

Abstract
Adenovirus (Ad) is an airborne, nonenveloped virus infecting respiratory epithelium. To study the mechanism of Ad entry, we used alveolar adenocarcinoma A549 cells, which have retained the ability of alveolar epithelial type II cells to synthesize the major component of pulmonary surfactant, disaturated phosphatidylcholine. Stimulation of phosphatidylcholine secretion by calcium ionophore or phorbol ester augmented the susceptibility of these cells to Ad. Both Ad infection and recombinant-Ad-mediated transfection increased in the presence of dipalmitoyl phosphatidylcholine (DPPC) liposomes in culture medium. Importantly, in the presence of DPPC liposomes, virus penetrates the cells independently of virus-specific protein receptors. DPPC vesicles bind Ad and are efficiently incorporated by A549 lung cells, serving as a virus vehicle during Ad penetration. To identify the viral protein(s) mediating Ad binding, a flotation of liposomes preincubated with structural viral proteins was employed, showing that the only Ad protein bound to DPPC vesicles was a hexon. The hexon preserved its phospholipid-binding properties upon purification, confirming its involvement in virus binding to the phospholipid. Given that disaturated phosphatidylcholine not only covers the inner surface of alveoli in the lungs but also reenters alveolar epithelium during lung surfactant turnover, Ad binding to this phospholipid may provide a pathway for virus entry into alveolar epithelium in vivo.
Adenovirus (Ad) is an nonenveloped animal virus whose virion contains one molecule of linear double-stranded DNA and at least 11 proteins. The capsid of Ad is an icosahedron with 20 facets built from hexons and 12 vertices occupied by penton complexes. The penton complex, made of an elongated trimeric fiber and a pentameric penton base, mediates virus attachment and internalization. The head domain of the fiber binds to the primary receptor, which for certain human serotypes is a 46-kDa membrane protein, the coxsackie-Ad receptor (CAR) (5). The penton base interacts with a secondary receptor, integrins αvβ3 and αvβ5 (34). Receptor-bound Ad enters the cells via receptor-mediated endocytosis (31).
The majority of data concerning the molecular mechanisms of Ad entry have been obtained with immortalized cells that are easily infected. Due to the application of recombinant Ad (rAd) as a vector for gene therapy, the mechanism of Ad entry in respiratory epithelium in vivo has recently gained more attention. Surprisingly, it has been found that differentiated tissues and, in particular, ciliated airway epithelia, a target tissue for the rAd-based therapy of cystic fibrosis, are resistant to rAd-mediated gene transfer (32, 36). The low efficiency of virus entry correlates with the low numbers of CAR (32) and α-integrins (22) present on the apical surface of differentiated epithelium. These receptors, abundant on the surface of undifferentiated cells, become redistributed during cell differentiation and concentrate on the basolateral surface of the cells. In addition, cellular glycocalyx (21) and, in particular, mucin (2), produced and secreted by airway epithelium, create a physical barrier preventing virus access to cells. The mechanism of in vivo Ad entry into bronchial ciliated epithelia remains therefore unclear.
The process of Ad entry into another potential site of virus replication, alveolar epithelium, is even less understood. By using animal models, it has been demonstrated that intratracheally instilled rAd poorly penetrates distal alveoli but that virus entry into alveolar epithelium can be significantly improved by the addition of pulmonary surfactant, a phospholipid-rich mixture produced by type II pneumocytes which stabilizes alveoli at the air-liquid interface (13, 15). Human lung adenocarcinoma A549 cells have retained the properties of type II alveolar epithelium to synthesize a major component of human lung surfactant, disaturated phosphatidylcholine (16). Using these cells, we show that the stimulation of phosphatidylcholine secretion results in enhanced Ad entry. Studying the mechanism behind this phenomenon, we found that Ad binds dipalmitoyl phosphatidylcholine (DPPC) through its hexon protein and enters A549 lung epithelial cells in complex with this phospholipid independently of virus-specific cellular receptors.
MATERIALS AND METHODS
Cells and virus.
A549 lung carcinoma, HeLa cervical cancer, and 293 embryo kidney cells were obtained from the European Collection of Cell Cultures and grown as a monolayer in medium recommended by them. To amplify wild-type Ad serotype 2 (Ad2), A549 or HeLa cells were infected at a multiplicity of infection (MOI) of 5, harvested 2 days later, and extracted with Freon 113 (trichloro-trifluoroethane) and virus was purified by banding twice on CsCl gradient (14). Replication-deficient rAd5 expressing β-galactosidase (rAd5-βgal; a kind gift of F. Graham) was purified similarly after propagation on 293 cells.
Surfactant secretion.
Surfactant secretion was monitored by the release of [3H]phosphatidylcholine (37). Cells (2 × 106) were labeled overnight with 1 mCi of [3H]choline, rinsed with serum-free medium, and incubated for 30 min in fresh þßcomplete medium, after which A23187 (5 μM final concentration) or phorbol myristate acetate (PMA; 15 nM final concentration) was added. The medium was collected 2 h later and centrifuged for 5 min to remove floating cells. Lipids were extracted from the medium with chloroform-methanol (2:1, vol/vol) in the presence of DPPC as a carrier lipid (6). Extracted lipids were dried under a vacuum and analyzed for radioactive phosphatidylcholine by scintillation counting. All lipids were from Sigma.
Infection assay.
Cells in a 96-well plate (2 × 104/well) were infected for 45 min with serially diluted Ad2. Following incubation, virus suspension was removed and the medium was replaced with fresh medium supplemented with 5% fetal calf serum. Cells were fixed 20 h later with methanol and stained with polyclonal anti-Ad2 antibody (1:300) and fluorescein isothiocyanate-labeled goat anti-rabbit antibody (1:100). Ad2 infection was quantified by measuring the fluorescein isothiocyanate fluorescence of the cellular monolayer (excitation at 485 nm, emission at 535 nm) with a Wallac 1420 counter (Perkin-Elmer).
