

Abstract
Retroviral late-budding (L) domains are required for the efficient release of nascent virions. The three known types of L domain, designated according to essential tetrapeptide motifs (PTAP, PPXY, or YPDL), each bind distinct cellular cofactors. We and others have demonstrated that recruitment of an ESCRT-I subunit, Tsg101, a component of the class E vacuolar protein sorting (VPS) machinery, is required for the budding of viruses, such as human immunodeficiency virus type 1 (HIV-1) and Ebola virus, that encode a PTAP-type L domain, but subsequent events remain undefined. In this study, we demonstrate that VPS28, a second component of ESCRT-I, binds to a sequence close to the Tsg101 C terminus and is therefore recruited to the plasma membrane by HIV-1 Gag. In addition, we show that Tsg101 exhibits a multimerization activity. Using a complementation assay in which Tsg101 is artificially recruited to sites of HIV-1 assembly, we demonstrate that the integrity of the VPS28 binding site within Tsg101 is required for particle budding. In addition, mutation of a putative leucine zipper or residues important for Tsg101 multimerization also impairs the ability of Tsg101 to support HIV-1 budding. A minimal multimerizing Tsg101 domain is a dominant negative inhibitor of PTAP-mediated HIV-1 budding but does not inhibit YPDL-type or PPXY-type L-domain function. Nevertheless, YDPL-type L-domain activity is inhibited by expression of a catalytically inactive mutant of the class E VPS ATPase VPS4. These results indicate that all three classes of retroviral L domains require a functioning class E VPS pathway in order to effect budding. However, the PTAP-type L domain appears to be unique in its requirement for an intact, or nearly intact, ESCRT-I complex.
The final step in the life cycle of enveloped viruses is a membrane fusion event that releases the nascent virion from the host cell. Studies on the morphogenesis of human immunodeficiency virus type 1 (HIV-1) were the first to suggest that a virus-encoded function within the Gag protein was necessary for this late budding step to proceed efficiently (15). Subsequent investigations have shown that all retroviruses examined encode a so-called late-budding (L) domain within Gag whose disruption results in a phenotype characterized by normal virion assembly, with the exception of particle release (27, 37-39, 41). Moreover, at least some otherwise unrelated rhabdo-, filo-, and possibly reoviruses also encode sequences with proven or putative (based on sequence homology) L-domain activity (11, 22, 33, 34).
Viral L domains fall into one of three classes, based on characteristic sequence motifs. The majority contain either PT/SAP or PPXY tetrapeptide motifs (15, 18, 38, 39, 41). In some, both motifs are present within a relatively short linear sequence. Conversely, equine infectious anemia virus (EIAV) encodes an unusual L-domain sequence that does not conform to the PT/SAP or PPXY consensus sequences but instead contains a critical YPDL motif that is essential for function (27).
In several cases, viral L domains are functionally interchangeable and can exert their activity when positioned at different locations within a retroviral Gag protein (22, 24, 40). In fact, an HIV-1 strain that is noninfectious due to inactivating mutations in the PTAP-type L domain can be complemented in trans by coexpression of a Gag protein fused to either a homologous (PTAP) or heterologous (YPDL) L domain (22). These observations strongly suggest that L domains act by recruiting host cell factors to facilitate egress, rather than by directly influencing virion morphogenesis. In fact, specific host cell factors have been implicated in mediating the activity of each class of viral L domain. For EIAV, the YPDL motif conforms to the YXXL consensus sequence that mediates endocytosis of certain transmembrane proteins via interaction with the AP2 clathrin adaptor (10). Accordingly, EIAV p9 was shown to bind to the AP50 subunit of the AP-2 complex in vitro and induce AP-2 relocalization to sites of viral assembly in infected cells (28). The PPXY L-domain motif found in some retroviruses, rhabdoviruses, and filoviruses is similar to the consensus motif that binds WW domains (13). In fact, the L-domain sequences of Rous sarcoma virus, Ebola virus, and vesicular stomatitis virus have each been reported to bind to a particular class of WW domain-containing proteins, namely, the Nedd4-like E2 ubiquitin ligases (16, 17, 20), and a physiological Nedd4 binding site has been reported to exhibit viral L-domain activity (33).
