

Abstract
DIABETIC KETOACIDOSIS AND THE HYPERGLYCEMIC hyperosmolar state are the most serious complications of diabetic decompensation and remain associated with excess mortality. Insulin deficiency is the main underlying abnormality. Associated with elevated levels of counterregulatory hormones, insulin deficiency can trigger hepatic glucose production and reduced glucose uptake, resulting in hyperglycemia, and can also stimulate lipolysis and ketogenesis, resulting in ketoacidosis. Both hyperglycemia and hyperketonemia will induce osmotic diuresis, which leads to dehydration. Clinical diagnosis is based on the finding of dehydration along with high capillary glucose levels with or without ketones in the urine or plasma. The diagnosis is confirmed by the blood pH, serum bicarbonate level and serum osmolality. Treatment consists of adequate correction of the dehydration, hyperglycemia, ketoacidosis and electrolyte deficits.
Diabetic ketoacidosis (DKA) and the hyperglycemic hyperosmolar state (HHS) appear as 2 extremes in the spectrum of diabetic decompensation.1 They remain the most serious acute metabolic complications of diabetes mellitus and are still associated with excess mortality. Because the approach to the diagnosis and treatment of these hyperglycemic crises are similar, we have opted to address them together.
The incidence of DKA is between 4.6 and 8.0 per 1000 person-years among patients with diabetes, whereas that of HHS is less than 1 per 1000 person-years.2 Based on the estimated diabetic population in Canada,3 we can anticipate that 5000–10 000 patients will be admitted to hospital because of DKA every year and 500–1000 patients because of HHS. The estimated mortality rate for DKA is between 4% and 10%, whereas the rate for HHS varies from 10% to 50%, the range most likely owing to underlying illnesses.2
Pathogenesis
In both DKA and HHS, the underlying metabolic abnormality results from the combination of absolute or relative insulin deficiency and increased amounts of counterregulatory hormones.
Glucose and lipid metabolism
When insulin is deficient, the elevated levels of glucagon, catecholamines and cortisol will stimulate hepatic glucose production through increased glycogenolysis and enhanced gluconeogenesis4 (Fig. 1). Hypercortisolemia will result in increased proteolysis, thus providing amino acid precursors for gluconeogenesis. Low insulin and high catecholamine concentrations will reduce glucose uptake by peripheral tissues. The combination of elevated hepatic glucose production and decreased peripheral glucose use is the main pathogenic disturbance responsible for hyperglycemia in DKA and HHS. The hyperglycemia will lead to glycosuria, osmotic diuresis and dehydration. This will be associated with decreased kidney perfusion, particularly in HHS, that will result in decreased glucose clearance by the kidney and thus further exacerbation of the hyperglycemia.
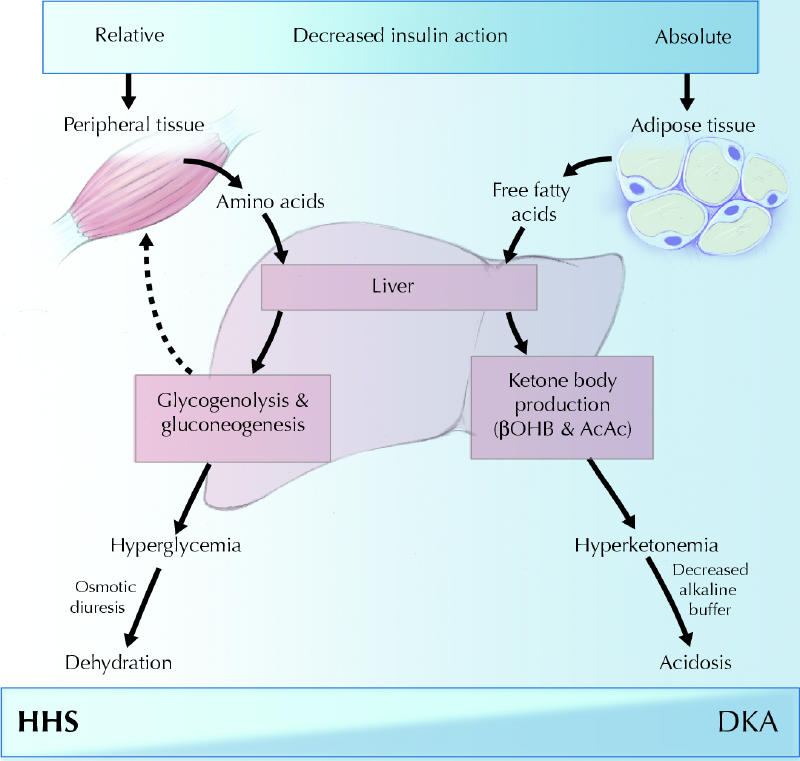
Fig. 1: Schematic of the pathogenesis of diabetic ketoacidosis (DKA) and the hyperglycemic hyperosmolar state (HHS). Relative or absolute insulin deficiency stimulates hepatic glucose production, which results in hyperglycemia, osmotic diuresis and dehydration. In severe insulin deficiency, the liver will augment ketone body production, culminating in hyperketonemia and, eventually, acidosis. βOHB = β-hydroxybutyric acid; AcAc = acetoacetic acid. Photo: Chesley Sheppard