Transfection assay.
Cells in a 24-well plate (2 × 105/well) were infected with rAd5-βgal (at an MOI of 40 to 200) for 45 min. Virus-containing medium was replaced with fresh complete medium, and cells were cultured for an additional 24 h. To quantify the expression of β-galactosidase, methanol-fixed cells were stained in situ with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; 1 mg/ml) and the optical density at 630 nm was measured either directly in the cell monolayer or, when the cell monolayer revealed nonhomogenous staining, after the extraction of blue stain with dimethyl sulfoxide.
Liposome uptake.
N-(7-nitro-2-1,3-benzoxadiazol-4-yl) phosphatidylethanolamine (NBD-PE) synthesized and purified according to reference 29 was kindly provided by Maxim Balakirev. Fluorescently labeled unilamellar liposomes were prepared by the sonication of 10 mg of either DPPC or egg yolk phosphatidylcholine (EYPC) with 0.1 mg of NBD-PE in 1 ml of 25 mM HEPES-NaOH, pH 7.0, containing 100 mM NaCl and 5 mM KCl (HBS). EYPC-NBD-PE or DPPC-NBD-PE liposomes were added to A549 cells in 24-well plates. After 1-h incubation at 37°C, cells were thoroughly washed with phosphate-buffered saline (PBS) and cell-associated fluorescence was measured with a Wallac 1420 counter. Radioactive choline-loaded liposomes were prepared by the sonication of 10 mg of lipid in 1 ml of HBS, pH 7.0, with [3H]choline (50 μCi/ml). The nonencapsulated [3H]choline was removed by gel filtration on G-25 Sepharose. The radioactively labeled liposomes were added to cells in serum-free medium for 1 h, after which the cells were thoroughly washed with PBS and cell-associated radioactivity was measured with a MicroBeta counter (PerkinElmer).
Phospholipid-binding assays.
Prior to being used in sedimentation assays, fluorescently labeled ultrasonicated liposomes were collected by centrifugation at 10,000 × g for 15 min and resuspended in fresh HBS, pH 7.0. A new lipid concentration was calculated by measuring the fluorescence of NBD-PE. Purified Ad2 (4 to 10 μg of protein) was incubated with liposomes (5 to 75 μg) for 15 min in a total volume of 100 μl at room temperature. After centrifugation at 10,000 × g for 15 min at 4°C, the proteins in supernatant and pellet fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 12.5% acrylamide) followed by Coomassie brilliant blue staining or by Western blotting with anti-Ad2 serum (1:2,500). The amount of virus cosedimented with liposomes was quantified by densitometric scanning of the hexon band (110 kDa) of the Coomassie blue-stained gels by using Molecular Analyst software (Bio-Rad).
In flotation experiments, purified Ad2 (9 μg) was incubated with unfractionated, ultrasonicated, fluorescently labeled liposomes (20 μg) for 15 min in 100 μl of HBS, pH 7.0; mixed with 60% sucrose to yield a 40% final concentration; placed in the bottom of a 2-ml ultracentrifuge tube; and covered with 900-μl portions of 25 and 4% sucrose in HBS, pH 7.0. After centrifugation in a Beckman TLS55 rotor at 100,000 × g for 2 h, 200-μl fractions were collected from the top of the tube into a 96-well plate, where NBD-PE fluorescence was quantified with a Wallac counter (excitation at 485 nm, emission at 535 nm). Aliquots (20 μl) of each fraction were analyzed by SDS-PAGE (12.5% acrylamide), followed by Western blotting with anti-Ad2 or anti-penton polyclonal antibody (1:2,500).
Identification of viral proteins bound to DPPC liposomes.
The pool of soluble viral proteins was obtained after first virus banding on CsCl gradient. It was incubated with sodium deoxycholate for 30 min (1%, wt/vol) to solubilize aggregates and dialyzed against 20 mM Tris-HCl, pH 7.5, until optical clarity of the solution was reached (∼24 h). After centrifugation at 10,000 × g for 15 min, clarified dialyzate was used for the binding assay. Proteins (100 μg) were incubated with fluorescently labeled liposomes (400 μg) in a total volume of 240 μl of HBS, pH 7.0, overlaid onto the discontinuous sucrose gradient (6 to 40% in HBS, pH 7.0), and centrifuged at 100,000 × g for 16 h. Gradient fractions were analyzed for the presence of lipids and proteins as described above. The hexon was purified from the fraction of soluble viral proteins by chromatography on DEAE-Sepharose (20). Coflotation of hexon (100 μg) with DPPC-NBD-PE liposomes (400 μg) on a sucrose density gradient was performed as described above.
Recombinant viral proteins.
The head domains of Ad2 fiber and Ad3 penton base proteins were expressed in the baculovirus system and purified as described previously (9, 17).
Electron microscopy.
Ad2 (6.25 μg) or purified hexon (4 μg) was incubated with sonicated liposomes (5 μg) in 25 μl of HBS, pH 7.0. Aliquots of the obtained mixtures were applied on thin carbon films and negatively stained with 2% uranyl acetate. Electron microscopy was carried out in 1200EXII JEOL microscope at a nominal magnification of ×40,000.
RESULTS
Secretagogues A23187 and PMA promote infection of A549 cells by Ad.
The A549 cell line initiated from a human lung adenocarcinoma has retained the properties of type II alveolar epithelium (16). The cells are polarized, and they synthesize disaturated phosphatidylcholine, which is accumulated in multilamellar cytoplasmic inclusion bodies prior to release into the medium. A549 cells thus furnish an in vitro model to study the mechanism of entry of various airborne pathogens in alveolar epithelium.
