

Abstract
Retroviral recombinants result from template switching between copackaged viral genomes. Here, marker reassortment between coexpressed vectors was measured during single replication cycles, and human immunodeficiency virus type 1 (HIV-1) recombination was observed six- to sevenfold more frequently than murine leukemia virus (MLV) recombination. Template switching was also assayed by using transduction-type vectors in which donor and acceptor template regions were joined covalently. In this situation, where RNA copackaging could not vary, MLV and HIV-1 template switching rates were indistinguishable. These findings argue that MLV's lower intermolecular recombination frequency does not reflect enzymological differences. Instead, these data suggest that recombination rates differ because coexpressed MLV RNAs are less accessible to the recombination machinery than are coexpressed HIV RNAs. This hypothesis provides a plausible explanation for why most gammaretrovirus recombinants, although relatively rare, display evidence of multiple nonselected crossovers. By implying that recombinogenic template switching occurs roughly four times on average during the synthesis of every MLV or HIV-1 DNA, these results suggest that virtually all products of retroviral replication are biochemical recombinants.
Genetic recombination contributes significantly to the genetic variability observed in populations of retroviruses such as human immunodeficiency virus type 1 (HIV-1) and Moloney murine leukemia virus (MLV) (6, 25, 28, 35). Retroviral recombination occurs shortly after cell infection and results from template switching between RNA or DNA replication intermediates during reverse transcription (17, 25, 50). One factor that contributes to the frequency of recombination is retroviruses' property of copackaging two intact genomes, which provides unusual access to a genetic recombination substrate (28). Another contributing factor is reverse transcriptase's propensity to perform recombinogenic switching, which may reflect selective pressure to maintain a polymerase capable of performing the strong-stop template switches that are required during retroviral DNA synthesis (8, 48).
How frequently retroviral recombination occurs in replicating virus populations is difficult to assess due to the influence of selective pressures. Recombination frequencies can be examined with single-cycle recombination assays that monitor marker cosegregation during individual rounds of DNA synthesis (51). Such assays have yielded estimated spleen necrosis virus (SNV) recombination frequencies of about one recombination event during the synthesis of every three to seven viral DNAs (18, 19, 22), and similar rates have been reported for the related gammaretrovirus MLV (4). Surprisingly, recombination occurs one to three times on average during the synthesis of every HIV-1 DNA (21, 43), a rate which is perhaps 10-fold higher than calculated SNV or MLV rates.
What accounts for this dramatic difference between HIV-1 and gammaretrovirus recombination rates? One possibility is that retroviruses' reverse transcriptases may possess different enzymatic properties and that template switching rates vary among viruses (21). Possible support for this notion comes from the observation that although average missense mutation rates for HIV-1 and gammaretroviruses appear to be similar, retroviruses differ in how frequently they generate errors in response to specific template features (2, 38).
Alternatively, the untested assumption that gammaretroviruses and HIV-1 both randomly select coexpressed RNAs for copackaging may be incorrect. Reported recombination frequencies have been calculated by dividing the frequency of scorable recombinants by the “recombining population” size (18). The size of this population, which consists of virions that contain two different RNAs, has not been measured directly but has been inferred by assuming random cosegregation of coexpressed RNAs into nascent virions (18, 52). Models for random copackaging of genomic RNAs are based on the assumptions that among unspliced viral RNAs that reach the cytoplasm, some will become mRNAs and others will become genomes and that RNA copackaging results from random pairing among packaging-competent cytoplasmic RNAs. However, there is strong evidence that gammaretrovirus unspliced RNAs reside in two functionally separate pools (11, 26). Additionally, how retroviruses export unspliced RNAs to the cytoplasm, how RNAs are partitioned between progeny genome and mRNA functions, and what intracellular processes precede dimeric RNA packaging all appear to differ among retroviruses (5, 7, 9, 11, 12, 26, 41). If the randomness of RNA copackaging also varied among retroviruses, then species-specific differences in genetic marker reassortment might reflect differences in the fractions of virions that had copackaged two different RNAs rather than differences in template switching rates. This hypothesis suggests that recombinogenic template switching may occur more frequently during the replication of certain retroviruses than is evident from an examination of their reverse transcription products. Furthermore, the frequencies of genetic recombination (evident from the reassortment of genetic markers) and of biochemical recombination (the occurrence of recombinogenic template switches) might be more similar for some retroviruses than for others.
Here, the frequencies of marker reassortment for HIV-1 and MLV were compared directly, using the same producer and recipient cell types for each virus, virus-specific replication proteins, and recombination assay vectors that differed only in virus-specific cis-acting sequences. For both HIV-1 and MLV, the frequencies of recombination between two different coexpressed RNAs were compared to template switching rates observed when donor and acceptor sequences were parts of a single RNA and thus uniformly copackaged.
MATERIALS AND METHODS
Plasmids and cells.
The helper function plasmids used were pCMVΔR8.2 (30), which encodes all HIV-1 trans-acting proteins including Vpu but not envelope, and pMLV Ψ− for MLV (34). In control experiments to test whether or not transfection-related recombination contributed to results, the mammalian lacZ expression plasmid pCH110 (15) was used.
One recombination assay vector was pHR′CMVlacZ (29). The other vectors were derivatives of the HIV-1 vector pHIVlac (2) or the MLV vector pLacPuro (34), which have previously been described. The previously described (2) pHIVlac contains a cytomegalovirus promoter-driven lacZ gene followed by a simian virus 40 (SV40) early promoter and puromycin resistance gene in an HIV-1-based retroviral vector. pLacPuro is an MLV-based vector with the same structure as pHIVlac, except that lacZ expression is driven by the MLV long terminal repeat (LTR) and the HIV-1 cis-acting sequences necessary for genome metabolism are replaced with the cognate sequences from MLV. To reflect the relationships among vectors, pHR′CMVlacZ (29) is referred to as HIV Lac in this report, and pHIVlac (2) is called HIV LacPuro. MLV L∧cPuro and HIV L∧cPuro were derivatives of pLacPuro and pHIVlac, respectively, and contain a 117-bp deletion between the EcoRV and SspI sites in lacZ. The entire puromycin expression cassette was removed from pLacPuro to make MLV Lac. To make MLV L∧cPuro-ac and HIV L∧cPuro-ac, MLV and HIV L∧cPuro were modified in their downstream LTRs and flanking regions by deletion of polyadenylation sequences in R. For both viruses, this was achieved by truncating the downstream R at a restriction site (SacI for HIV and Asp718 for MLV) upstream of the polyadenylation addition signal and about 40 nucleotides downstream of the U3/R junction. A 2.6-kbp FspI-BsiWI fragment of lacZ, flanked on both ends with polylinker and adjacent sequences derived from passage through Litmus 38 (New England Biolabs), was appended downstream of the polyadenylation-inactivated R regions. These fragments of lacZ were flanked at their 3′ ends with SV40 polyadenylation sequences derived from pRep8 (Invitrogen). The repeat deletion vectors were derivatives of pHIVlac that was first engineered to contain EcoRI and XhoI restriction sites flanking the 156-bp coding segment that was mutagenized (1). The degenerate replacement fragment was synthesized by using two lengthy 3′-terminal complementary oligonucleotides, which were filled out with Klenow and digested with XhoI and EcoRI. The tandem repeat vectors contained a 1.6-kb XhoI-to-MfeI linker fragment derived from MLV coding sequences between the two coding repeats; spacers have previously been implicated as increasing repeat deletion without altering high negative interference for simple retroviruses (10). Construction details for all plasmids are available upon request.