The most compelling evidence that a specific cellular protein mediates the activity of viral L domains exists for Tsg101. A variety of physiological activities have been attributed to this protein, but it appears likely that its primary function is in endosomal protein sorting (3, 7, 8, 19). Tsg101 has been demonstrated to interact with the HIV-1 PTAP-type L domain by using various protein-protein interaction assays (14, 22, 35), and we and others have established that Tsg101 recruitment is indeed the mechanism by which the HIV-1 L domain mediates particle budding. Specifically, there is an almost perfect correlation between the abilities of HIV-1 p6 mutants to bind Tsg101 and to mediate virion production (14, 18, 22, 35). In addition, depletion of Tsg101 by using small interfering RNA results in an HIV-1 budding defect that recapitulates the phenotype of a PTAP-defective virus, and reintroduction of Tsg101 restores viral budding (14). Moreover, tethering Tsg101 to sites of HIV-1 particle assembly by fusion to Gag reverses the HIV-1 budding defect that results from mutational inactivation of the PTAP L domain (22). It is also evident that the PTAP-type L domain encoded by Ebola virus, a filovirus, recruits Tsg101 to particle assembly sites to facilitate budding (22).
Tsg101 is a member of a family of mammalian proteins whose orthologs in yeast are required for the formation of multivesicular bodies. In yeast, the Tsg101 ortholog is termed Vps23, and genetic elimination of this factor, or any one of approximately 15 other factors, results in the so-called class E phenotype, in which aberrant enlarged endosomes lacking internal vesicles accumulate (29). Several interactions between members of this group of proteins have been documented, and class E factors participate in the formation of at least three protein complexes, termed ESCRT-I, -II and -III (1, 2, 19). These complexes select a subset of transmembrane proteins for delivery to the endosomal lumen and mediate intraluminal vesicle budding. Tsg101 is one subunit of mammalian ESCRT-I (3), which also includes VPS28 (8) and, possibly, an as-yet-unidentified mammalian ortholog of Vps37, an additional component of the yeast ESCRT-I complex (19). The precise mechanisms by which class E Vps factors mediate vesicle budding are not well understood, but ESCRT-I, -II, and -III may act sequentially. Ultimately, an ATPase, Vps4, binds to components of ESCRT-III and mediates dissociation of this and perhaps other class E factors from the endosome (4, 5). Catalytically inactive VPS4 mutants have a dominant negative (DN) phenotype and induce the formation of aberrant endosomes lacking internal vesicles on which class E factors accumulate in both yeast and mammalian cells (4, 5, 8).
Despite the fact that Tsg101 depletion does not affect the activity of the PPXY-type L domain encoded by murine leukemia virus (MuLV), evidence that both PTAP- and PPXY-type L domains utilize class E vacuolar protein sorting (VPS) proteins derives from the observation that the budding of both HIV-1 and MuLV is arrested at late stage by DN VPS4 (14). Thus, the processes of vesicle budding at endosomes and retroviral budding at the plasma membrane are topologically equivalent and can both be blocked by DN VPS4, and at least one viral L-domain type recruits a known component of the ESCRT-I complex. Because of this, an attractive but as-yet-unproven hypothesis is that the same set of factors mediates both processes in a mechanistically analogous manner.
We have previously shown that a C-terminal Tsg101 domain (residues 250 to 390) is largely sufficient to mediate HIV-1 budding, provided that it is artificially recruited to sites of particle assembly (22). Thus, it appears that the N-terminal 250 amino acids are dispensable and serve only to mediate Tsg101 recruitment to particle budding sites. In this study, we present evidence that HIV-1 particle budding is highly dependent on the integrity of the Tsg101 C-terminal domain. Sequences that mediate VPS28 binding and a putative leucine zipper are each required. The integrity of other C-terminal Tsg101 sequences that mediate a newly described homomultimerization activity is also important for HIV-1 budding. Together, these findings suggest that an intact ESCRT-I complex is necessary for PTAP-type L-domain function. Moreover, minimal Gag binding and multimerizing subdomains of Tsg101 act as DN inhibitors of the HIV-1 p6 (PTAP-type) late budding domain but do not affect budding mediated by EIAV (YPDL-type) or MuLV (PPXY-type) L domains. However, because the EIAV L domain is inhibited by DN VPS4, these data suggest that each of the three known classes of L domains accesses a common pathway involving class E VPS factors but that the PTAP type is unique in requiring an intact ESCRT-I complex to exploit this cellular function during viral budding.