In DKA, the low insulin levels combined with increased levels of catecholamines, cortisol and growth hormone will activate hormone-sensitive lipase, which will cause the breakdown of triglycerides and release of free fatty acids. The free fatty acids are taken up by the liver and converted to ketone bodies that are released into the circulation. The process of ketogenesis is stimulated by the increase in glucagon levels.5 This hormone will activate carnitine palmitoyltransferase I, an enzyme that allows free fatty acids in the form of coenzyme A to cross mitochondrial membranes after their esterification into carnitine. On the other side, esterification is reversed by carnitine palmitoyltransferase II to form fatty acyl coenzyme A, which enters the β-oxidative pathway to produce acetyl coenzyme A. Most of the acetyl coenzyme A is used in the synthesis of β-hydroxybutyric acid and acetoacetic acid, 2 relatively strong acids responsible for the acidosis in DKA. Acetoacetate is converted to acetone through spontaneous nonenzymatic decarboxylation in linear relation to its concentration. β-Hydroxybutyric acid, acetoacetic acid and acetone are filtered by the kidney and partially excreted in urine.6 Therefore, progressive volume depletion leading to a reduced glomerular filtration rate will result in greater ketone retention.6 The reason for the absence of ketosis in the presence of insulin deficiency in HHS remains unknown.1 The current hypothesis is that the absence may be due to the lower levels of free fatty acids or the higher portal vein insulin levels, or both.1,7,8
Acid–base balance, fluids and electrolytes
Acidosis in DKA is due to the overproduction of β-hydroxybutyric acid and acetoacetic acid. At physiological pH, these 2 ketoacids dissociate completely, and the excess hydrogen ions bind the bicarbonate, resulting in decreased serum bicarbonate levels. Ketone bodies thus circulate in the anionic form, which leads to the development of anion gap acidosis that characterizes DKA. The anion gap can be calculated using the following formula: Na+ – (Cl– + HCO3–). According to this formula, the normal anion gap is 12 (standard deviation [SD] 2) mmol/L. In DKA, bicarbonate is replaced by β-hydroxybutyric acid and acetoacetic acid, so that the sum of bicarbonate and chloride concentrations is reduced and the anion gap is thus increased. Despite substantial losses of ketoacids in the urine, the decrease in serum bicarbonate concentration and the increase in the anion gap observed in DKA are about equal.9
Under normal circumstances, β-hydroxybutyric acid levels are 2 to 3 times higher than acetoacetic acid levels, the difference reflecting the mitochondrial redox state. Any increase in the mitochondrial redox state, such as occurs in DKA, will increase the ratio of β-hydroxybutyric acid to acetoacetic acid. If the disproportionately augmented β-hydroxybutyric acid is not measured, this could lead to an underestimation of ketones.7
Metabolic acidosis will induce hyperventilation through stimulation of peripheral chemoreceptors and the respiratory centre in the brainstem, which will elicit a decrease in the partial pressure of carbon dioxide. This will partially compensate for the metabolic acidosis.
Hyperglycemia-induced osmotic diuresis results in severe fluid loss. The total body deficit of water is usually about 5–7 L in DKA and 7–12 L in HHS (Table 1), which represents a loss of about 10%–15% of body weight. The osmotic diuresis is associated with large losses of electrolytes in the urine.