The increase in cytosolic calcium, cAMP, or the direct activation of protein kinase C is known to provoke the secretion of surfactant by type II epithelial cells (8). Measuring [3H]phosphatidylcholine secretion by A549 cells labeled with [3H]choline, we observed that, similar to cells from which they were derived, A549 cells respond to the calcium ionophore A23187 and the phorbol ester PMA by increased secretion of phosphatidylcholine into the medium (Fig. 1A). When the effect of A23187 and PMA on Ad2 infection was studied, a 1.5- to 1.8-fold increase in virus infectivity was observed for A549 cells (Fig. 1B). When similar experiments were performed with HeLa cells derived from cervical cancer, the level of Ad infection was unchanged (PMA) or diminished (A23187), suggesting that neither an increase in intracellular calcium nor direct activation of protein kinase C is sufficient to promote Ad2 infection.
FIG. 1.

Effect of A23187 and PMA on phosphatidylcholine secretion and Ad2 infectivity. (A) A549 and HeLa cells prelabeled with [3H]choline were incubated with A23187 or PMA, and 2 h later, the amount of [3H]phosphatidylcholine ([3H]PC) in culture medium was measured as described in Materials and Methods. Data are percentages of [3H]PC secreted under control conditions taken as 100% (0.02% dimethyl sulfoxide or 15 nM α-PMA) and in the presence of 5 μM A23187 or 15 nM PMA, respectively. (B) A549 and HeLa cells were exposed to A23187 (5 μM) or PMA (15 nM) for 1 h, followed by the addition of Ad2 (MOI = 0.5). One hour later, virus was removed and cells were cultured for an additional 20 h. The synthesis of late viral proteins was estimated after immunostaining as described in Materials and Methods. Data are given in arbitrary fluorescence units (a.f.u.) associated with the cell monolayer. Bars indicate the means ± standard errors (n = 6).
DPPC liposomes promote Ad2 infection and rAd5-βgal-mediated transfection.
To find out whether the stimulating effect of A23187 and PMA on Ad2 infection in A549 cells could be due to cell-secreted disaturated phosphatidylcholine, we studied the effect of liposomes made of disaturated and monounsaturated phosphatidylcholine on Ad2 infection. A549 cells were incubated with Ad2 in the presence of either DPPC liposomes or liposomes made of EYPC containing one saturated (usually palmitic) and one unsaturated (arachidonic or oleic) fatty acid. As shown in Fig. 2A, the synthesis of late viral proteins was 1.5- to 2-fold more efficient when cells were infected in the presence of DPPC vesicles (0.2 to 0.5 mg/ml). On the contrary, EYPC liposomes inhibited Ad infection.
FIG. 2.

DPPC liposomes enhance the infectivity of wild-type Ad2 (wtAd2) and augment the efficiency of rAd-mediated transfection. (A) Serially diluted Ad2 was added to A549 cells together with EYPC or DPPC liposomes (0.62 mM). At the end of a 45-min incubation period at 37°C, the virus-liposome suspension was removed and cells were cultured for an additional 20 h before being processed for indirect immunofluorescence staining. Bars indicate the means ± standard errors (n = 2). Curves representative of three independent experiments are shown. (B and C) A549 cells were transduced with rAd5-βgal (MOI = 50) in the presence of increasing concentrations of liposomes (0.025 to 0.625 mM) for 45 min at 37°C. Virus was removed, and the expression of β-galactosidase was measured 24 h later, either as the optical density of the cell monolayer measured at 630 nm (B) or after in situ X-Gal staining of methanol-fixed cells (C). Bars in panel B indicate the means ± standard errors (n = 2).
Upon transfection of A549 cells with the recombinant replication-deficient Ad5 containing the β-galactosidase reporter gene (rAd5-βgal), we observed that, in the presence of DPPC, the activity of β-galactosidase was 1.5- to 2.5-fold higher than that in cells transfected in the absence of DPPC (Fig. 2B and C). Again, EYPC vesicles inhibited rAd-mediated transfection. Since rAd5-βgal was unable to replicate in A549 cells, this implies that DPPC promotes an early step of infection such as virus entry. In addition, these data support the hypothesis that the enhancement of Ad infection by secretagogue addition is due to the presence of disaturated phosphatidylcholine in culture medium.
DPPC-promoted Ad entry is independent of primary and secondary receptors.
Next, we studied the role of the primary and secondary receptors of Ad2 and Ad5 (CAR [5] and αvβ3 and αvβ5 integrins [34]) in DPPC-mediated Ad entry. Receptors were saturated with an excess of ligand: recombinant penton base or fiber head domain. This inhibited rAd5-βgal-mediated transfection by 65 and 100%, respectively. However, in the presence of DPPC liposomes, Ad-mediated gene transfer increased above the level of control transfection (Fig. 3), suggesting that DPPC vesicles provide a pathway of virus entry that is independent of virus-specific receptors. It should be noted that DPPC-promoted transfection is not due to the absorption of viral proteins on liposomes that could release cell receptor inhibition, since neither fiber nor penton base proteins interacted directly with DPPC vesicles (see below).
FIG. 3.