ET cells, which are a 293T derivative that constitutively express ecotropic envelope (34), were transfected to produce virus. mATRC1/293 cells, which are derived from human 293 cells and stably express ecotropic receptor (27), were used in infections. These and derivative cell lines were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gemini). NIH 3T3 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% calf serum (Gibco).
Transfection and virus harvesting.
Vector-containing virions were generated by transiently cotransfecting ET cells with pCMVΔR8.2 (30) or pMLV Ψ− (34) helper function plasmid and the appropriate assay vector plasmid(s) by calcium phosphate precipitation (40). When two vectors were cotransfected, similar molar equivalents of each plasmid DNA were used, with a total of 8 μg of DNA per 6-cm-diameter plate. As a result, all virions, whether HIV-1 or MLV derived, possessed ecotropic envelope and could infect cells that express ecotropic receptor. Virus was harvested by replacing the culture medium at 16 to 24 h after calcium phosphate exposure and collecting medium at 36 to 48 h posttransfection. Residual cells were removed by filtration. Virus was stored at −70°C prior to use.
Infection and functional assay for LacZ activity.
mATRC1/293 cells and NIH 3T3 cells in 6-cm-diameter dishes were infected for 1 h in the presence of 8 μg of hexadimethrine bromide (Polybrene; Sigma) per ml at low multiplicities of infection (<0.01). Puromycin (Sigma) selection, cell fixation, and 5-bromo-4-chloro-β-d-galactopyranoside (X-Gal) (Sigma) staining were performed as previously reported (34).
Southern blotting.
At least 200 independent puromycin-resistant colonies were pooled for each vector, and genomic DNA was isolated by using the Wizard genomic DNA purification kit (Promega), DNA was digested with ClaI and MluI, separated on an agarose gel, blotted, and probed with a random-primed radiolabeled probe generated from a PCR fragment that hybridized to both the lengthy (5.2-kb) and the shorter (478- and 316-bp) bands visible in the blot, using standard approaches (33). Note that the length of probe that hybridized to the 316-bp band was the same as that which hybridized to the 478-bp band.
Virus RNA quantification.
Virion RNA was purified from filtered culture medium from transfected cells as previously described (42) and used in RNase protection assays. Vector RNAs were differentiated in RNase protection assays, and their ratios were quantified with a PhosphorImager (Molecular Dynamics). To allow differentiation of both vectors with a single probe, the probe was complementary to the regions between ClaI and SspI in lacZ and thus included sequences complementary to the region deleted in pL∧cPuro and related vectors. To quantify vector RNAs, RNase protection assay background values were subtracted and molarities were normalized by dividing protected product values by the number of radiolabeled nucleoside monophosphate (CMP) residues in each.
Calculation of the recombining population size and number of crossovers for two-vector recombination assays.
To calculate the proportion of virions predicted to contain one each of coexpressed vector RNAs, the equation D2 + 2DA + A2 = 1, which assumes random copackaging, was used as previously described (19). D stands for the molar proportion of total virion RNA comprised of the donor lacZ vector (MLV or HIV Lac) as determined by RNase protection assay, and A stands for the corresponding amount of puromycin resistance-conferring acceptor vector (MLV or HIV L∧cPuro). Only one of each heterozygous RNA pair contained a puromycin resistance cassette. Thus, assuming that whether a product DNA contained 3′ end sequences from the puromycin vector or from the other vector was random, the portion of heterozygous virions capable of generating puromycin colonies was half of 2DA. DA + A2 represented the total number of puromycin resistant colonies. One half the 2DA term was the recombining population, whose portion of the total puromycin resistant colony population could be calculated from DA/DA + A2, which equals D, the molar proportion of total virion RNA that was the donor.
Generating LacZ-positive puromycin-resistant proviruses required at least one recombination event in the recombination target region, which was the 1.8-kb 3′ portion of lacZ. For the calculations in Fig. 3C, the observed frequency of blue colonies (25.7%) for the HIV-1-based vectors was divided by 0.94 to correct for colonies that would have scored blue but became white due to lacZ mutational inactivation (in this case, 27.4%). The inactivation frequency was from control data in experiments here (not shown) and reported in reference 2. Based on donor and acceptor RNA proportions, the recombining population for this example was calculated to be about 57%. The corrected frequency of blue colonies per recombining population (47.4%) reflected the percentage of single plus other odd-numbered crossovers in the target region minus the percentages of double and other even-numbered crossovers in this region, since the latter did not contribute to the blue colony population because colonies would be white. Thus, the following calculation was used to determine crossover frequencies as in Fig. 3: 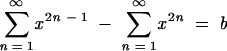 , where b is the corrected blue colony frequency (47.4% in the example in Fig. 3). In the case of the HIV-1 vector example in Fig. 3, this yielded a target region crossover frequency (X) of roughly 0.86. Note, however, that as a result of the calculation methods used, values close to one become somewhat inaccurate and thus these values are approximations.
, where b is the corrected blue colony frequency (47.4% in the example in Fig. 3). In the case of the HIV-1 vector example in Fig. 3, this yielded a target region crossover frequency (X) of roughly 0.86. Note, however, that as a result of the calculation methods used, values close to one become somewhat inaccurate and thus these values are approximations.
FIG. 3.

RNase protection assay to quantify virion RNAs. Positions corresponding to the mobilities of undigested probe, probe protecting a fragment of undisrupted lacZ, and probe protecting a fragment of the disrupted lacZ allele are indicated by probe, lac, and l∧c, respectively, at the left. (A) RNase protection assay of virion RNAs harvested from ET producer cells that had been cotransfected with the indicated HIV-1 based vector, the HIV-1 virion protein helper construct (pCMVΔR8.2), and/or control plasmids. Lane 1, undigested RNA probe used for both panels. Lane 2, digested products of virion RNA from cells cotransfected with the HIV-1 helper plasmid and HIV LacPuro. Lane 3, products from cells with helper, HIV L∧cPuro, and the nonretroviral lacZ mammalian expression plasmid pCH110 (15). Lane 4, helper, HIV L∧c, Puro, and HIV Lac. Lane 5, helper and HIV L∧cPuro-ac. Lane 6, helper alone; lane 7, helper and pCH110. (B) MLV-based vector products analyzed by RNase protection assay. The same probe as in panel A was used. Lane 1, virion RNA from cells cotransfected with the MLV helper pMLV Ψ−, MLV Lac, and the nonretroviral lacZ mammalian expression plasmid pCH110 (15). Lane 2, helper, MLV L∧cPuro, and pCH110; lane 3, helper, MLV L∧cPuro, and MLV Lac; lane 4, helper and MLV L∧cPuro-ac; lane 5, helper and MLV L∧cPuro; lane 6, helper alone; lane 7, size standards. (C) Comparison of crossover frequencies for MLV- and HIV-1-based vectors. The values presented are from two independent experiments for each virus and were calculated as described in the text.