Table 1
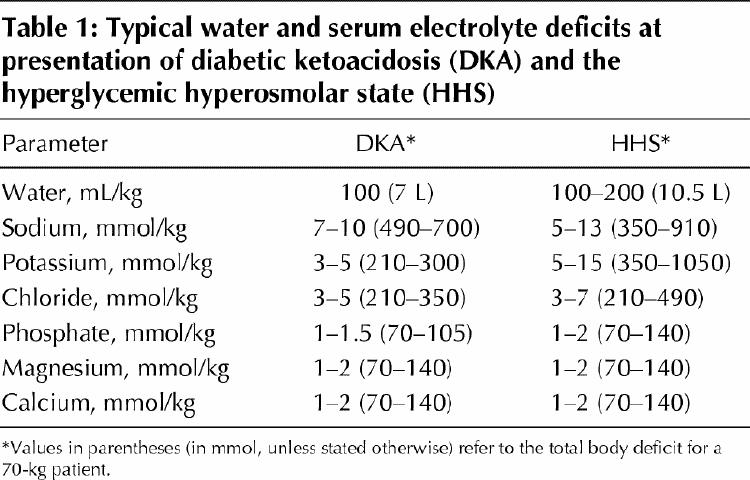
The sodium chloride deficit in DKA and HHS is usually 5–13 mmol/kg of body weight for sodium and 3–7 mmol/kg for chloride1,4,7 (Table 1). Initially, increased glucose concentration is restricted to the extracellular space, which forces water from the intracellular to the extracellular compartment and induces dilution of the plasma sodium concentration. Subsequently, further increases in the plasma glucose concentration will lead to osmotic diuresis, with losses of water and sodium chloride in the urine; the water loss usually exceeds that of the sodium chloride.4,9 Eventually, the loss of water from the intracellular and extracellular compartments will become quantitatively similar. Because of the osmotic shift of water, plasma sodium concentrations are usually low or normal in DKA and can be slightly increased in HHS, despite extensive water loss.9,10 In this context, the plasma sodium concentration should be corrected for hyperglycemia by adding 1.6 mmol to the reported sodium level for every 5.6 mmol/L increase in glucose above 5.6 mmol/L.4 The plasma sodium concentration may also be factitiously lowered by the presence of severe hyperlipidemia.
DKA and HHS are also associated with profound total body potassium depletion, ranging from 3 to 15 mmol/kg of body weight,7,11,12 (Table 1). However, plasma potassium concentrations are typically normal or elevated at the time of presentation. As with sodium, the presence of hyperglycemia leads to a shift of water and potassium from the intracellular to the extracellular space. This shift of potassium is further enhanced in the presence of acidosis, intracellular proteolysis and insulinopenia.13 Potassium depletion is due to excessive urinary potassium loss secondary to osmotic diuresis, and it leads to increased delivery of fluid and sodium to potassium secretory sites in the distal nephron.4 This can be further exacerbated by poor oral intake of potassium, vomiting and secondary hyperaldosteronism.4
Phosphate, magnesium and calcium are other elements excreted in excess in urine during the development of DKA and HHS owing to osmotic diuresis, for a deficit of 1–2 mmol/kg on average7,11 (Table 1).
Precipitating factors
Infection remains the most important precipitating factor in the development of DKA and HHS. In 20%–25% of cases, infections are the first manifestations of previously undiagnosed diabetes mellitus.13 Omissions or inadequate insulin doses are frequent precipitating factors, particularly for DKA.7
Other precipitating factors, especially for HHS, are silent myocardial infarction, cerebrovascular accident, mesenteric ischemia, acute pancreatitis and use of medications such as steroids, thiazide diuretics, calcium-channel blockers, propranolol and phenytoin.14 In 2%–10% of cases of DKA, no obvious precipitating factor can be identified.13
Diagnosis
Clinical presentation
In general, DKA and HHS differ in presentation. If physical examination reveals dehydration along with a high capillary blood glucose level with or without urine or increased plasma ketone bodies, acute diabetic decompensation should be strongly suspected. A definitive diagnosis of DKA or HHS must be confirmed through laboratory investigation.
The clinical presentation can provide helpful information for the preliminary bedside diagnosis.15 DKA usually occurs in younger, lean patients with type 1 diabetes and develops within a day or so, whereas HHS is more likely to occur in older, obese patients with type 2 diabetes and can take days or weeks to fully develop. In addition, HHS usually occurs in elderly diabetic patients, often those with decreased renal function who do not have access to water.16 Both DKA and HHS often present with polyuria and polydypsia, although polydypsia may be absent in elderly patients with HHS. In both conditions, abdominal pain with nausea and vomiting can develop owing to acidosis per se or to decreased mesenteric perfusion and can be mistaken for an acute surgical abdomen. Kussmaul–Kien respiration (rapid and deep respiration) with breath acetone is typical of DKA but is absent in HHS. Although dehydration occurs in both conditions, it is often more pronounced in HHS. Because DKA and HHS are usually accompanied by hypothermia, a normal or elevated temperature may indicate underlying infection.
