENCES
- 1.Baddour, L. M., G. D. Christensen, W. A. Simpson, and E. H. Beachey. 1990. Microbial adherence, p. 9-25. In J. E. Bennet (ed.), Principles and practice of infectious disease, 3 ed., vol. 2. Churchill Livingston, New York, N.Y.
- 2.Baneyx, F., and G. Georgiou. 1990. In vivo degradation of secreted fusion proteins by the Escherichia coli outer membrane protease OmpT. J. Bacteriol. 172:491-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes, W. M., M. Bevan, and P. H. Son. 1983. Kilo-sequencing: creation of an ordered nest of asymmetric deletions across a large target sequence carried on phage M13. Methods Enzymol. 101:98-122. [DOI] [PubMed] [Google Scholar]
- 4.Barnhart, M. M., J. S. Pinkner, G. E. Soto, F. G. Sauer, S. Langermann, G. Waksman, C. Frieden, and S. J. Hultgren. 2000. PapD-like chaperones provide the missing information for folding of pilin proteins. Proc. Natl. Acad. Sci. USA 97:7709-7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blomfield, I. C., M. S. McClain, and B. I. Eisenstein. 1991. Type 1 fimbriae mutants of Escherichia coli K12: characterization of recognized afimbriate strains and construction of new fim deletion mutants. Mol. Microbiol. 5:1439-1445. [DOI] [PubMed] [Google Scholar]
- 6.Bock, K., M. E. Breimer, A. Brignole, G. C. Hansson, K.-A. Karlsson, G. Larson, H. Leffler, B. E. Samuelsson, N. Strömberg, C. Svanborg-Edén, and J. Thurin. 1985. Specificity of binding of a strain of uropathogenic Escherichia coli to Galα(1-4)Gal-containing glycosphingolipids. J. Biol. Chem. 260:8545-8551. [PubMed] [Google Scholar]
- 7.Bullitt, E., and L. Makowski. 1995. Structural polymorphism of bacterial adhesion pili. Nature 373:164-167. [DOI] [PubMed] [Google Scholar]
- 8.Choudhury, D., A. Thompson, V. Stojanoff, S. Langermann, J. Pinkner, S. J. Hultgren, and S. D. Knight. 1999. X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic Escherichia coli. Science 285:1061-1066. [DOI] [PubMed] [Google Scholar]
- 9.Deléage, G., and C. Geourjon. 1993. An interactive graphic program for calculating the secondary structure content of proteins from circular dichroism spectrum. Comput. Appl. Biosci. 9:197-199. [DOI] [PubMed] [Google Scholar]
- 10.Dodson, K. W., F. Jacob-Dubuisson, R. T. Striker, and S. J. Hultgren. 1993. Outer membrane PapC usher discriminately recognizes periplasmic chaperone-pilus subunit complexes. Proc. Natl. Acad. Sci. USA 90:3670-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dodson, K. W., J. S. Pinkner, T. Rose, G. Magnusson, S. J. Hultgren, and G. J. Waksman. 2001. Structural basis of the Interaction of the pyelonephritic E. coli adhesin to Its human kidney receptor. Cell 105:733-743. [DOI] [PubMed] [Google Scholar]
- 12.Harms, N., W. C. Oudhuis, E. A. Eppens, Q. A. Valent, M. Koster, J. Luirink, and B. Oudega. 1999. Epitope tagging analysis of the outer membrane folding of the molecular usher FaeD involved in K88 fimbriae biosynthesis in Escherichia coli. J. Mol. Microbiol. Biotechnol. 1:319-325. [PubMed] [Google Scholar]
- 13.Holmgren, A., and C. Brändén. 1989. Crystal structure of chaperone protein PapD reveals an immunoglobulin fold. Nature 342:248-251. [DOI] [PubMed] [Google Scholar]
- 14.Jacob-Dubuisson, F., R. Striker, and S. J. Hultgren. 1994. Chaperone-assisted self-assembly of pili independent of cellular energy. J. Biol. Chem. 269:12447-12455. [PubMed] [Google Scholar]
- 15.Jones, C. H., P. N. Danese, J. S. Pinkner, T. J. Silhavy, and S. J. Hultgren. 1997. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 16:6394-6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones, C. H., J. S. Pinkner, A. V. Nicholes, L. N. Slonim, S. N. Abraham, and S. J. Hultgren. 1993. FimC is a periplasmic PapD-like chaperone that directs assembly of type 1 pili in bacteria. Proc. Natl. Acad. Sci. USA 90:8397-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones, C. H., J. S. Pinkner, R. Roth, J. Heuser, A. V. Nicholoes, S. N. Abraham, and S. J. Hultgren. 1995. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc. Natl. Acad. Sci. USA 92:2081-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klemm, P., and G. Christiansen. 1990. The fimD gene required for cell surface localization of Escherichia coli type 1 fimbriae. Mol. Gen. Genet. 220:334-338. [DOI] [PubMed] [Google Scholar]
- 19.Klemm, P., B. J. Jorgensen, B. Kreft, and G. Christiansen. 1995. The export systems of type 1 and F1C fimbriae are interchangeable but work in parental pairs. J. Bacteriol. 177:621-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuehn, M. J., J. Heuser, S. Normark, and S. J. Hultgren. 1992. P pili in uropathogenic E. coli are composite fibres with distinct fibrillar adhesive tips. Nature 356:252-255. [DOI] [PubMed] [Google Scholar]