Genome-wide recombination frequency calculations.
The number of recombination events per genome were calculated based on transduction-type recombination assays. Assumptions included the following. (i) Two template switching events were required to generate functional lacZ. (ii) The region between FspI and BsiW1 of the acceptor template (2,633 bases of the sequence between the LTRs) was the recombination target. (iii) Native HIV-1 genomes are ∼3.5 times this length, and switching could occur at similar frequencies per distance for all genome segments. (iv) Some lacZ genes that would have been functional were inactivated by mutation (2). (v) Only half of the double crossovers were detected, because whether donor or acceptor RNA served as a template for the deletion region was random. (vi) The frequency of each recombination event was independent of others. (vii) The inability to score abortive products that would have resulted from a switch to the 3′ donor and failure to switch back before reaching acceptor noncomplementary sequences between R and the internal lacZ fragment would not appreciably affect values, nor would omission of calculated crossovers at frequencies other than two. (viii) Crossovers between the two RNAs that involved only sequences between the two Rs of each RNA, which were not detectable because they did not change the lacZ allele, did not occur at a significant frequency.
With the use of these assumptions to calculate recombination frequencies with transduction-type assay values, the average frequency of blue colony formation (36.4%, using the MLV values) was divided by 0.92 to correct for lacZ inactivation by the MLV replication machinery as assayed in mATRC1/293 cells (2) and was doubled to account for the possibility of either allele in the deletion region (yielding 0.79). Because this is the frequency of two crossovers in 2.6 kb and each is assumed to be independent, the frequency of one crossover in this interval is the square root of that value (0.89). This value is then multiplied by 3.5 to account for genome length, to yield a final value of 3.1 crossovers per genome per replication cycle. Note, however, that we believe that some of these parameters, most notably assumptions vii and viii, are most likely incorrect. Because these events probably did occur and did reduce apparent recombination rates, we believe that these calculations likely underestimate template switching frequencies.
Recombination frequency calculations based on HIV-1 two-vector assays.
RNA copackaging was assumed to be random. The averaged HIV-1 crossover frequency value of 0.85 for the 1.8-kb interval can be multiplied by 5.1 to account for the whole genome length, to yield a genome-wide frequency of approximately 4.3 crossovers per replication cycle.
Calculation of predicted deletion frequencies for reciprocal repeat vectors.
Assuming equal expression levels when reciprocal vectors were coexpressed and random copackaging of RNAs, the recombining population in these experiments would be half of the total population. For each vector (LaacPuro and LaacPuro), any attempt at intramolecular repeat deletion would be unsuccessful (1). The assumption was made that for reasons such as the limited time spent copying the downstream repeat, the replication machinery would have an opportunity to consider only one potential acceptor region. If the replication machinery was equally likely to seek an acceptor template region on the same vector as on the copackaged vector, then only the half that sought an acceptor in the copackaged vector would have the potential of generating a deletion product. Therefore, the predicted repeat deletion frequency for coexpression of two reciprocal vectors would be half of one half, or 25%, of the frequency of single, identical repeat vector deletion.
RESULTS
Genetic recombination frequencies for HIV-1 and for MLV.
The evidence that HIV-1 recombines more frequently than gammaretroviruses is derived from a comparison of reports about MLV and SNV to results for HIV-1 (4, 18, 19, 21, 22, 43, 52, 53). However, each of these previous studies involved a single type of retrovirus, and because the work was performed in several separate laboratories which each used different assay vectors, it remained possible that some components of the reported differences were assay specific or due to variation in experimental approaches among researchers. Thus, our initial studies were aimed at determining whether or not HIV-1's higher rate of recombination would remain evident when both viruses were assayed under parallel conditions.
Virus-specific recombination assay vectors were developed to allow a direct comparison of recombination frequencies during HIV-1 replication to those for MLV. Two different vectors, each derived from previously described LacPuro vectors (Fig. 1A) (2, 34) were used in each virus system (Fig. 1B and C). The first contained a 117-base coding region deletion within a lacZ expression cassette, followed by a puromycin resistance cassette (MLV L∧cPuro and HIV L∧cPuro for MLV and HIV, respectively [Fig. 1B]). The second vector contained intact lacZ alone (MLV Lac or HIV Lac [Fig. 1C]). The rationale for using these vectors was as follows. Reverse transcription of the first vector would yield a puromycin-resistant provirus that did not express functional β-galactosidase; the second vector alone would yield a lacZ provirus that did not confer puromycin resistance. During reverse transcription of virions that had copackaged one of each vector RNA, template switching within regions of sequence identity could yield recombinant viral DNAs, some of which would contain functional versions of both lacZ and puromycin resistance genes (Fig. 2A). The frequency of such recombinants could be scored as that fraction of the total puromycin resistant colonies that stained blue when incubated with X-Gal. Because they provided the scored genetic material, MLV Lac and HIV Lac were referred to as the donor template vectors; L∧cPuro vectors with disrupted lacZ sequences were the acceptor vectors. This system allowed us to compare recombination rates for HIV-1 and MLV by using the same producer and recipient cell types for each virus, virus-specific replication proteins, and recombination assay vectors that differed only in virus-specific cis-acting sequences such as those in the LTRs and packaging regions.
FIG. 1.

Inter- and intramolecular recombination assay vectors. The vectors as drawn were for MLV. Shaded boxes represent intact functional lacZ genes. PSV40, SV40 promoter; puroR, gene that confers puromycin resistance. Positions of restriction sites used in construction and analysis are indicated. MLV-based vectors MLV LacPuro (A), MLV L∧cPuro (B), MLV Lac (C), and MLV L∧cPuro-ac (D) are indicated at the left, with the name of the corresponding HIV-1-based vector in parentheses. The regions containing virus-specific cis-acting sequences, which differ between the vectors for MLV and those for HIV-1, are indicated by hatched bars over the diagrams. Note that the vector called HIV Lac here is pHR′CMVlacZ (29). The bar marked probe indicates the location of sequences complementary to the RNA probe used for the RNase protection assays presented in Fig. 3.