Laboratory findings
Typical laboratory findings are listed in Tables 1 and 2.17,18 Most patients presenting with DKA have a plasma glucose level of 14 mmol/L or greater. However, most patients with type 1 diabetes who have such a plasma glucose level do not have ketoacidosis. On the other hand, ketoacidosis may develop in patients with a plasma glucose level below 14 mmol/L. In HHS, hyperglycemia is usually more severe than in DKA, and a plasma glucose level of 34 mmol/L or greater is arbitrarily one of the diagnostic criteria. Glucose is the main osmole responsible for the hyperosmolar syndrome. The increased serum osmolality can be calculated as follows: (2 х serum Na) + serum glucose, with normal values being 290 (SD 5) mmol/kg water. Blood urea nitrogen is not included in the calculation of effective osmolality because it is freely permeable in and out of the intracellular compartment.1,19 By definition, the osmolality must be greater than 320 mmol/kg to be diagnostic of HHS. However, it is not uncommon in DKA to have increased osmolality. DKA will have a pH of 7.30 or less, and HHS in isolation will have a pH greater than 7.30. Venous blood can be used to measure pH and bicarbonate levels, unless information on oxygen transport is required. It must be remembered that venous blood, without arterial blood gas values, does not permit the identification of mixed acid–base disorders.20 In DKA, a lower pH will usually be associated with a decrease in bicarbonate to 15 mmol/L or less, although a milder form of DKA may present with a bicarbonate level between 15 and 18 mmol/L. Less severe DKA is always accompanied by moderate to large amounts of ketones in the blood and urine. Trace amounts may also be found in cases of HHS.21 It is now possible to measure blood β-hydroxybutyric acid levels at the bedside, using a reagent strip and a reflectance meter.22
Table 2

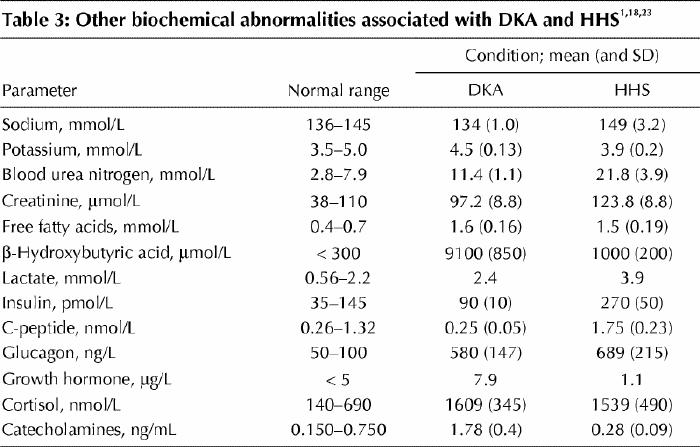
Other biochemical abnormalities associated with DKA and HHS are listed in Table 3.1,18,23 The majority of patients presenting with DKA and HHS will have an elevated leukocyte count, usually in the range of 10.0–15.0 х 109/L. This has been attributed to stress and dehydration.13 Amylase levels are often elevated in patients with DKA, but represent enzyme activity from nonpancreatic tissues such as the parotid gland. Lipase levels will usually be normal. Additional laboratory tests should include blood culture, urinalysis and urine culture, chest radiography and electrocardiography, as well as measurement of the lactate level if indicated. Because a high fetal mortality rate is associated with ketoacidosis, it is important to eliminate the possibility of pregnancy in women of reproductive age.
Table 3
Treatment
The success of treatment of DKA and HHS depends on adequate correction of dehydration, hyperglycemia, ketoacidosis and electrolyte deficits24 (Fig. 2). Any comorbid precipitating event should be identified and treated appropriately. Both DKA and HHS are medical emergencies, and patients with these conditions must be admitted to hospital.
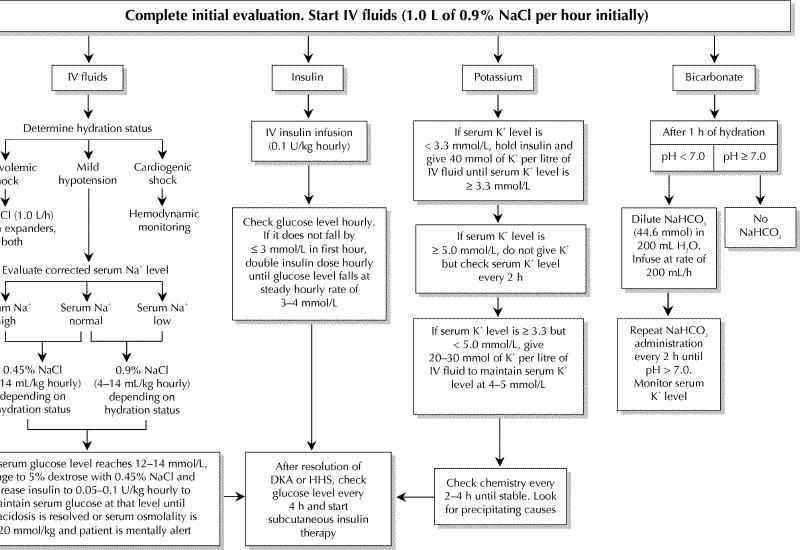
Fig. 2: Management of adult patients with DKA and HHS. Copyright ©2003 American Diabetes Association. From Diabetes Care 2003;26(1 Suppl): S109-17. Reprinted and adapted with permission from The American Diabetes Association.