MATERIALS AND METHODS
Plasmid construction and mutagenesis.
Tsg101-, VPS28-, and VPS4-coding sequences were amplified from previously described expression plasmids (22) or from HeLa cDNA. Deletion and substitution mutants of Tsg101 were derived by using PCR-based methods, as was an active-site mutant of VPS4 (E228Q). For expression in mammalian cells, wild-type and mutant forms of Tsg101, as well as VPS28, were fused to the C terminus of yellow or cyan fluorescent protein (YFP or CFP, respectively) and inserted into pCR3.1 to generate pCR3.1/YFP/Tsg101 and pCR3.1/CFP/VPS28. In addition, VPS28 was inserted into pCR3.1/myc, as were wild-type and mutant forms of VPS4. Plasmids expressing fusion proteins consisting of Tsg101 and VPS28 fused to the TAP tag (31), which contains two copies of the immunoglobulin G (IgG) binding domain of protein A (pA) of Staphylococcus aureus, were generated by insertion of these sequences into pCR3.1 to generate pCR3.1/Tsg101-pA and pCR3.1/VPS28-pA, respectively. Codon optimized HIV-1 full-length and truncated Gag sequences encoded by pSYN-GP (21) were amplified by PCR and inserted into pCR3.1 (Invitrogen) to generate pCR3.1/Gag and pCR3.1/Gagδp6.
An HIV-1 proviral plasmid containing two substitutions in the PTAP sequence (P7L and P10L), named pNL(LTAL), has been previously described (22). The pNLδp6 proviral plasmid was derived from pNL/HXB and contains a translation termination codon in place of the N-terminal p6 residue; this mutation introduces changes in the overlapping Pol frame that do not affect particle release or infectivity (18). The plasmids pGagδp6/p6, pGagδp6/p9, and pGagδp6/Tsg101 are derived from the trans-complementing plasmid pENX (22), which is in turn derived from the proviral plasmid pNL/HXB but expresses a truncated HIV-1 Gag protein lacking p6 (Gagδp6) and lacks a pol gene.
MuLV proviral plasmids pNCS and pNCS P6-PY, which contain full-length viral genomes that encode the endogenous L domain (MuLV) or in which the PPXY motive is replaced by an HIV-1 PTAP motif [MuLV(PY/p6)] (40), were a gift from Stephen Goff. An MuLV-based retroviral vector that expresses HIV-1 Tat was derived by inserting sequences derived from pcTat into pMSCV-Neo (Clontech) to generate pMSCV/Tat.
For yeast two-hybrid assays, wild-type and mutant Tsg101, HIV-1 Gag, and VPS28 were inserted into the GAL4 DNA binding domain expression vector pGBKT7 (Clontech) and/or the VP16 transcription activation domain expression vector pVP16/HA (9).
Coprecipitation experiments.
To analyze Tsg101-Tsg101 and Tsg101-VPS28 interactions in mammalian cells, approximately 105 293T cells in 12-well plates were transfected with 500 ng of pCR3.1/YFP/Tsg101 or pCR3.1/myc/VPS28 and 500 ng of either pCR3.1/Tsg101-pA or pCR3.1/VPS28-pA. At 24 h after transfection, the cells were lysed in a buffer containing 50 mM Tris (pH 7.4), 5% glycerol, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and a protease inhibitor cocktail (Roche). Cell lysates were clarified by centrifugation, incubated with 20 μl of rabbit IgG agarose beads for 3 h, and washed four times with the same buffer, and bound proteins were eluted by boiling in sodium dodecyl sulfate (SDS) sample buffer. Both unfractionated cell lysates and the IgG-bound fraction were analyzed by Western blotting with anti-green fluorescent protein (anti-GFP), anti-Tsg101, or anti-Myc antibodies, as described below.
Yeast two-hybrid assays.