FIG. 2.

Two-vector (intermolecular) genetic recombination assay. (A) Schematic representation of how template switching during reverse transcription could yield a puromycin-resistant vector with functional lacZ. The line with the arrowhead represents minus-strand DNA synthesis after minus-strand strong-stop transfer (47). (B) Comparison of MLV- and HIV-1-based two-vector assay results, presented as blue colonies per total puromycin-resistant colonies. Hatched boxes, values for MLV vectors in human cells; open boxes, HIV-1 results; black boxes, values for MLV vectors assayed in murine cells. All values shown are results from four separate cotransfection and infection experiments, and at least 400 puromycin-resistant colonies were tallied to yield each value. Note that although the four MLV vector titers from murine cells are grouped with data for both MLV and HIV in human cells, the murine cell data shown were collected from experiments performed after the human cell experiments. The data shown for human cells are from the same experiments as those used to generate the data in Fig. 3. Although they were performed later, the reported murine cell experiments were controlled with and normalized to human cell experiments that were performed in parallel.
To perform these experiments, producer cells were cotransfected with helper function plasmids and approximately equimolar amounts of plasmids encoding the two recombination vectors. Fresh cells were infected, and puromycin-resistant colonies were selected and stained with X-Gal. Results from four independent cotransfection experiments for each virus followed by infection experiments that generated more than 200 colonies on each of two or three plates for each virus stock are shown in Fig. 2B. In each experiment, four- to sevenfold more of the total puromycin-resistant colonies stained blue (indicative of recombination) for the HIV-1-based vectors than for the MLV products.
To more accurately quantify recombination rates, the portion of colonies produced by virions predicted to contain two different RNAs was calculated by first determining virion RNA ratios and then using these ratios to deduce the size of the heterozygous virion recombining population. RNA ratios within the population of virion RNA were determined by RNase protection assays, using RNA prepared from aliquots of the same virion stocks used in the infection experiments described above (Fig. 3A and B). The crossover frequency in the assayed 1.8-kb interval (Fig. 3C) was calculated by using observed blue colony frequencies and virion RNA ratios as follows. First, the recombining population, which was the proportion of virions predicted to contain one of each vector, was calculated with the assumption of random RNA copackaging (see Materials and Methods). Next, the fraction of this recombining population that generated functional lacZ was calculated. Finally, values were adjusted to reflect the fact that puromycin-resistant products with odd numbers of crossovers in the assayed interval would yield blue colonies, while those with even numbers of crossovers would yield white colonies (see Materials and Methods). The results are summarized in Fig. 3C. Comparing the averages of the values calculated for two independent assays for MLV to those for HIV-1 suggested that the total number of crossovers per presumptive recombining population member for HIV-1 was roughly 6.3-fold higher than that for MLV.
Several controls specific to each set of experiments were performed to address the possibility that recombination of plasmid DNA during transfection, which is known to occur and which might generate products that inaccurately resembled virus replication-associated recombinants (31, 39), might contribute to the frequencies of recombination reported in this paper. The first of these, which was performed as a parallel control for every repetition of every transfection experiment reported in this paper, was to cotransfect assay vectors with a large molar excess of the nonretroviral mammalian lacZ expression plasmid pCH110 (15). All assays described here score retroviral recombination by screening for LacZ function. Thus, the rationale for this control was that although pCH110 RNAs are Ψ− and should not be encapsidated, the extended region of homology to vector genes suggests that pCH110 plasmid DNA should be at least as capable of generating plasmid DNA-level recombinants as any of the vector plasmids utilized. Virions harvested from these cotransfections were used to infect fresh cells, and puromycin resistant colonies were screened for β-galactosidase activity. Although blue-staining colonies were detectable in some control repetitions, these arose at a frequency of significantly less than 1% of the white colony titer, which suggests that DNA-level recombination did not affect outcomes measurably.
Two additional controls also addressed the potential impact of DNA-level recombination on this assay's results. If plasmid DNAs had recombined during transfection to generate a transcription template for a product that would erroneously score as a retroviral recombinant, a recombinant RNA in virions would result. One control that suggests that such products did not arise at a frequency high enough to affect assay outcomes is shown in Fig. 3A, lane 3. If DNA-level recombination had generated recombinants to a detectable level, this RNase protection assay lane would contain a protected product with the mobility indicated by “lac” at the panel's left. However, this autoradiogram and darker exposures (not shown) indicate clearly that if any such product existed, it was present at a level significantly lower than 1% of the nonrecombinant product with mobility indicated by “l∧c” in Fig. 3A, lane 3.
In a separate test for preexisting recombinant RNAs in virions, we examined the structures of the RNAs in the virions used to test intermolecular recombination. This involved RNase protection assays with a lengthy (∼3.0-kb) riboprobe that was predicted to protect different-length products for each vector (2.22 kb for L∧cPuro, 2.27 kb for Lac, and 2.62 kb for any recombinants or for LacPuro). We consistently saw protected products of the correct size for the L∧cPuro-, Lac-, and LacPuro-alone control virions. We consistently did not detect any recombinant RNAs (e.g., recombinant-sized product was not visible and appeared to be more than 10-fold less abundant than parental vector products) within virions from producer cells cotransfected with pLac and pL∧cPuro. Instead, a prominent single band containing the closely migrating products for the two parental vectors was observed. Results from these assays are not shown because they suffered from numerous additional bands that appeared to be background products, since they, and not the vector-specific bands, were present in lanes with digested probe alone. Nonetheless, because recombinants that arose at the frequency calculated for HIV should have been clearly detectable in these assays, these results provide a third source of information suggesting that the majority of observed recombinant products cannot be attributed to plasmid-level recombination. However, these findings cannot rule out the possibility that some DNA-level recombination occurred during transfection.
Other controls were performed to verify the differences between HIV-1 and MLV intermolecular recombination frequencies. To address whether or not the lower frequency of recombination observed for MLV could be attributed to the lack of host species-specific factors in human cells, MLV recombination frequencies were measured in murine cells as well as in human cells. As in the human cells, the blue/white colony ratios observed in murine cells were low, thus confirming that the higher recombination rate of HIV was not due to host effects on MLV recombination (2B)> . To address the possibility that recombination was as high for MLV as it was for HIV but that most colonies with MLV recombination products aberrantly remained unstained, the structures of pooled MLV reverse transcription products were examined by Southern blotting (Fig. 4). The absence of a detectable recombination-diagnostic Lac band among products of virions from vector-coproducing cells, whereas parental L∧cPuro vector product was clearly visible (lane 4), is consistent with expectations if MLV recombination was much lower than that of HIV.
FIG. 4.