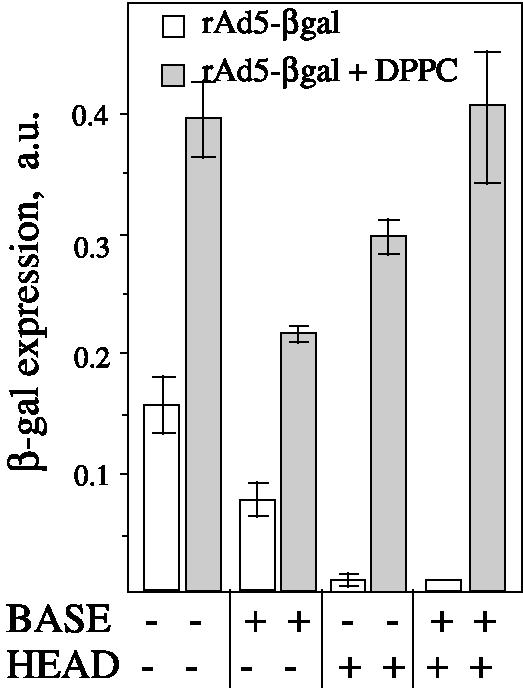
DPPC liposomes mediate Ad entry via a receptor-independent pathway. (A) A549 cells were preincubated with the recombinant viral proteins either penton base (80 μg/ml) or fiber head domain (8 μg/ml) at 4°C for 1 h. DPPC liposomes (0.5 mg/ml) and rAd5-βgal (at an MOI of 100) were then added for 45 min. The expression of β-galactosidase was measured 24 h later. Error bars represent the standard deviations around the means for three independent measurements. a.u., arbitrary units.
Ad binds to phosphatidylcholine liposomes.
Looking for the explanation of the opposite effects of DPPC and EYPC liposomes on Ad infection, we compared the uptake of these vesicles by A549 cells. To this aim, a fluorescent derivative of phosphatidylethanolamine, NBD-PE, was inserted into the lipid bilayer and its fluorescence was used as a measure of the cell-associated phospholipid. Figure 4 shows that DPPC vesicles are incorporated by A549 cells ∼5 times more efficiently than EYPC liposomes. This preferential uptake of DPPC may reflect the ability of alveolar type II cells to reincorporate secreted disaturated phosphatidylcholine (10, 30), which has been preserved by A549 cells. In addition, it is conceivable that DPPC-promoted Ad entry might result from virus binding to this phospholipid, readily incorporated by A549 cells.
FIG. 4.

Liposome uptake by A549 cells. A549 cells were incubated with DPPC-NBD-PE or EYPC-NBD-PE liposomes (A) or with [3H]choline-loaded DPPC or EYPC liposomes (B) for 1 h and thoroughly rinsed with PBS, and cell-associated fluorescence or radioactivity was measured. Bars indicate the means ± standard errors (n = 2). a.u., arbitrary units.
To determine Ad binding to phosphatidylcholine, we employed a centrifugation assay originally developed to study protein-lipid interaction (12). When purified Ad2 was incubated with EYPC or DPPC liposomes at pH 7.0, subsequent pelleting of vesicles removed a major part of the virus from the suspension, suggesting virus binding to liposomes (Fig. 5A). Both EYPC and DPPC were able to precipitate Ad when used at a 1.25 mM lipid concentration (1 mg/ml). A pattern of viral proteins found in the liposomal pellet was indistinguishable from that of the virus, suggesting that complete virions have been sedimented with liposomes. Similar results were obtained with Ad of the closely related serotype 5 (data not shown). At a lower lipid concentration (100 μM), EYPC vesicles coprecipitated virus less efficiently than DPPC (Fig. 5B). Liposomes made of the mixture of DPPC with the second most abundant surfactant lipid, dipalmitoyl phosphatidylglycerol, bound less virus than pure DPPC vesicles (Fig. 5B). EYPC-phosphatidylserine, EYPC-phosphatidethanolamine, and EYPC-phosphatidylinositol vesicles were even less efficient than those made of pure EYPC (data not shown). These results show the importance of the phosphocholine group for virus-lipid interaction. Approximately half of the virus was recovered in the pellet at a ∼150 μM concentration of DPPC (Fig. 5C). Assuming that roughly half of the lipid is exposed on the surface of unilamellar liposomes, the real concentration of DPPC would be about 75 μM, which can be used as an approximate estimation of the strength of the virus-lipid interaction.
FIG. 5.

Cosedimentation of purified Ad with liposomes. (A) Liposomes (100 μg) were incubated with 10 μg of Ad2 in 100 μl of HBS, pH 7.0, and processed as described in Materials and Methods. (B) Similar analyses performed at lower concentrations of lipid (8 μg/100 μl) and Ad2 (2 μg/100 μl) were revealed by Western blotting with polyclonal anti-Ad2 antibodies (only the hexon band is shown). (C) Ad2 (3.7 μg) was incubated with increasing amounts of DPPC liposomes and centrifuged. The pellet fractions were analyzed by SDS-PAGE. The amount of Ad precipitated by DPPC liposomes was estimated by densitometric scanning of the hexon protein after Coomassie staining (upper insert). OD, optical density. (D) Effect of pH on the formation of Ad2-liposome complexes. Ad2 (100 μg/ml) was incubated with DPPC liposomes (0.5 mM) in 50 mM morpholineethanesulfonic acid-OH (pH below 6) or 50 mM HEPES-OH (pH above 6.0) and recovered by centrifugation, and its amount was estimated as shown in panel B. The representative data from at least two independent experiments are shown.
From the previous studies on Ad-phospholipid interaction, it is known that Ad permeabilizes artificial phospholipid membranes upon acidification, implying that Ad can bind and perturb the phospholipid bilayer (7, 23). However, it remains unclear whether low pH is a condition required for Ad binding to phospholipids rather than for subsequent events leading to membrane permeabilization by virus. At the same time, the acidification is known to induce strong conformational changes in Ad structure, leading to the dissociation of viral pentons and peripentonal hexons from virions and to the aggregation of virus particles (20). The latter phenomenon limits the application of any centrifugation or optical assay to study the role of pH on virus functions. Indeed, using a sedimentation assay, we have found that at pH 6.0 and below, virus aggregates and sediments even in the absence of liposomes (Fig. 5D). However, at measurable range of pH 6.5 to 7.5 approximately, constant amounts of virus cosediment with liposomes.