Assays of protein-protein interaction in yeast were done with GAL4 and VP16 fusion protein expression plasmids in Y190 cells as described previously (6, 9). To isolate mutants with mutations in the Tsg101 domain from residue 303 to 390 [Tsg101(303-390) domain] that were defective for multimerization, the corresponding DNA sequence was randomly mutagenized by using a GeneMorph kit (Stratagene) under conditions predicted to give one to three nucleotide substitutions per kilobase. A library of these mutants was constructed in the pVP16/HA vector and used to transform Y190 cells carrying a GAL4-Tsg101 expression plasmid. Approximately 10,000 transformants were screened by overlaying plates with agarose containing 0.5 mg of X-Gal (isopropyl-β-d-thiogalactopyranoside) per ml, and 40 white colonies were expanded and tested by Western blotting for expression of an intact VP16-HA-Tsg101(303-390) protein. Plasmids were recovered from two transformants that expressed intact proteins and retransformed into Y190 cells along with GAL4-Tsg101, and one clone that expressed a protein that failed to bind Tsg101 was sequenced and designated Tsg101(EP4).
Microscopy.
293T cells were plated on coverslips and transfected with 20 ng of pCR3.1/CFP or pCR3.1/CFP/VPS28, 80 ng of pCR3.1/YFP/Tsg101, and 400 ng of either pCR3.1, pCR3.1/Gag, or pCR3.1/Gagδp6. Cells were fixed with paraformaldehyde 24 h after transfection, and images were collected and deconvolved with a Deltavision microscope and software (Applied Precision).
HIV-1 and MuLV particle formation assays.
Approximately 5 × 104 293T or HOS cells in 24-well plates were transfected by using Lipofectamine Plus (Invitrogen). For Gagδp6-Tsg101 trans-complementation assays, HOS cells were transfected with 800 ng of pNL(LTAL) and 200 ng of a wild-type or mutant Gagδp6-Tsg101 expression plasmids. For analysis of the effects of truncated Tsg101 proteins on L-domain function in the context of MuLV, 293T cells were transfected with 200 ng of pMSCV/Tat, 50 ng of vesicular stomatitis virus G expression plasmid, 200 ng of pNCS or pNCS P6-PY, and 300 ng of pCR3.1/YFP, pCR3.1/YFP-1-157 or pCR3.1/YFP-303-360. For similar analyses in the context of HIV-1, 293T cells were transfected with 150 ng of pNLδp6, 100 ng of pGagδp6/p6 or pGagδp6/p9, and 300 ng of pCR3.1/YFP, pCR3.1/YFP-1-157, or pCR3.1/YFP-303-360. Alternatively, 100 ng of wild-type or mutant pCR3.1/myc-VPS4 was used in place of the YFP-Tsg101 expression plasmid. In each case, culture supernatants were collected 48 h after transfection and clarified by low-speed centrifugation. Viral particles were harvested by centrifugation through a 20% sucrose cushion at 100,000 × g for 1.5 h. Viral proteins in cell and virion lysates were analyzed by Western blotting.
Western blot analysis.
Samples were separated on 10 or 12% polyacrylamide gels and transferred to nitrocellulose membranes. The blots were sequentially probed with antibodies against HIV-1 p24 (183-H12-5C), MuLV capsid (NCI 79S-117), Myc tag (9E10), Tsg101 (C-2; Santa Cruz), or GFP (Roche) followed by a peroxidase-conjugated secondary antibody. The blots were developed with chemiluminescent substrate reagents (Pierce).
MuLV and HIV-1 infectivity assays.
P4/R5 cells were used as target cells for both HIV-1 and MuLV infection. This cell line expresses CD4, contains an integrated HIV-1 long terminal repeat-LacZ reporter construct, and expresses high levels of β-galactosidase in the presence of HIV-1 Tat. Cells were infected with supernatants derived from transfected 293T cells, and β-galactosidase activity in cell lysates was measured with chemiluminescent detection reagents (Galactostar; Tropix) 48 h later.
RESULTS
We have previously demonstrated that recruitment of a C-terminal domain of Tsg101 (residues 250 to 390) to sites of HIV-1 assembly is sufficient to restore the budding of a viral construct that is rendered defective by mutation of its PTAP L domain (22). This Tsg101 domain is predicted to be predominantly helical and has previously been shown to bind to a second mammalian ESCRT-I component, VPS28 (8). A putative leucine zipper domain whose function is unknown but, based on findings in yeast, may bind to a third putative ESCRT-I component, VPS37, is also contained within this domain (19). We therefore constructed a series of mutant Tsg101 expression plasmids to delineate which, if any, of the activities that reside in this domain are necessary to effect viral budding. Figure 1 shows the sequence of this Tsg101 domain and the mutations that were introduced to determine which sequences are required for VPS28 binding, Tsg101 multimerization, and HIV-1 budding activity.