Southern blot of pooled integrated MLV vector products. Genomic DNA was collected from pooled MLV provirus-containing colonies for each of the vectors or vector pairs indicated at the top of the autoradiogram. This DNA was digested with ClaI and MluI, which produced a 5.6-kb band for all vectors as well as a shorter band diagnostic of either the intact (478 bp from Lac) or the corresponding deleted (316 bp from L∧c) ClaI-to-MluI region of lacZ. Lengths of predicted products are indicated at the left. The products in lane 3 are from transduction-type vectors used in the experiments described in Fig. 5.
Template switching independent of copackaging variables.
The results described above confirmed that marker segregation was more frequent for HIV-1 than for MLV, but the calculated recombination frequencies relied heavily on the assumption that RNA copackaging was random. Thus, a second assay, which relied on a single vector with both the disrupted lacZ acceptor and the source of recombinational repair on a single RNA, was developed (Fig. 1D and Fig. 5). Every virion that packaged this single vector RNA copackaged template switch donor and acceptor sequences in equal amounts.
FIG. 5.

One-vector transduction-style recombination assay. (A) Transduction-patch repair process that would generate recombination-repaired lacZ (shaded box) during reverse transcription of L∧cPuro-ac transduction-type vectors (see Fig. 1D). Only those steps of minus-strand synthesis that follow a minus-strand strong-stop template switch are shown. (B) Comparison of MLV- and HIV-1-based transduction-type assay results, presented as blue colonies per total puromycin-resistant colonies. Each value shown is the result of a separate transfection and infection experiment, and at least 400 puromycin-resistant colonies were tallied to yield each value. Shaded boxes, values for MLV; hatched boxes, HIV-1 results; black boxes, results for the HIV double-poly(A) transduction vector, as described in the text.
The vector design was based on putative retroviral transduction intermediates (44). It has been postulated that retroviral transduction of host sequences results from a series of rare events that start when a lengthened RNA, which results from read-through of retroviral polyadenylation signals into adjacent cellular sequences, is packaged into virions. The cellular sequences in the 3′ ends of these RNAs are believed to subsequently become patched into the retroviral genome via rare nonhomologous recombination events (44, 55).
In transduction-type vectors used in this study, polyadenylation signals of MLV and HIV L∧cPuro (Fig. 1B) were removed to allow constitutive 3′ extension into an appended internal portion of the lacZ coding sequence (indicated by ΔpA in the L∧cPuro-ac vectors [Fig. 1D]). RNase protection assays confirmed that both disrupted sequences (from the body of the vectors) and nondisrupted sequences (from the region downstream of the inactivated polyadenylation signals) were present in equimolar amounts in virions that contained L∧cPuro-ac RNAs (Fig. 3A, lane 5, and B, lane 4). Despite the unusual structures of these vectors, they were not excessively lengthy: at ∼9.7 kb, the HIV-1-based transduction vector was only about 5% longer than native HIV-1, and the MLV version was actually shorter than MLV.
It has been demonstrated that reverse transcription of a polyadenylation read-through transcript can proceed normally despite the RNA's elongated 3′ end (45). Thus, our expectation was that reverse transcription of the transduction vectors ordinarily would involve copying only those sequences that were between the two R repeats. This would generate puromycin-resistant transductants with lacZ genes containing the engineered disruption. However, each encapsidated vector RNA would bring into virions a source of recombinational patch repair in its extended 3′ end. Thus, if two template switches occurred between the disrupted lacZ gene in a vector's body (the acceptor template region) and the fragment of lacZ at the 3′ end of either the same or the copackaged RNA (the recombination donor sequences), a functional lacZ gene could be generated (Fig. 5A).
Using these vectors, template switching frequencies for HIV-1 and MLV could be compared by a direct determination of the portion of puromycin-resistant colonies that stained blue with X-Gal. These experiments were performed by cotransfecting vector plasmid with the cognate helper plasmid into producer cells, infecting fresh target cells with the resulting virus, and determining ratios of X-Gal-stained to total (stained and unstained) puromycin-resistant colonies. The results of three independent transfection and infection experimental repetitions for each virus are shown in Fig. 5B. Surprisingly, blue colony formation was frequent: about 32% for HIV-1 and a similar average frequency of 36% for the MLV vector. The ratios of Lac to L∧c bands observed in the blot of transduction vector products (Fig. 4, lane 3) appear to be roughly consistent with these values.
Controls were performed in which a molar excess of pCH110, a nonretroviral mammalian lacZ expression plasmid (15), was cotransfected along with helper and the deletion-containing lacZ expression vectors that lacked 3′ extended regions (MLV L∧cPuro and HIV L∧cPuro [Fig. 1B]). The resulting virions generated no X-Gal-staining colonies (<0.01% blue colonies) for the HIV-1-based vector and very low levels of blue colonies (≤0.5%) for the MLV-based vector, thus suggesting that DNA-level recombination during transfection did not contribute appreciably to the observed findings. A second control provided additional support for the assertion that plasmid-level recombination did not cause transduction-related patch repair. For this control, a second HIV transduction-type vector was constructed, which contained both the native HIV polyadenylation signal and a second polyadenylation signal derived from SV40, in the downstream LTR between the vector body and the appended 3′ end lacZ sequences (not shown). Data on the fraction of puromycin-resistant products of this double-poly(A) HIV transduction-type vector that stained blue are presented in Fig. 5B. Because of the similarity of the plasmid encoding this vector to that for the assayed HIV L∧cPuro-ac vector, these values likely represent the maximum approximate contribution of plasmid DNA-level recombination to the reported transduction values.
Intermolecular recombination with reciprocal repeat vectors.
By demonstrating that recombination rates differed when acceptor and donor were on separate RNAs but were similar when acceptor and donor were covalently joined, the results described above suggested that what differed between HIV-1 and MLV was the accessibility of coexpressed acceptor and donor vector sequences to the viral recombination machinery. A prediction from this hypothesis was that if both switching between two RNAs and switching between two positions on a single RNA were possible, more intermolecular switching would be detectable for HIV-1 than for MLV. Here, this notion was tested with reciprocal repeat vectors. Repeated sequences are deleted frequently during reverse transcription, in a process presumed to be related to recombinogenic template switching (24, 37, 54). Because the coexpressed vectors used in this set of experiments were highly similar to one another, these experiments also addressed whether or not the differences in intermolecular template switching frequencies described above resulted because the coexpressed vectors differed significantly. As in the work described above, experiments to monitor possible DNA-level recombination by using the pCH110 control were included with these experiments, and the results suggested that DNA-level recombination did not significantly affect the results of the following assays.
Aided by the degeneracy of the genetic code, a lacZ substitution variant of HIV LacPuro (Fig. 1A) was constructed. This variant contained a 156-bp gene segment that was identical to the parental allele in coding capacity but that differed by 27% in nucleic acid sequence due to the substitution of about one nucleotide per codon throughout the 156-bp region. As assayed by the production of X-Gal-staining puromycin-resistant colonies, the degenerate gene segment served as well as the wild type as a functional part of lacZ (data not shown).