Fluid therapy
The objective of initial fluid therapy is to expand extracellular volume (intravascular and extravascular) and restore renal perfusion. In the absence of major heart problems, it is suggested that treatment start with infusion of isotonic saline (0.9% sodium chloride) at a rate of 15–20 mL/kg per hour during the first hour (1–1.5 L in the average adult) to rapidly expand the extracellular space. The subsequent choice of fluid replacement depends on the state of hydration, electrolyte levels and urinary output. In general, it can be infusion of 0.45% sodium chloride at a rate of 4–14 mL/kg per hour if the serum sodium level is normal or elevated. The administration of hypotonic saline leads to gradual replacement of the intracellular and extracellular compartments. As soon as renal function is assured, potassium must be added to every litre of fluid. When the plasma glucose level reaches 12–14 mmol/L, each litre of fluid should contain 5% dextrose. Fluid replacement should correct the estimated water deficit over the first 24 hours. It is important that the change in osmolality not exceed 3 mmol/kg per hour.1,10,11,25,26 In patients with kidney and heart problems, their heart, kidney and mental status must be assessed frequently, with regular serum osmolality monitoring during rehydration to avoid iatrogenic water overload.1,10,11,25,26
Insulin therapy
There is general consensus that, in cases of DKA and HHS, regular insulin should be administered by means of continuous intravenous infusion in small doses through an infusion pump.4,5,25,27 Such low-dose insulin therapy provides insulin concentrations that are more physiologic and produce a more gradual and steady fall in plasma glucose levels,28,29 and it decreases the risk of hypoglycemia and hypokalemia.4 The available data do not support the subcutaneous or intramuscular route for insulin administration.4 Although most of the protocols proposed suggest that a loading dose of insulin should be given at the initiation of insulin therapy, there are no data to support any advantage for such a recommendation.4,29
As soon as hypokalemia (potassium concentration < 3.3 mmol/L) has been excluded, continuous infusion of regular insulin can be started at a dose of 0.1 U/kg per hour (Fig. 2). This should produce a gradual decrease in the plasma glucose level of 3–4 mmol/L per hour.28 If the glucose level does not decline by 3 mmol/L in the first hour, the hydration status should be checked; if it is acceptable, the insulin dose should be doubled every hour until a decrease of 3–4 mmol/L per hour in the plasma glucose level is observed. When the plasma glucose level reaches 12–14 mmol/L, the insulin infusion rate may be decreased by 50% as the 5% dextrose is added. Thereafter, the insulin infusion dose must be adjusted to maintain the plasma glucose values until the acidosis in DKA or the clouded consciousness and hyperosmolality in HHS have been resolved.
It takes longer to correct ketonuria than hyperglycemia. Because β-hydroxybutyric acid is the prevalent ketoacid and is gradually converted to acetoacetic acid, the correction of ketonuria is underestimated when measured by the nitroprusside method. Measurement of serum β-hydroxybutyric acid levels using a reagent strip and a reflectance meter22,30 has been validated and offers the possibility of bedside diagnosis with better follow-up parameters of hyperketonemia during treatment.
Once the ketoacidosis in DKA has been corrected (plasma glucose level < 11.0 mmol/L, serum bicarbonate level ≥ 18 mmol/L, venous pH > 7.3 and anion gap < 12 mmol/L), the clouded consciousness and hyperosmolality in HHS have resolved, and patients are able to take fluids orally, a multidose insulin regimen may be initiated based on the patient's treatment before DKA or HHS developed (Fig. 2).
Potassium therapy
The treatment of DKA and HHS with rehydration and insulin is typically associated with a rapid decline in the plasma potassium concentration, particularly during the first few hours of therapy.9,13 This rapid decrease is due to several factors, the most significant being the insulin-mediated re-entry of potassium into the intracellular compartment. Other factors are extracellular fluid volume expansion, correction of acidosis and continued potassium loss owing to osmotic diuresis and ketonuria.
Despite major potassium depletion in the whole body, mild to moderate hyperkalemia is not uncommon in patients in hyperglycemic decompensation. Because treatment will rapidly induce decreased serum potassium concentrations, potassium replacement must be initiated as soon as levels fall below 5.0 mmol/L, assuming urine output is adequate (Fig. 2). It is recommended that 20–30 mmol of potassium be added to each litre of infusion fluid to maintain the serum potassium concentration between 4 and 5 mmol/L.24 If the serum potassium level is less than 3.3 mmol/L, potassium replacement therapy should be started immediately with fluid therapy, and the initiation of insulin therapy should be delayed until the potassium concentration is restored to above 3.3 mmol/L, in order to avoid arrhythmia, cardiac arrest and respiratory muscle weakness.24 Initially, the serum potassium level should be measured every 1–2 hours because the most rapid change occurs during the first 5 hours of treatment. After that, it should be measured every 4–6 hours as indicated clinically.