Virus binding to liposomes was also followed by vesicle flotation on a sucrose density gradient. In the absence of liposomes, virus localized in the bottom fractions (Fig. 6A, upper panel). In the presence of DPPC liposomes, a portion of virus was found floating on the sucrose cushion in the fractions containing fluorescently labeled liposomes, which shows the formation of virus-liposome complexes (Fig. 6A, lower panel, and B). The presence of both large capsid proteins and low-molecular-mass core proteins in liposome fractions again implied that the entire viral particles are recovered bound to liposomes. The predominant part of the viral proteins was, however, found at the bottom of the gradient, together with some fluorescent lipids, suggesting that liposomes cause virus aggregation.
FIG. 6.

Coflotation of Ad with DPPC liposomes. (A) Ad2 (9 μg) was incubated with DPPC-NBD-PE liposomes (20 μg) for 15 min. (B) Next, it was processed for Western blot analysis as described in Materials and Methods. a.u., arbitrary units. (C) Electron microscopy of the Ad2-DPPC liposome mixture was done. Scale bar, 100 nm.
The formation of complexes between Ad and liposomes was further confirmed by electron microscopy. The predominant part of viral particles was found associated with liposomes upon incubation with 250 μM DPPC (Fig. 6C) or EYPC (data not shown) liposomes. Contact with liposomes did not lead to any evident morphological change in the virion structure. The simultaneous interaction of two distinct virions with the same vesicle resulted in the formation of large multiviral complexes.
Results of sedimentation assays and electron microscopy show that at lipid concentrations exceeding 250 μM, no obvious difference could be seen in Ad binding to EYPC or DPPC liposomes (Fig. 5A). The opposite effects of DPPC and EYPC vesicles observed at such a lipid concentration on Ad entry (Fig. 2B) reflect, therefore, the difference in the rate of cellular uptake of these liposomes. Under these conditions, both types of liposomes bound virus, but DPPC vesicles (alone or in complex with Ad) were incorporated by A549 lung epithelial cells whereas EYPC vesicles were not. In addition, EYPC liposomes might diminish Ad entry by sequestering virus particles and/or by saturating the phospholipid-binding sites present on the capsid and essential for virus entry.
Identification of viral protein responsible for phosphatidylcholine binding.
Proteins exposed on the Ad virion surface and therefore potentially able to interact with the liposomal membrane include hexons, penton bases, fibers, and, as it has been recently shown, the C-terminal domain of protein IX (1). To identify the viral protein(s) involved in Ad interaction with phosphatidylcholine, we performed sucrose gradient flotation of liposomes preincubated with the mixture of soluble viral proteins. For this, we used Ad structural proteins produced by infected cells and separated from assembled viral particles by centrifugation on a CsCl gradient. Compared to complete capsid, this fraction was poor in low-molecular-mass core proteins pVII and pVIII but rich in the capsid proteins hexon, base, and fiber (data not shown). It was demonstrated earlier that penton base protein plays an important role in Ad-induced membrane permeabilization (26) and can therefore have an affinity for phospholipids. This prompted us to study the distribution of penton proteins in gradient fractions by using the antibody against whole penton. There was no change in the penton component mobility in the presence of DPPC (Fig. 7B). However, the viral protein appearing in liposome fractions (5 through 9) was found to be a hexon, a protein of approximately 110 kDa visualized with anti-Ad2 serum (Fig. 7C).
FIG. 7.

Identification of viral proteins bound to DPPC liposomes. Soluble viral proteins were incubated with DPPC-NBD-PE liposomes in HBS as described in Materials and Methods. Gradient fractions were analyzed for the presence of lipid by measuring NBD-PE fluorescence (in arbitrary units [a.u.]) (A). The protein content of each fraction was analyzed by Western blot analysis using anti-penton (B) or anti-Ad2 (C) antibody. Gels representative of three independent experiments are shown.
The ability of hexon to bind DPPC was further confirmed with purified protein (Fig. 8). Usually found in bottom fractions (Fig. 8B, upper panel), in the presence of liposomes, hexon was displaced toward the upper part of the gradient, colocalizing with fluorescent lipid vesicles (Fig. 8A and B, fractions 4 through 11). It appears that liposomes become denser upon incubation with hexon (Fig. 8A), consistent with the formation of protein-liposome complexes. Electron microscopy analysis of gradient fractions has demonstrated the presence of liposomes carrying attached hexons on their surfaces (Fig. 8C). The presence of specific multihexon structures known as “groups-of-nine” (20) permits unequivocal recognition of the hexon as the protein bound to the vesicle membrane. Thus, these data demonstrate that the hexon, a major structural protein of Ad, is directly involved in Ad binding to DPPC vesicles.
FIG. 8.

Coflotation of purified hexon with DPPC liposomes. Hexon purified by DEAE-Sepharose chromatography was mixed with DPPC liposomes and centrifuged on a sucrose density gradient (6 to 40%), and gradient fractions were analyzed for NBD-PE fluorescence (A) or by Western blotting with anti-Ad2 antibody (B). a.u., arbitrary units. (C) Electron microscopy analysis of fraction 9 (with liposomes) was carried out after negative staining. Note groups-of-nine hexons (arrowheads) on the liposome surface. Scale bar, 100 nm.
DISCUSSION
We demonstrate here that the binding of Ads of respiratory serotypes 2 and 5 to phospholipid vesicles improves virus entry into lung epithelial cells in culture. Among the phospholipids tested, DPPC demonstrated the highest affinity towards purified Ad. A549 lung epithelial cells constantly produce and reincorporate DPPC (16), and we showed that reincorporated DPPC liposomes deliver the attached virus. Given the respiratory tropism of Ad and that DPPC, a major constituent of pulmonary surfactant, covers the surface of lung epithelium and undergoes constant turnover, our results raise the possibility that this phospholipid may be involved in Ad entry into alveolar epithelium in vivo.