Coding-region-internal duplications were incorporated into lacZ to generate tandem repeat derivatives (Fig. 6). Repeat deletion during reverse transcription of these vectors was assayed by the restoration of lacZ function (34) (Fig. 6A). Vectors were generated that contained exact duplications of the wild-type or the degenerate gene segment (HIV LaacPuro and HIV LaacPuro; the mutant repeat allele is indicated by an italic underlined letter a in Fig. 6C). As observed previously, when repeats were identical to one another, repeat deletion occurred at similar frequencies regardless of whether the wild-type or the degenerate repeat was studied (Fig. 6C) (1). When the two different synonymous repeats were placed in tandem in a single vector, this introduced sufficient genetic distance between repeats that homologous recombination, as measured by repeat deletion, did not occur (HIV LaacPuro vector [Fig. 6C]). The reciprocal vector (HIV LaacPuro), which differed from HIV LaacPuro only in the orientation of the two repeats, also showed no evidence of deletion among more than 2,000 colonies examined. However, when HIV LaacPuro and HIV LaacPuro were coexpressed and cells were infected with the resulting virions, blue-staining colonies were observed (Fig. 6C). These findings suggest that the two different vector RNAs were copackaged and template switching occurred between them (Fig. 6B). The frequency with which reciprocal, intermolecular repeat-deletion recombinants arose was similar to values predicted by models in which repeat deletion between randomly copackaged RNAs and repeat deletion within a single RNA were equally likely (see Materials and Methods and Discussion).
FIG. 6.

Reciprocal repeat recombination assay. (A) Schematic representation of LaacPuro-type repeat deletion assay vector as described in the text. Template switching between repeated sequences would result in a single copy of the repeat, which would yield a colony that expresses functional β-galactosidase (functional gene indicated with shaded box). Backwards slashes and the underscore and italics indicate that the repeated region in this vector was engineered to differ 27% from the wild-type sequence (see text). (B) Schematic representation of template switching between copackaged RNAs that would result in the generation of a functional lacZ gene. The “a” box without backwards slashes is the wild-type sequence. (C) Observed frequencies of functional lacZ among vector products. Values presented are averages from three to five independent experimental repetitions for each vector. Vector designations reflect the repeats in vectors with the structures shown in panels A and B. For the first vector, both repeats contained the parental wild-type (wt) lacZ sequence. The repeat vector for which both repeats contained silent mutations that resulted in a 27% difference from the parental allele was designated LaacPuro. Where LaacPuro plus LaacPuro is indicated, the value presented was for products of virions that resulted from cells cotransfected with helper and both of these vector plasmids. The values presented for both LaacPuro and LaacPuro alone indicate that no blue colonies were observed for either virus with any of these vectors in any experimental repetitions, and thus there are no differences among these; the values as presented differ slightly due to the different numbers of colonies scored.
These repeats were also incorporated into MLV-based vectors (Fig. 6C). As was the case for HIV-1 based vectors, identical repeats were efficiently deleted. Also similar to results with HIV-1 vectors, no repeat deletion was detected when the two different repeats were assayed in single vectors. However, reciprocal repeat deletion for coexpressed MLV vectors was about sevenfold lower than that observed for HIV-1. Overall, these MLV findings were consistent with previous genetic studies of gammaretrovirus repeat deletion (10, 53), but they demonstrate that HIV-1 and gammaretroviruses differ in that template switching events which genetically appear to be principally intramolecular for MLV can readily occur intermolecularly for HIV-1.
DISCUSSION
HIV-1 and MLV virus-specific assay vectors were developed to directly compare frequencies of genetic recombination during replication by these viruses. Despite the fact that the experiments presented here described results for virions generated by transient transfection, the results mirrored findings implicit in a comparison of previous reports performed with virions generated by stable vector-producing cells (19, 21), namely, that recombination between two viral genomes occurs significantly more frequently for HIV-1 than for the gammaretrovirus MLV. However, when transduction-type assays that ensured copackaging of recombination donor and acceptor sequences were employed, template switching was as frequent for MLV as it was for HIV-1. Results with reciprocal repeat vectors confirmed that template switching events which others have shown are principally intramolecular for MLV (10, 53) can occur intermolecularly for HIV-1 and showed that the differences in recombination frequency observed with our initial set of recombination vectors could not be attributed to sequence dissimilarity of donor and acceptor vectors. These findings demonstrate that when RNA availability is a constant, MLV and HIV-1 template switching occur at the same frequency, and they provide genetic evidence that coexpressed vectors are less accessible to the recombination machinery during MLV replication than is the case for HIV-1.
The cause of this difference in RNA accessibility was not studied directly in this work, but the speculation we provide here outlines a few of many possible causes of this difference. The simplest explanation for our findings may be that recruitment of genomic RNA for encapsidation differs between MLV and HIV-1, with HIV-1 apparently selecting RNAs for copackaging from among coexpressed RNAs in a more random fashion than that employed by MLV. It has generally been assumed that the way coexpressed RNAs become copackaged is as partners in mixed RNA heterodimers. Whether or not two different RNAs can heterodimerize has not been measured directly, and attempts to visualize heterozygote RNA dimers by electron microscopy have been unsuccessful (20). Our results raise the untested possibility that coexpressed MLV RNAs may not readily heterodimerize. Although on first consideration this seems unlikely, the literature provides examples of differences between HIV-1 and MLV on which speculative hypotheses that support this notion can be based. RNA trafficking and processing for lentiviruses such as HIV-1 are known to differ in several ways from the analogous processes for simple retroviruses (36), and the findings presented here may suggest that MLV and HIV-1 also differ in how RNAs are selected for copackaging. Our data might indicate a preferential affinity of one dimerizable MLV RNA for another RNA for which the same provirus acts as a template in coinfected cells. Speculatively, MLV might accomplish this by such means as a cotranscriptional commitment to dimerization or a more stable dimer precursor, in contrast to an apparent more random selection of dimerization partner at a later replication phase or a dimerization intermediate that is more susceptible to RNA exchange for HIV-1.
The present findings cannot rule out the alternate possibility that HIV-1 and MLV differ when recombination occurs between two vectors but are the same when recombination occurs between two regions on a single vector. In other words, MLV may be less capable of switching between copackaged RNAs than is HIV-1. However, because extended retroviral genome-length RNAs are more than 50 times as long as virions are wide, virion RNA must be highly condensed. Thus, there seems to be no a priori reason to anticipate that the acceptor and donor sequences in the transduction-style vectors, which are separated by half a genome length, would be in closer proximity during reverse transcription than would two sequences on two different RNAs. It is also noteworthy that genomic RNA is degraded by RNase H during retroviral DNA synthesis (46). As a result, the acceptor and donor regions of our transduction-type vectors were probably parts of separate RNA molecules at the time of the assayed template switches. However, other virus-specific differences may prove to be the cause of our apparent copackaged RNA accessibility differences. For example, HIV-1 and MLV are known to differ in viral components that are retained in postentry nucleoprotein complexes (13, 14). It is possible that our observed recombination properties reflect differences in how copackaged HIV-1 and MLV RNAs are related to one another within intracellular reverse transcription complexes.