FIG. 1.

Domain organization of Tsg101. (A) Schematic representation of the 390-residue Tsg101 protein, showing the amino-terminal UBC-like, proline rich, leucine zipper (Leu-Z) and carboxy-terminal (C-Term) domains. The UBC-like domain is necessary and sufficient for PTAP binding, while the Leu-Z and C-Term domains are necessary and sufficient to mediate HIV-1 particle budding when artificially recruited to HIV-1 assembly sites. (B) Amino acid sequence of the Leu-Z and C-Term domains, showing mutations that were used in this study.
Fine mapping of a VPS28 binding site on Tsg101.
To map a binding site for VPS28 on Tsg101, we performed a series of coprecipitation experiments. Truncated or mutant forms of Tsg101 fused to YFP were coexpressed in mammalian cells with a pA-tagged form of VPS28, and complexes were precipitated following incubation with IgG-agarose beads. As shown in Fig. 2, the full-length YFP-Tsg101 protein was efficiently coprecipitated with VPS28-pA by using IgG beads but was not precipitated when a control VPS28 protein lacking the pA tag was used in place of VPS28-pA. A C-terminal deletion analysis showed that Tsg101 residues 360 to 390 were required for binding to VPS28, as none of the C-terminally truncated YFP-Tsg101 fusion proteins [YFP-Tsg101(1-360), YFP-Tsg101(1-330), or YFP-Tsg101(1-303)] were coprecipitated with VPS28-pA (Fig. 2A). Conversely, N-terminal Tsg101 sequences were dispensable for VPS28 binding: truncated proteins consisting of Tsg101 residues 157 to 390, 250 to 390, or 303 to 390 bound to VPS28 as efficiently as the full-length protein (Fig. 2B). Although Tsg101 residues 360 to 390 were necessary for VPS28 binding (Fig. 2A), they were not in themselves sufficient to bind VPS28 with high affinity (Fig. 2B). Thus, this analysis shows that a VPS28 binding site on Tsg101 resides within residues 303 to 390.
FIG. 2.
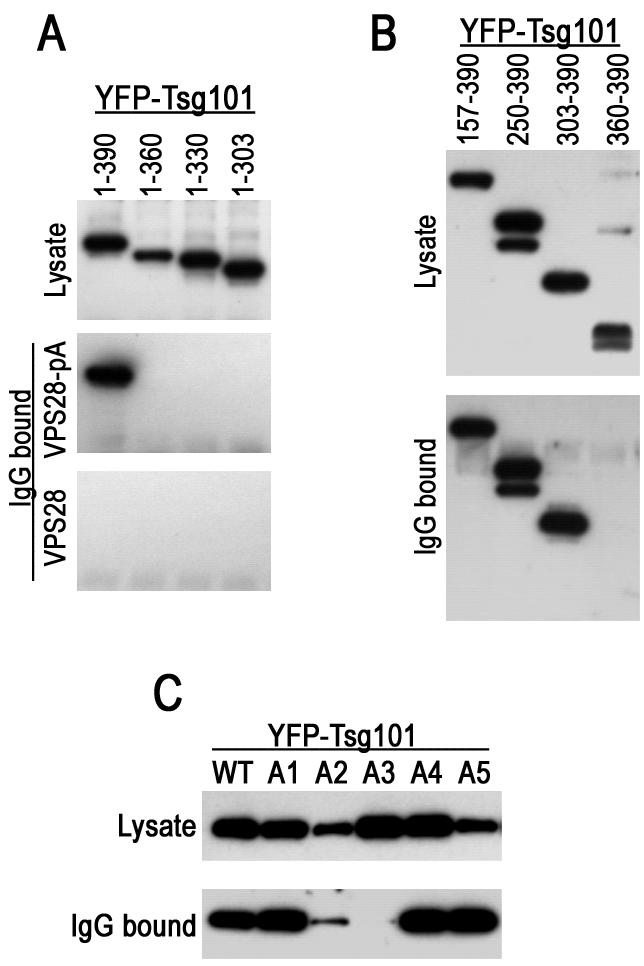
Mapping of a VPS28 binding site on Tsg101. (A) The indicated C-terminal Tsg101 deletion mutants were expressed in 293T cells as YFP fusion proteins along with either VPS28 or a VPS28-pA fusion protein. Cytoplasmic lysates (top panel) or proteins bound to IgG agarose beads (middle and lower panels) were analyzed by SDS-polyacrylamide gel electrophoresis and Western blotting with an anti-GFP antibody (B) N-terminally truncated Tsg101 proteins fused to YFP were coexpressed with VPS28-pA and analyzed as for panel A. (C) Mutant YFP-Tsg101 proteins A1 to A5 were coexpressed with VPS28-pA and analyzed for coprecipitation as for panels A and B.