Pulmonary surfactant is a phospholipid-rich mixture stabilizing alveoli at the air-liquid interface. It is synthesized and stored in alveolar type II epithelial cells and is secreted by these cells upon increase in cytosolic calcium or cAMP (24). A549 lung cells represent a cell culture model of type II alveolar epithelium in that they overproduce disaturated phosphatidylcholine and store it in inclusion bodies (16). Like their ancestors, these cells respond to the rise in intracellular calcium and/or the activation of protein kinase C by increased secretion of phosphatidylcholine into the medium (Fig. 1A). We have found that both calcium ionophore and phorbol ester enhance Ad infection in A549 cells. However, the failure of these compounds to enhance Ad infection in HeLa epithelial cells leads us to hypothesize that increased Ad infection was due to the stimulated secretion of DPPC by A549 cells rather than to the direct activation of the cellular Ca2+-dependent systems common for both cell types. This idea was supported by experiments in which both Ad infection and Ad-mediated transfection were improved in the presence of DPPC liposomes in culture medium. In contrast, liposomes prepared from monounsaturated species of phosphatidylcholine (EYPC [Fig. 2A] or POPC [data not shown]) inhibited Ad entry. At the lipid concentration used, both disaturated and monounsaturated species of phosphatidylcholine bound Ad. However, only DPPC vesicles were efficiently incorporated by lung epithelial cells and can therefore deliver attached virus. Importantly, the described pathway was found to be independent of known Ad primary and secondary receptors (Fig. 3).
In the lipid-binding assay performed in the presence of phosphatidylcholine liposomes, Ad was recovered in the pellet together with phospholipid vesicles (Fig. 5). At lipid concentrations exceeding 250 mM, both disaturated and monounsaturated phosphatidylcholine species bound Ad, but at the lower lipid concentration, DPPC was a stronger ligand. Sedimentation of about 50% Ad was achieved at ∼150 μM DPPC, showing medium-strength interaction (Kd ≈ 75 μM). It should be noted, however, that this first quantitative estimation of Ad affinity to DPPC has been obtained without any attempts to improve this value by varying the concentration of monovalent and bivalent cations.
The ability of Ad to bind phosphatidylcholine liposomes was confirmed by direct observation under an electron microscope (Fig. 6B). At the 250 μM lipid concentration, practically all virions were bound to liposomes prepared from either DPPC or EYPC, forming large virus-liposome aggregates due to the simultaneous binding of distinct virions to the same vesicle. Liposome-induced aggregation of virus explains why, in the presence of ultrasonicated liposomes, virus could be recovered in the pellet even at the low-speed centrifugation under which these small vesicles do not sediment (unpublished data).
Although this is a first demonstration that DPPC can enhance Ad entry in cultured cells, it has already been shown that the addition of complete exogenous surfactant improves the rAd-mediated gene transfer in the lungs of rabbits or rats (13, 15). One explanation put forward was that surfactant facilitates the mechanical distribution of instilled virus solution into the distal parts of lungs. Our results suggest that the role of surfactant in Ad entry is more complex. Once mixed with surfactant rich in DPPC (disaturated species of phosphatidylcholine account for 46% of surfactant mass), Ad would bind to it. Since the DPPC of intratracheally instilled exogenous surfactant is rapidly incorporated by lung epithelial cells (10), it is likely that, similar to the results we obtained for cultured lung cells, this lipid could transport bound Ad into lung epithelium in vivo.
The phospholipid-binding activity of Ad is consistent with the known lytic activity of this virus towards natural and artificial membranes (7, 26, 27). Like some other nonenveloped viruses, Ad permeabilizes liposomes (7) and forms channels in planar phosphatidylcholine membranes (23), implying that some of its structural proteins interact with phospholipids. Both liposome leakage and channel formation require acidic pH, which, at the same time, is known to induce the dissociation of pentons and virus aggregation. The results presented in Fig. 5C indicate that, contrary to membrane lysis, Ad binding to phosphatidylcholine membranes seems to be acid independent. Although a detailed study of the effect of pH on virus phospholipid-binding properties was complicated by virus aggregation at a pH below 6.0, our results clearly demonstrate that direct virus interaction with phospholipids occurs at a neutral pH and does not require acid-induced conformational changes in the capsid. Acidification (for example, inside the endosomes) could be required to trigger the membrane permeabilization essential for virus escape into the cytoplasm; however, recent data on the intracellular trafficking of Ad2 and Ad5 show that the virus remains at or only slightly below neutrality during infection (18).
To isolate the phospholipid-binding protein(s) of Ad, we first followed the distribution of viral proteins upon the centrifugation of virus-liposome mixtures on sucrose density gradients. However, the presence of a number of viral proteins in liposomal fractions, including those of the core, suggested that the integrity of the virions is preserved upon ultracentrifugation. To bypass the problems imposed by the strong protein-protein interaction within viral capsids, we used the fraction of the soluble structural proteins produced in virus-infected cells. Using a liposome flotation technique, we could observe that the only viral protein appearing in liposome-rich fractions was the hexon (Fig. 7), a major structural protein of Ad which forms the facets of the Ad icosahedral virion. The ability of hexons to bind DPPC was confirmed with a purified protein by liposome flotation and electron microscopy (Fig. 8).