One attractive feature of the model that HIV-1 and simple retroviruses differ in accessibility of coexpressed RNAs is that it provides a plausible explanation for the genetic phenomenon of high negative interference. High negative interference, which has been observed both for replication-competent retroviruses (8) and among gammaretroviral vector products (3, 4), is the appearance in simple retroviruses of multiple nonselected crossovers at a far greater frequency than would be predicted if each recombination event were independent. Most gammaretrovirus DNAs produced by virions from coinfected cells are nonrecombinant, but the relatively rare recombinants often contain evidence of multiple template switches. To explain negative interference, it has been suggested that only a subset of virions are recombination competent or else that although recombination generally does not occur, when it does, one recombination event predisposes the replication machinery to performing additional template switches (16, 32). Our findings are consistent with an alternate explanation for negative interference: that instead of the virions containing two different equally usable RNAs, only one of two coexpressed RNAs is accessible to the recombination machinery in most virions produced by cells coinfected by two different gammaretroviruses. HIV-1 does not appear to display negative interference (21).
In addition to explaining negative interference, it seems likely that differences in virion RNA availability account for reported differences between HIV-1 and simple retroviruses in whether or not strong-stop switching can occur between two RNAs (19, 23, 49, 52). For HIV-1, evidence of intermolecular strong-stop switching is common (49, 52). However, for gammaretroviruses, genetic evidence suggests that strong-stop switching generally occurs only between the two ends of a single kind of RNA (19, 23). The findings presented here suggest that these differences between HIV and simple retroviruses may reflect differences in the accessibility of genetically distinct RNAs rather than enzymological differences.
Although calculations still require some untested assumptions (most of which likely lead to underestimates), the transduction vector data here provide new information on which estimated retroviral recombination rate calculations can be based (see Materials and Methods). Factoring in the measured frequencies of marker assortment, lacZ-inactivating mutation rates (2), and the assumption that template switching can occur at a similar frequency at any genome position (56), our transduction vector data suggest that recombinogenic template switching occurs approximately three times during the synthesis of every retroviral DNA. Applying similar assumptions to the two-vector data yields an estimated crossover frequency of about four per replication cycle (see Materials and Methods). These values represent a significantly higher recombination frequency than those previously reported for gammaretroviruses (4, 18, 19, 23), but they are similar to some previous values for HIV-1 (21, 52). Based on our findings that HIV-1 and MLV template switching rates are indistinguishable when assayed with transduction-type vectors, we hypothesize that the biochemical act of recombinogenic template switching is as frequent for MLV as it is for HIV-1. However, the genetic consequences of recombinogenic switching are greater for HIV-1 than for MLV due to differences in which coexpressed RNAs can serve as templates during reverse transcription.
Acknowledgments
We thank Mark Benson, Wes Dunnick, Tris Parslow, and John Moran for helpful comments on the manuscript. We also thank Denise Kirschner for help with mathematics, Inder Verma for providing pCMVΔR8.2 and pHR′CMVlacZ, and Lorraine Albritton for providing mATRC1/293 cells.
This project was supported by grant GM64479 from the National Institutes of Health, with assistance in the early stages from grant R29 CA69300, and a Searle Scholars Award from the Chicago Community Trust.
REFERENCES
- 1.An, W., and A. Telesnitsky. 2002. The effects of varying sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 76:7897-7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An, W., and A. Telesnitsky. 2001. Frequency of direct repeat deletion in a human immunodeficiency virus type I vector during reverse transcription in human cells. Virology 286:475-482. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, J. A., E. H. Bowman, and W.-S. Hu. 1998. Retroviral recombination rates do not increase linearly with marker distance and are limited by the size of the recombining subpopulation. J. Virol. 72:1195-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson, J. A., V. K. Pathak, and W. S. Hu. 2000. Effect of the murine leukemia virus extended packaging signal on the rates and locations of retroviral recombination. J. Virol. 74:6953-6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bray, M., S. Prasad, J. Dubay, E. Hunter, K. Jeang, D. Rekosh, and M. Hammarskjold. 1994. A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev-independent. Proc. Natl. Acad. Sci. USA 91:1256-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke, D. S. 1997. Recombination in HIV: an important viral evolutionary strategy. Emerg. Infect. Dis. 3:253-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butsch, M., and K. Boris-Lawrie. 2002. Destiny of unspliced retroviral RNA: ribosome and/or virion? J. Virol. 76:3089-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coffin, J. M. 1979. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 42:1-26. [DOI] [PubMed] [Google Scholar]
- 9.Cullen, B. R. 2000. Nuclear RNA export pathways. Mol. Cell. Biol. 20:4181-4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delviks, K. A., and V. K. Pathak. 1999. Effect of distance between homologous sequences and 3′ homology on the frequency of retroviral reverse transcriptase template switching. J. Virol. 73:7923-7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorman, N., and A. Lever. 2000. Comparison of viral genomic RNA sorting mechanisms in human immunodeficiency virus type 1 (HIV-1), HIV-2, and Moloney murine leukemia virus. J. Virol. 74:11413-11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupont, S., N. Sharova, C. DeHoratius, C. A. Virbasius, X. Zhu, A. G. Bukrinskaya, M. Stevenson, and M. R. Green. 1999. A novel nuclear export activity in HIV-1 matrix protein required for viral replication. Nature 402:681-685. [DOI] [PubMed] [Google Scholar]
- 13.Fassati, A., and S. Goff. 2001. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 75:3626-3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fassati, A., and S. P. Goff. 1999. Characterization of intracellular reverse transcription complexes of Moloney murine leukemia virus. J. Virol. 73:8919-8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall, C. V., P. E. Jacob, G. M. Ringold, and F. Lee. 1983. Expression and regulation of Escherichia coli lacZ gene fusions in mammalian cells. J. Mol. Appl. Genet. 2:101-109. [PubMed] [Google Scholar]
- 16.Hu, W.-S., E. H. Bowman, K. A. Delviks, and V. K. Pathak. 1997. Homologous recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J. Virol. 71:6028-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu, W.-S., V. Pathak, and H. M. Temin. 1993. Role of reverse transcriptase in retroviral recombination, p. 251-274. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratories, Cold Spring Harbor, N.Y.