- 21.Kuehn, M. J., S. Normark., and S. J. Hultgren. 1991. Immunoglobulin-like PapD chaperone caps and uncaps interactive surfaces of nascently translocated pilus subunits. Proc. Natl. Acad. Sci. USA 88:10586-10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leong, J. M., R. S. Fournier, and R. R. Isberg. 1991. Mapping and topographic localization of epitopes of the Yersinia pseudotuberculosis invasin protein. Infect. Immun. 59:3424-3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maurer, L., and P. E. Orndorff. 1985. A new locus, pilE, required for the binding of type 1 piliated Escherichia coli to erythrocytes. FEMS Microbiol. Lett. 30:59-66. [Google Scholar]
- 24.Mol, O., W. C. Oudhuis, R. Oud, R. Sijbrandi, J. Luirink, N. Harms, and B. Oudega. 2001. Biosynthesis of K88 fimbriae in Escherichia coli: interaction of tip-subunit FaeC with the periplasmic chaperone FaeE and the outer membrane usher FaeD. J. Mol. Microbiol. Biotechnol. 3:135-142. [PubMed] [Google Scholar]
- 25.Nikaido, H. 1994. Isolation of outer membranes. Methods Enzymol. 235:225-234. [DOI] [PubMed] [Google Scholar]
- 26.Norgren, M., M. Baga, J. M. Tennent, and S. Normark. 1987. Nucleotide sequence, regulation and functional analysis of the papC gene required for cell surface localization of Pap pili of uropathogenic Escherichia coli. Mol. Microbiol. 1:169-178. [DOI] [PubMed] [Google Scholar]
- 27.Pugsley, A. 1993. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57:50-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rost, B., and C. Sander. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232:584-599. [DOI] [PubMed] [Google Scholar]
- 29.Sauer, F. G., K. Fütterer, J. S. Pinkner, K. W. Dodson, S. J. Hultgren, and G. Waksman. 1999. Structural basis of chaperone function and pilus biogenesis. Science 285:1058-1061. [DOI] [PubMed] [Google Scholar]
- 30.Saulino, E. T., E. Bullitt, and S. J. Hultgren. 2000. Snapshots of usher-mediated protein secretion and ordered pilus assembly. Proc. Natl. Acad. Sci. USA 97:9240-9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saulino, E. T., D. G. Thanassi, J. S. Pinkner, and S. J. Hultgren. 1998. Ramifications of kinetic partitioning on usher-mediated pilus biogenesis. EMBO J. 17:2177-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schifferli, D. M., and M. A. Alrutz. 1994. Permissive linker insertion sites in the outer membrane protein of 987P fimbriae of Escherichia coli. J. Bacteriol. 176:1099-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schirmer, T., and S. W. Cowan. 1993. Prediction of membrane-spanning β-strands and its application to maltoporin. Protein Sci. 2:1361-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnaitman, C. A. 1973. Outer membrane proteins of Escherichia coli. I. Effect of preparative conditions on the migration of protein in polyacrylamide gels. Arch. Biochem. Biophys. 157:541-552. [DOI] [PubMed] [Google Scholar]
- 35.Schulz, G. E. 2000. β-Barrel membrane proteins. Curr. Opin. Struct. Biol. 10:443-447. [DOI] [PubMed] [Google Scholar]
- 36.Slonim, L. N., J. S. Pinkner, C. I. Branden, and S. J. Hultgren. 1992. Interactive surface in the PapD chaperone cleft is conserved in pilus chaperone superfamily and essential in subunit recognition and assembly. EMBO J. 11:4747-4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stathopoulos, C., D. R. Hendrixson, D. G. Thanassi, S. J. Hultgren, J. W. St. Geme III, and R. Curtiss III. 2000. Secretion of virulence determinants by the general secretory pathway in Gram-negative pathogens: an evolving story. Microbes Infect. 2:1061-1072. [DOI] [PubMed] [Google Scholar]
- 38.Sugawara, E., M. Steiert, S. Rouhani, and H. Nikaido. 1996. Secondary structure of the outer membrane proteins OmpA of Escherichia coli and OprF of Pseudomonas aeruginosa. J. Bacteriol. 178:6067-6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thanassi, D. G., and S. J. Hultgren. 2000. Multiple pathways allow protein secretion across the bacterial outer membrane. Curr. Opin. Cell Biol. 12:420-430. [DOI] [PubMed] [Google Scholar]
- 40.Thanassi, D. G., E. T. Saulino, and S. J. Hultgren. 1998. The chaperone/usher pathway: a major terminal branch of the general secretory pathway. Curr. Opin. Microbiol. 1:223-231. [DOI] [PubMed] [Google Scholar]
- 41.Thanassi, D. G., E. T. Saulino, M.-J. Lombardo, R. Roth, J. Heuser, and S. J. Hultgren. 1998. The PapC usher forms an oligomeric channel: implications for pilus biogenesis across the outer membrane. Proc. Natl. Acad. Sci. USA 95:3146-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valent, Q. A., J. Zaal, F. K. de Graaf, and B. Oudega. 1995. Subcellular localization and topology of the K88 usher FaeD in Escherichia coli. Mol. Microbiol. 16:1243-1257. [DOI] [PubMed] [Google Scholar]