The adenoviral hexon is a 110-kDa trimeric protein which accounts for over 60% of the total protein mass of the virion. Our data are the first showing the phospholipid-binding activity of this capsid protein, implying that hexons have a role in virus entry. Two other nonenveloped animal viruses studied for their interaction with liposomes, rotavirus and poliovirus, require either capsid proteolysis to start membrane permeabilization (24) or conformational changes in the capsid to initiate liposome binding (11). In particular, the amino terminus of poliovirus VP1, which is entirely internal in the native virion, becomes externalized and enables virions to attach to liposomes (11). Similarly, based on the neutralization capacity of antibodies specific to the N-terminal segment, it has been suggested that the N-terminal end of the Ad hexon plays a critical role in virus entry. The recently improved three-dimensional hexon structure (25) shows that the N terminus localized on the protein exterior, with the first 43 amino acids folded in a discontinuous α-helix and hydrophobic amino acids localized on one side. Such an N-terminal amphipathic helix could mediate hexon attachment to the phospholipid membrane. Further argument for the N-terminal role of hexon in phospholipid binding could be furnished by the structural resemblance between the Ad hexon and capsid protein P3 of bacteriophage PRD1 (4). Similar in their genome structure and in the overall architecture of their capsids and major capsid proteins, bacteriophage and Ad differ by the internal lipid membrane underlying the capsid of PRD1 phage. It seems that the N-terminal segment of P3 protein organized in an α-helix similar to that of the N-terminal part of Ad hexon is a likely site of membrane contact (3). However, the N termini of both proteins are localized firmly at the base of each protein (25, 3) and such localization is incompatible with the binding of liposomes to the external surface of Ad.
Preferential binding of Ad to phosphatidylcholine compared to other phospholipids implies an important role of the phosphocholine group for this interaction. It prompted us to compare the structures of several phosphocholine-binding proteins that have been deposed in the Protein Data Base database: C2 domain-containing proteins, hemolysins, and pentraxins (19, 28, 35). Their phosphocholine-binding domains are all folded into an antiparallel β-sandwich of variable topology, with the phosphocholine-binding site formed by amino acids located in extended loops. It might be conceivable that some of the loops, which protrude from the hexon body folded in an antiparallel β-barrel toward the capsid exterior, could attach phosphatidylcholine. Clearly, new experiments are required to identify the hexon domain(s) involved in phospholipid binding.
Ad is a very efficient respiratory pathogen. However, a low concentration of primary and secondary Ad receptors on the apical surface of airway epithelium raises the question of the mechanism of the primary Ad infection in vivo. The receptor localization on the basolateral surface of differentiated epithelium seems to be more adapted to the secondary, more advanced infection once the infected cells undergo some morphological changes. It has been shown quite recently that the interaction of the Ad fiber protein with CAR intervenes rather than participates in virus escape (33), which substantiates our results showing Ad entry independent of known protein receptors (Fig. 3). It is conceivable that initial airway infection may begin with hexon-mediated Ad interaction with DPPC, a major constituent of the pulmonary surfactant. Constant turnover of pulmonary surfactant will then bring the incoming virus in contact with airway epithelium, resulting in Ad infection, without the need for primary or secondary receptors. These results are relevant to Ad entry into lungs and may be used to improve the efficiency of Ads as vectors for gene therapy.
Acknowledgments
This work was supported in part by the French Cystic Fibrosis Society and a Region Rhône-Alpes fellowship (to L.B.) and by a grant from the French Cancer Society (ARC).
We are indebted to Rob Ruigrok for discussions and to Richard Wade for editorial comments.
REFERENCES
- 1.Akalu, A., H. Liebermann, U. Bauer, H. Granzow, and W. Seidel. 1999. The subgenus-specific C-terminal region of protein IX is located on the surface of the adenovirus capsid. J. Virol. 73:6182-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arcasoy, S. M., J. Latoche, M. Gondor, S. C. Watkins, R. A. Henderson, R. Hughey, O. J. Finn, and J. M. Pilewski. 1997. MUC1 and other sialoglycoconjugates inhibit adenovirus-mediated gene transfer to epithelial cells. Am. J. Respir. Cell. Mol. Biol. 17:422-435. [DOI] [PubMed] [Google Scholar]
- 3.Benson, S. D., J. K. Bamford, D. H. Bamford, and R. M. Burnett. 2002. The X-ray crystal structure of P3, the major coat protein of the lipid-containing bacteriophage PRD1, at 1.65 Ä resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 58:39-59. [DOI] [PubMed] [Google Scholar]
- 4.Benson, S. D., J. K. H. Bamford, D. H. Bamford, and R. M. Burnett. 1999. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell 98:825-833. [DOI] [PubMed] [Google Scholar]
- 5.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 6.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911-917. [DOI] [PubMed] [Google Scholar]
- 7.Blumenthal, R., P. Seth, M. C. Willingham, and I. Pastan. 1986. pH-dependent lysis of liposomes by adenovirus. Biochemistry 25:2231-2237. [DOI] [PubMed] [Google Scholar]
- 8.Chander, A., and A. B. Fisher. 1990. Regulation of lung surfactant secretion. Am. J. Physiol. 258:L241-L253. [DOI] [PubMed] [Google Scholar]
- 9.Fender, P., R. W. H. Ruigrok, E. Gout, S. Buffet, and J. Chroboczek. 1997. Adenovirus dodecahedron, a new vector for human gene transfer. Nat. Biotechnol. 15:52-56. [DOI] [PubMed] [Google Scholar]