Bicarbonate therapy
The use of bicarbonate in the treatment of DKA remains controversial.31 The rationale for bicarbonate therapy is the theoretical assumption that severe acidosis could contribute to organ malfunction, such as of the liver, heart and brain. However, there are few prospective, randomized studies of the use of bicarbonate in DKA. Furthermore, bicarbonate therapy is associated with risks: (a) a heightened risk of hypokalemia, (b) induction of paradoxical central nervous system acidosis, (c) worsening of intracellular acidosis owing to increased carbon dioxide production and (d) prolongation of ketoanion metabolism. Retrospective reviews have not demonstrated any difference in the severity of acidosis, in mental status improvement or in the correction of hyperglycemia, whether or not bicarbonate therapy was used.32 Nevertheless, it is still recommended that, if the arterial pH is below 7.0 after 1 hour of hydration, bicarbonate therapy should be used. Sodium bicarbonate should then be administered in hypotonic fluid (44.6 mmol/L) every 2 hours until the pH is at least 7.0.13 If the arterial pH is 7.0 or higher, no bicarbonate therapy is recommended (Fig. 2).
Phosphate therapy
The beneficial effect of phosphate therapy is purely theoretical. It would be expected to prevent potential complications associated with hypophosphatemia, such as respiratory depression, skeletal muscle weakness, hemolytic anemia and cardiac dysfunction. Furthermore, it would be expected to restore the 2,3-diphosphoglycerate level, which is decreased in DKA, shifting the oxygen dissociation curve to the right and enhancing oxygen delivery to the tissues.33 On the other hand, excessive phosphate administration can lead to hypocalcemia, tetanus and soft-tissue calcification.4 Finally, the majority of controlled, randomized trials have been unable to demonstrate any clinical benefit of routine phosphate therapy. Kitabchi and Wall4 still recommend that one-third of potassium replacement be given as potassium phosphate. There are no studies on the use of phosphate therapy for HHS.
Clinical and laboratory follow-up
Meticulous follow-up of patients' vital signs, clinical conditions and laboratory parameters is important. Vital signs should be monitored every half hour for the first hour, hourly for the next 4 hours and then every 2–4 hours until resolution of the condition. An accurate record of hourly urine output is necessary to monitor kidney function. On admission, a comprehensive profile will include at least arterial or venous blood gas values, levels of plasma glucose, electrolytes, blood urea nitrogen and creatinine, ketone levels in serum or urine, or both, and serum osmolality. Capillary blood glucose levels should be monitored every hour to allow adjustment of the insulin infusion dose. Electrolyte levels should be measured every 1–2 hours initially, and every 4 hours thereafter. Measurement of venous pH can replace that of arterial pH and should be undertaken every 4 hours until the DKA has been corrected.
Treatment-related complications
Cerebral edema
Fortunately, symptomatic cerebral edema occurs rarely in adults with diabetes who receive treatment for DKA or HHS. There is evidence, however, from electroencephalograms and CT scans in adults and pediatric patients, that the development of subclinical cerebral edema is not uncommon during the first 24 hours of DKA therapy.4 Multiple factors in the treatment of DKA and HHS may contribute to the cerebral edema. These include (a) the idiogenic osmoles, which cannot be dissipated rapidly during rehydration, thus creating a gradient and a shift of water into the cells;4 (b) insulin therapy per se, which may promote the entry of osmotically active particles into the intracellular space; and (c) rapid replacement of sodium deficits.11,12 To reduce the risk of cerebral edema, it is recommended that physicians correct sodium and water deficits gradually and avoid the rapid decline in plasma glucose concentration.23,34
Adult respiratory distress syndrome
Adult respiratory distress syndrome, or noncardiogenic pulmonary edema, is a potentially fatal complication of DKA that fortunately occurs rarely.4 The partial pressure of oxygen, which is normal on admission, decreases progressively during treatment to unexpectedly low levels. This change is believed to be due to increased water in the lungs and reduced lung compliance. These changes may be similar to those occurring in brain cells leading to cerebral edema, which suggests that it is a common biological phenomenon in tissues.4
Hyperchloremic metabolic acidosis
This phenomenon is not uncommon during the treatment of DKA.9 A major mechanism is the loss of substrates (ketoanions) in the urine that are necessary for bicarbonate regeneration.9,35 Other mechanisms include (a) intravenous fluids containing chloride concentrations exceeding that of plasma,6,35 (b) volume expansion with bicarbonate-free fluids6,35 and (c) intracellular shift of sodium bicarbonate during correction of DKA.36 This acidosis usually has no adverse effect and is corrected spontaneously in the subsequent 24–48 hours through enhanced renal acid excretion.6,35,37
Vascular thrombosis
Many features of DKA and HHS predispose the patient to thrombosis: dehydration and contracted vascular volume, low cardiac output, increased blood viscosity and the frequent presence of underlying atherosclerosis.5,19 In addition, a number of hemostatic changes favour thrombosis.38 This complication is more likely to happen when osmolality is very high. Low-dose or low-molecular-weight heparin therapy should be considered for prophylaxis in patients at high risk of thrombosis. However, there are no data demonstrating its safety or efficacy.