These data are consistent with a previous analysis which showed that the C-terminal helical domain of Tsg101 binds to VPS28 in vitro (8). However, to more precisely determine the sequence requirements for VPS28 binding by Tsg101, we constructed a series of Tsg101 mutants in which groups of four contiguous amino acids were replaced with alanine. The mutant YFP-Tsg101 proteins that were well expressed in an intact form in mammalian cells were designated A1 through A5 (Fig. 1). As is shown in Fig. 2C, coprecipitation analysis revealed that the YFP-Tsg101 mutants A1, A4, and A5 bound efficiently to VPS28-pA, while the A2 mutant bound inefficiently and A3 mutant binding to VPS28 could not be detected. Overall, the results in Fig. 2 indicate that the VPS28 binding to Tsg101 requires the integrity of residues within the tetrapeptide L368KHV371. In addition, flanking sequences, most notably contained within residues 303 to 368, contribute to affinity, perhaps by providing additional protein-protein contacts or by influencing the conformation of the core VPS28 binding site.
VPS28 is recruited to the plasma membrane by the HIV-1 p6 L domain via its interaction with Tsg101.
We have previously shown that the Ebola virus matrix protein (EbVp40) recruits Tsg101 to sites of particle budding at the plasma membrane via a PTAP motif (22), and the same is true of HIV-1 Gag (unpublished observations). To determine whether VPS28 is recruited along with Tsg101, we coexpressed YFP-Tsg101 and CFP-VPS28 with HIV-1 Gag and examined Tsg101 and VPS28 localization by deconvolution microscopy. Importantly, each of the wild-type and mutant forms of YFP-Tsg101 was expressed in an undegraded form (Fig. 2), as was CFP-VPS28 (data not shown), indicating that the localization of the fluorescent signal accurately reflects that of the fusion protein. As is shown in Fig. 3, when overexpressed alone or with unfused CFP, YFP-Tsg101 accumulated in bright fluorescent cytoplasmic speckles (Fig. 3A). These speckles appeared to be predominantly spherical, clustered together, and were less fluorescent in their centers, suggesting that YFP-Tsg101 accumulated on an intracellular vesicular compartment. It is unclear at present whether this reflects a preexisting cellular structure or was induced by overexpression of Tsg101. In contrast, CFP-VPS28 exhibited a diffuse pattern of localization in both the nucleus and the cytoplasm (Fig. 3B). Because the size of the CFP-VPS28 fusion protein is less than that required for exclusion from nuclear pores, its presence in the nucleus likely results from passive diffusion. When they were coexpressed together, the localizations of both YFP-Tsg101 and CFP-VPS28 were altered (Fig. 3C). Tsg101 no longer localized to cytoplasmic vesicular structures and instead was distributed in a diffuse pattern throughout the cytoplasm. Moreover, CFP-VPS28 was at least partially excluded from the nucleus and was predominantly colocalized with Tsg101 in the cytoplasm. These observations suggest that overexpressed YFP-Tsg101 and CFP-VPS28 form a complex in mammalian cells that resides primarily in the cytoplasm. At present, it is unclear why the localization of overexpressed Tsg101 differs according to whether or not it is bound to VPS28, but since the majority of endogenous Tsg101 is associated with VPS28 under physiological conditions (3), we titrated YFP-Tsg101 and CFP-VPS28 expression plasmids to give equivalent levels of fusion protein expression. Thereafter, we coexpressed both proteins in subsequent localization experiments. Upon coexpression with HIV-1 Gag, both YFP-Tsg101 and CFP-VPS28 were redistributed and colocalized at the plasma membrane (Fig. 3D). This relocalization was entirely dependent on the p6 domain of HIV-1 Gag, which binds to Tsg101 (Fig. 3E), and on the N-terminal 156 residues of Tsg101, which bind to the PTAP motif in