- 10.Fisher, A. B., C. Dodia, and A. Chander. 1991. Alveolar uptake of lipid and protein components of surfactant. Am. J. Physiol. 261:L334-L340. [DOI] [PubMed] [Google Scholar]
- 11.Fricks, C. E., and J. M. Hogle. 1990. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harlan, J. E., P. J. Hajduk, H. S. Yoon, and S. W. Fesik. 1994. Structural characterization of the interaction between a pleckstrin homology domain and phosphatidylinositol 4,5-bisphosphate. Nature 371:168-170. [DOI] [PubMed] [Google Scholar]
- 13.Jobe, A. H., T. Ueda, J. A. Whitsett, B. C. Trapnell, and M. Ikegami. 1996. Surfactant enhances adenovirus-mediated gene expression in rabbit lungs. Gene Ther. 3:775-779. [PubMed] [Google Scholar]
- 14.Kanegae, Y., M. Makimura, and I. Saito. 1994. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn. J. Med. Sci. Biol. 47:157-166. [DOI] [PubMed] [Google Scholar]
- 15.Katkin, J. P., R. C. Husser, C. Langston, and S. E. Welty. 1997. Exogenous surfactant enhances the delivery of recombinant adenoviral vectors to the lung. Hum. Gene Ther. 20:171-176. [DOI] [PubMed] [Google Scholar]
- 16.Lieber, M., B. Smith, A. Szakal, W. Nelson-Rees, and G. Todaro. 1976. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer 17:62-70. [DOI] [PubMed] [Google Scholar]
- 17.Louis, N., P. Fender, A. Barge, P. Kitts, and J. Chroboczek. 1994. Cell-binding domain of adenovirus serotype 2 fiber. J. Virol. 68:4104-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyazawa, N., R. G. Crystal, and P. L. Leopold. 2001. Adenovirus serotype 7 retention in a late endosomal compartment prior to cytosol escape is modulated by fiber protein. J. Virol. 75:1387-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olson, R., H. Nariya, K. Yokota, Y. Kamio, and E. Gouaux. 1999. Crystal structure of staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat. Struct. Biol. 6:134-140. [DOI] [PubMed] [Google Scholar]
- 20.Pereira, H. G., and N. G. Wrigley. 1974. In vitro reconstitution, hexon bonding and handedness of incomplete adenovirus capsid. J. Mol. Biol. 85:617-630. [DOI] [PubMed] [Google Scholar]
- 21.Pickles, R. J., J. A. Fahrner, J. M. Petrella, R. C. Boucher, and J. M. Bergelson. 2000. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 74:6050-6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickles, R. J., D. McCarty, H. Matsui, P. J. Hart, S. H. Randell, and R. C. Boucher. 1998. Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer. J. Virol. 72:6014-6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenkranz, A. A., Y. N. Antonenko, O. A. Smirnova, G. K. Yurov, B. S. Naroditsky, and A. S. Sobolev. 1997. Avian adenovirus induces ion channels in model bilayer lipid membranes. Biochem. Biophys. Res. Commun. 236:750-753. [DOI] [PubMed] [Google Scholar]
- 24.Ruiz, M.-C., S. R. Alonso-Torre, A. Charpilienne, M. Vasseur, F. Michelangeli, J. Cohen, and F. Alvarado. 1994. Rotavirus interaction with isolated membrane vesicles. J. Virol. 68:4009-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rux, J. J., and R. M. Burnett. 2000. Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol. Ther. 1:18-30. [DOI] [PubMed] [Google Scholar]
- 26.Seth, P. 1994. Adenovirus-dependent release of choline from plasma membrane vesicles at an acidic pH is mediated by the penton base protein. J. Virol. 68:1204-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seth, P., M. C. Willingham, and I. Pastan. 1984. Adenovirus-dependent release of 51Cr from KB cells at an acidic pH. J. Biol. Chem. 259:14350-14353. [PubMed] [Google Scholar]
- 28.Shrive, A. K., G. M. T. Cheetham, D. Holden, D. A. A. Myles, W. G. Turnell, J. E. Volanakis, M. B. Pepys, A. C. Bloomer, and T. J. Greenhough. 1996. The three-dimensional structure of calcium-depleted human C-reactive protein from perfectly twinned crystals. Nat. Struct. Biol. 3:346-353. [DOI] [PubMed] [Google Scholar]
- 29.Struck, D. K., D. Hoekstra, and R. Pagano. 1981. Formation of asymmetric phospholipid membranes via spontaneous transfer of fluorescent lipid analogues between vesicle populations. Biochemistry 20:4093-4099. [DOI] [PubMed] [Google Scholar]
- 30.Tsuzuki, A., Y. Kuroki, and T. Akino. 1993. Pulmonary surfactant protein A-mediated uptake of phosphatidylcholine by alveolar type II cells. Am. J. Physiol. 265:L193-L199. [DOI] [PubMed] [Google Scholar]
- 31.Varga, M. J., C. Weibull, and E. Everitt. 1991. Infectious entry pathway of adenovirus type 2. J. Virol. 65:6061-6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walters, R. W., T. Grunst, J. M. Bergelson, R. W. Finberg, M. J. Welsh, and J. Zabner. 1999. Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J. Biol. Chem. 274:10219-10226. [DOI] [PubMed] [Google Scholar]
- 33.Walters, R. W., P. Freimuth, T. O. Moninger, I. Ganske, J. Zabner, and M. J. Welsh. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110:789-799. [DOI] [PubMed] [Google Scholar]
- 34.Wickham, T. J., P. Mathias, D. A. Cheresh, and G. R. Nemerow. 1993. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 73:309-319. [DOI] [PubMed] [Google Scholar]
- 35.Xu, G. Y., T. McDonagh, H. A. Yu, E. A. Nalefski, J. D. Clark, and D. A. Cumming. 1998. Solution structure and membrane interactions of the C2 domain of cytosolic phospholipase A2. J. Mol. Biol. 280:485-500. [DOI] [PubMed] [Google Scholar]
- 36.Zabner, J., P. Freimuth, A. Puga, A. Fabrega, and M. J. Welsh. 1997. Lack of high affinity fiber receptor activity explains the resistance of ciliated airway epithelia to adenovirus infection. J. Clin. Investig. 100:1144-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmerman, U. J., M. Wang, and L. Liu. 1995. Inhibition of secretion from isolated rat alveolar epithelial type II cells by the cell permeant calpain inhibitor II (N-acetyl-leucyl-leucyl-methioninal). Cell Calcium 18:1-8. [DOI] [PubMed] [Google Scholar]