Hypoglycemia and hypokalemia
These complications are less common with current low-dose insulin therapy.4,27,28 The potassium deficit should be adequately corrected and 5% dextrose should be added to infusion fluids as soon as the plasma glucose level decreases below 12–14 mmol/L.
Conclusion
Much remains to be done to lower the incidence of DKA and HHS and to improve the outcome of patients with these conditions. Although it has been suggested that the rate of death associated with these complications is decreasing, the rate is still excessive.39 The various factors that can precipitate hyperglycemic decompensation in patients with diabetes should alert the physician to early diagnosis and prompt therapy.
Footnotes
This article has been peer reviewed.
Contributors: Dr. Chiasson was responsible for drafting the article. Drs. Aris-Jilwan, Bélanger, Bertrand, Beauregard, Ékoé, Fournier and Havrankova contributed substantially to the concept of the paper and critical revisions. All authors approved the final version.
Competing interests: None declared.
Correspondence to: Dr. Jean-Louis Chiasson, Research Centre, Centre hospitalier de l'Université de Montréal, Rm. 8-202, Masson Pavilion, 3850 St-Urbain St., Montreal QC H2W 1T7; fax 514 412-7208; jean.louis.chiasson@umontreal.ca
References
- 1.Ennis ED, Stahl E, Kreisberg RA. The hyperosmolar hyperglycemic syndrome. Diabetes Rev 1994;2:115-26.
- 2.Fishbein H, Palumbo PJ. Acute metabolic complications in diabetes. In: National Diabetes Data Group. Diabetes in America. Bethesda (MD): National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 1995. p. 283-91.
- 3.Choi BC, Shi F. Risk factors for diabetes mellitus by age and sex: results of the National Population Health Survey. Diabetologia 2001;44(10):1221-31. [DOI] [PubMed]
- 4.Kitabchi AE, Wall BM. Diabetic ketoacidosis. Med Clin North Am 1995;79:9-37. [DOI] [PubMed]
- 5.Foster DW, McGarry JD. The metabolic derangements and treatment of diabetic ketoacidosis. N Engl J Med 1983;309:159-69. [DOI] [PubMed]
- 6.Adrogue HJ, Eknoyan G, Suki WK. Diabetic ketoacidosis: role of the kidney in the acid-base homeostasis re-evaluated. Kidney Int 1984;25:591-8. [DOI] [PubMed]
- 7.Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes mellitus. A fundamental and clinical text. Philadelphia: Lippincott Williams & Wilkins; 2000. p. 336-47.
- 8.Halperin ML, Marsden PA, Singer GG, West ML. Can marked hyperglycemia occur without ketosis? Clin Invest Med 1985;8:253-6. [PubMed]
- 9.Adrogue HJ, Wilson H, Boyd AE III, Suki WN, Eknoyan G. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med 1982;307:1603-10. [DOI] [PubMed]
- 10.Hillman K. Fluid resuscitation in diabetic emergencies — a reappraisal. Intensive Care Med 1987;13:4-8. [DOI] [PubMed]
- 11.Kitabchi AE, Fisher JN, Murphy MB, et al. Diabetic ketoacidosis and hyperglycemic, hyperosmolar nonketotic state. In: Kahn CR, Weir GC, editors. Joslin's diabetes mellitus textbook. Philadelphia: Lea & Febiger; 1993. p. 753-60.
- 12.Kreisberg RA. Diabetic ketoacidosis. In: Rifkin M, Porte D, editors. Diabetes mellitus: theory and practice. New York: Elsevier Science; 1990. p. 591-603.
- 13.Umpierrez GE, Khajavi M, Kitabchi AE. Review: diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Am J Med Sci 1996;311: 225-33. [DOI] [PubMed]
- 14.Hennis ED, Stahl E, Kreisberg RA. The hyperosmolar hyperglycemic syndrome. Diabetes Rev 1994;2:115.
- 15.Gonzalez-Campoy JM, Robertson RP. Diabetic ketoacidosis and hyperosmolar nonketotic state: gaining control over extreme hyperglycemic complications. Postgrad Med 1996;99:143-52. [PubMed]
- 16.Braaten JT. Hyperosmolar nonketotic diabetic coma: diagnosis and management. Geriatrics 1987;42:83-92. [PubMed]
- 17.American Diabetes Association. Hospital admission guidelines for diabetes mellitus. Diabetes Care 2002;25(Suppl 1):S109. [PubMed]
- 18.Normal laboratory values. In: Beers MH, Berkow R, editors. The Merck manual of diagnosis and therapy. 17th ed. Whitehouse Station (NJ): Merck & Co; 1999. Available: www.merck.com/pubs/mmanual/section21/chapter296/296a.htm (accessed 2003 Feb 28).