- 18.Hu, W.-S., and H. M. Temin. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 87:1556-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu, W.-S., and H. M. Temin. 1990. Retroviral recombination and reverse transcription. Science 250:1227-1233. [DOI] [PubMed] [Google Scholar]
- 20.Hunter, E. 1978. The mechanism for genetic recombination in the avian retroviruses. Curr. Top. Microbiol. Immunol. 79:295-309. [DOI] [PubMed] [Google Scholar]
- 21.Jetzt, A. E., H. Yu, G. J. Klarmann, Y. Ron, B. D. Preston, and J. P. Dougherty. 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 74:1234-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones, J. S., R. W. Allan, and H. M. Temin. 1993. Alteration of location of dimer linkage sequence in retroviral RNA: little effect on replication or homologous recombination. J. Virol. 67:3151-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones, J. S., R. W. Allan, and H. M. Temin. 1994. One retroviral RNA is sufficient for synthesis of viral DNA. J. Virol. 68:207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Julias, J. G., D. Hash, and V. K. Pathak. 1995. E-vectors: development of novel self-inactivating and self-activating retroviral vectors for safer gene therapy. J. Virol. 69:6839-6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katz, R. A., and A. M. Skalka. 1990. Generation of diversity in retroviruses. Annu. Rev. Genet. 24:409-445. [DOI] [PubMed] [Google Scholar]
- 26.Levin, J. G., P. M. Grimley, J. M. Ramseur, and I. K. Berezesky. 1974. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J. Virol. 14:152-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malhotra, S., A. G. Scott, T. Zavorotinskaya, and L. Albritton. 1996. Analysis of the murine ecotropic leukemia virus receptor reveals a common biochemical determinant on diverse cell surface receptors that is essential to retrovirus entry. J. Virol. 70:321-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malim, M. H., and M. Emerman. 2001. HIV-1 sequence variation: drift, shift, and attenuation. Cell 104:469-472. [DOI] [PubMed] [Google Scholar]
- 29.Naldini, L., U. Blomer, F. H. Gage, D. Trono, and I. M. Verma. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 93:11382-11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263-267. [DOI] [PubMed] [Google Scholar]
- 31.Olson, P., H. M. Temin, and R. Dornberg. 1992. Unusually high frequency of reconstitution of long terminal repeats in U3-minus retrovirus vectors by DNA recombination or gene conversion. J. Virol. 66:1336-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pathak, V. K., and W.-S. Hu. 1997. “Might as well jump!” template switching by retroviral reverse transcriptase, defective genome formation, and recombination. Semin. Virol. 8:141-150. [Google Scholar]
- 33.Pfeiffer, J. K., and A. Telesnitsky. 2001. Effects of limiting homology on intermolecular recombinogenic template switching during Moloney murine leukemia virus replication. J. Virol. 75:11263-11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfeiffer, J. K., R. Topping, N.-H. Shin, and A. Telesnitsky. 1999. Altering the intracellular environment increases the frequency of tandem repeat deletion during Moloney murine leukemia virus reverse transcription. J. Virol. 73:8441-8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Preston, B. D., and J. P. Dougherty. 1996. Mechanisms of retroviral mutation. Trends Microbiol. 4:16-21. [DOI] [PubMed] [Google Scholar]
- 36.Rabson, A. B., and B. J. Graves. 1997. Synthesis and processing of viral RNA, p. 205-262. In J. Coffin, S. Hughes, and H. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. [PubMed]
- 37.Rhode, B. W., M. Emerman, and H. M. Temin. 1987. Instability of large direct repeats in retrovirus vectors. J. Virol. 61:925-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricchetti, M., and H. Buc. 1990. Reverse transcriptases and genomic variability: the accuracy of DNA replication is enzyme specific and sequence dependent. EMBO J. 9:1583-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robert de Saint Vincent, B., and G. M. Wahl. 1983. Homologous recombination in mammalian cells mediates formation of a functional gene from two overlapping gene fragments. Proc. Natl. Acad. Sci. USA 80:2002-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robson, N. D., and A. Telesnitsky. 2000. Selection of optimal polypurine tract region sequences during Moloney murine leukemia virus replication. J. Virol. 74:10293-10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheifele, L. Z., R. A. Garbitt, J. D. Rhoads, and L. J. Parent. 2002. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc. Natl. Acad. Sci. USA 99:3944-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin, N. H., D. Hartigan-O'Connor, J. K. Pfeiffer, and A. Telesnitsky. 2000. Replication of lengthened Moloney murine leukemia virus genomes is impaired at multiple stages. J. Virol. 74:2694-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.St. Louis, D. C., D. Gotte, Sanders-Buell, D. W. Ritchey, M. O. Salminen, J. K. Carr, and F. E. McCutchan. 1998. Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J. Virol. 72:3991-3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swain, A., and J. M. Coffin. 1992. Mechanism of transduction by retroviruses. Science 255:841-845. [DOI] [PubMed] [Google Scholar]
- 45.Swain, A., and J. M. Coffin. 1989. Polyadenylation at correct sites in genome RNA is not required for retrovirus replication or genome encapsidation. J. Virol. 63:3301-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Telesnitsky, A., and S. P. Goff. 1997. Reverse transcriptase and the generation of retroviral DNA, p. 121-160. In J. Coffin, S. Hughes, and H. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. [PubMed]
- 47.Telesnitsky, A., and S. P. Goff. 1993. Strong-stop strand transfer during reverse transcription, p. 49-83. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 48.Temin, H. M. 1993. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci. USA 90:6900-6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Wamel, J. L. B., and B. Berkhout. 1998. The first strand transfer during HIV-1 reverse transcription can occur either intramolecularly or intermolecularly. Virology 244:245-251. [DOI] [PubMed] [Google Scholar]
- 50.Vogt, P. K. 1973. The genome of avian RNA tumor viruses: a discussion of four models, p. 35-41. In L. Sylvestri (ed.), Possible episomes in eukaryotes. Proceedings of the Fourth Lepetit Colloquium. North-Holland, Amsterdam, The Netherlands.
- 51.Yu, H., A. E. Jetzt, and J. P. Dougherty. 1997. Use of single-cycle analysis to study rates and mechanisms of retroviral mutation. Methods 12:325-336. [DOI] [PubMed] [Google Scholar]
- 52.Yu, H., A. E. Jetzt, Y. Ron, B. D. Preston, and J. P. Dougherty. 1998. The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem. 273:28384-28391. [DOI] [PubMed] [Google Scholar]
- 53.Zhang, J., and Y. Ma. 2001. Evidence for retroviral intramolecular recombinations. J. Virol. 75:6348-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, J., and C. M. Sapp. 1999. Recombination between two identical sequences within the same retroviral RNA molecule. J. Virol. 73:5912-5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, J., and H. M. Temin. 1993. Rate and mechanism of nonhomologous recombination during a single cycle of retroviral replication. Science 259:234-238. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, J., and H. M. Temin. 1994. Retrovirus recombination depends on the length of sequence identity and is not error prone. J. Virol. 68:2409-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]