- 19.Lorber D. Nonketotic hypertonicity in diabetes mellitus. Med Clin North Am 1995;79:39-52. [DOI] [PubMed]
- 20.Brandenburg MA, Dire DJ. Comparison of arterial and venous blood gas values in the initial emergency department evaluation of patients with diabetic ketoacidosis. Ann Emerg Med 1998;31:459-65. [DOI] [PubMed]
- 21.Wachtel TJ, Tetu-Mouradjian LM, Goldman DL, Ellis SE, O'Sullivan PS. Hyperosmolarity and acidosis in diabetes mellitus: a three-year experience in Rhode Island. J Gen InternMed 1991;6:495-502. [DOI] [PubMed]
- 22.Wiggam MI, O'Kane MJ, Harper R, Atkinson AB, Hadden DR, Trimble ER, et al. Treatment of diabetic ketoacidosis using normalization of blood 3-hydroxybutyrate concentration as the endpoint of emergency management. Diabetes Care 1997;20:1347-52. [DOI] [PubMed]
- 23.Kitabchi AE, Umpierrez GE, Murphy MB, Barrett EJ, Kreisberg RA, Malone JI, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care 2001;24:131-53. [DOI] [PubMed]
- 24.American Diabetes Association. Hyperglycemic crises in patients with diabetes mellitus. Diabetes Care 2003;26(Suppl 1):S109-17. [DOI] [PubMed]
- 25.Marshall SM, Walker M, Alberti KGM. Diabetic ketoacidosis and hyperglycemic non-ketotic coma. In: Alberti KGM, Zimmet P, DeFronzo RA, editors. International textbook of diabetes mellitus. New York: John Wiley; 1997. p. 1215-29.
- 26.Ennis ED, Stahl EJ, Kreisberg RA. Diabetic ketoacidosis. In: Porte D Jr, Sherwin RS, editors. Diabetes mellitus: theory and practice. Amsterdam: Elsevier; 1997. p. 827-44.
- 27.Fleckman AM. Diabetic ketoacidosis. Endocrinol Metab Clin North Am 1993; 22:181-207. [PubMed]
- 28.Kitabchi AE. Low-dose insulin therapy in diabetic ketoacidosis: Fact or fiction? Diabetes Metab Rev 1989;5:337-63. [DOI] [PubMed]
- 29.Burghen GA, Etteldorf JN, Fisher JN, Kitabchi AQ. Comparison of high-dose and low-dose insulin by continuous intravenous infusion in the treatment of diabetic ketoacidosis in children. Diabetes Care 1980;3:15-20. [DOI] [PubMed]
- 30.Fulop M, Murthy V, Michilli A, Nalamati J, Qian Q, Saitowitz A. Serum beta-hydroxybutyrate measurement in patients with uncontrolled diabetes mellitus. Arch Intern Med 1999;159:381-4. [DOI] [PubMed]
- 31.Barnes HV, Cohen RD, Kitabchi AE, Murphy MB. When is bicarbonate appropriate in treating metabolic acidosis including diabetic ketoacidosis? In: Gitnick G, Barnes HV, Duffy TP, et al, editors. Debates in medicine. Chicago: Yearbook; 1990. p. 172.
- 32.Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med 1986;105:836-40. [DOI] [PubMed]
- 33.Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab 1983;57:177-80. [DOI] [PubMed]
- 34.Harris GD, Fiordalisi I, Harris WL, Mosovich LL, Finberg L. Minimizing the risk of brain herniation during treatment of diabetic ketoacidemia: a retrospective and prospective study. J Pediatr 1990;117:22-31. [DOI] [PubMed]
- 35.Oh MS, Carroll HJ, Uribarri J. Mechanism of normochloremic and hyperchloremic acidosis in diabetic ketoacidosis. Nephron 1990;54:1-6. [DOI] [PubMed]
- 36.Madias NE, Homer SM, Johns CA, Cohen JJ. Hypochloremia as a consequence of anion gap metabolic acidosis. J Lab Clin Med 1984;104:15-23. [PubMed]
- 37.Oh MS, Banerji MA, Carroll HJ. The mechanism of hyperchloremic acidosis during the recovery phase of diabetic ketoacidosis. Diabetes 1981;30:310-3. [DOI] [PubMed]
- 38.Paton RC. Haemostatic changes in diabetic coma. Diabetologia 1981;21:172-7. [DOI] [PubMed]
- 39.Acute complications. In: Diabetes 1996 vital statistics. Alexandria (VA): American Diabetes Association; 1996. p. 29-44.