HIV-1 Gag (Fig. 3F). These data indicate that the relocalization of overexpressed CFP-VPS28 to the plasma membrane by HIV-1 Gag is dependent on the HIV-1 L domain. Moreover, VPS28 is not recruited independently, but instead requires a Gag-Tsg101 interaction for recruitment. Consistent with this, coexpression of CFP-VPS28 and YFP-Tsg101(A3), a mutant that does not bind VPS28, with HIV-1 Gag resulted in YFP-Tsg101(A3) localization at the plasma membrane while CFP-VPS28 remained diffuse throughout the cell (Fig. 3G). Conversely, in the presence of an HIV-1 Gag protein lacking p6 (or in the absence of Gag [data not shown]), YFP-Tsg101(A3) exhibited a punctate vesicular distribution, much like that of the overexpressed wild-type Tsg101 protein. However, unlike that of the wild-type YFP-Tsg101 protein, YFP-Tsg101(A3) localization was unaffected by coexpression of CFP-VPS28 (Fig. 3H). Curiously, a small fraction of CFP-VPS28 colocalized with YFP-Tsg101(A3) in cytoplasmic vesicular structures. It is unclear at present whether this was due to residual weak binding of CFP-VPS28 to YFP-Tsg101(A3) or, alternatively, to independent recruitment of CFP-VPS28 to Tsg101-induced vesicles.
FIG. 3.

VPS28 is recruited to the plasma membrane as a result of HIV-1 p6Gag-Tsg101 interaction. 293T cells were transfected with YFP-Tsg101, CFP, CFP-VPS28, and/or HIV-1 Gag expression plasmids, fixed, and examined by deconvolution microscopy. The images represent single optical sections acquired with a YFP filter set (left column), a CFP filter set (middle column), and overlaid images (right column). (A) YFP-Tsg101 plus CFP. (B) CFP-VPS28 only. (C) YFP-Tsg101 plus CFP-VPS28. (D) YFP-Tsg101 plus CFP-VPS28 plus HIV-1 Gag. (E) YFP-Tsg101 plus CFP-VPS28 plus HIV-1 Gagδp6. (F) YFP-Tsg101(157-390) plus CFP-VPS28 plus HIV-1 Gag. (G) YFP-Tsg101(A3) plus CFP-VPS28 plus HIV-1 Gag. (H) YFP-Tsg101(A3) plus CFP-VPS28 plus HIV-1 Gagδp6.
Independent domains in Tsg101 contribute to homomultimerization.
Gel filtration analysis has shown that Tsg101 and VPS28 and their yeast orthologs, Vps23 and Vps28, exist in the cell as part of a large (approximately 350-kDa) complex, termed ESCRT-I (3, 19). In yeast, this protein complex contains only Vps23, Vps28, and Vps37. This suggests that multiple copies of one or more components are present in the ESCRT-I complex or that the entire complex and/or individual ESCRT-I components are capable of multimerization. We therefore determined whether Tsg101 was capable of homomultimerization by expressing a Tsg101-pA fusion protein in mammalian cells and examining whether the endogenous Tsg101 protein could be coprecipitated. To ensure that all of the Tsg101 in the cell was saturated with VPS28 and to provide an internal control for coprecipitation, we coexpressed an excess of a Myc-tagged VPS28 protein. As shown in Fig. 4A, precipitation of the Tsg101-pA protein led to coprecipitation of both Myc-VPS28 and the endogenous Tsg101 protein, suggesting that Tsg101/VPS28 complexes formed in mammalian cells contain multiple copies of Tsg101. Multimerization of Tsg101 could also be observed by using a yeast two-hybrid assay (Fig. 4B). We used this assay to map Tsg101 domains that were capable of binding to the full-length Tsg101 protein and found that this activity resided primarily in the C-terminal domain of Tsg101 that is necessary and sufficient to mediate HIV-1 budding. Specifically, an N-terminal fragment of Tsg101(1-157) was incapable of binding to the full-length Tsg101 protein, and a fragment consisting of residues 1 to 250 did so only weakly. Conversely, Tsg101 fragments consisting of residues 1 to 303 and 1 to 330 exhibited substantial Tsg101 binding activity, and residues 1 to 360 appeared to be at least as active as the full-length Tsg101 protein. Fragments consisting of Tsg101 residues 157 to 390, 250 to 390, and 303 to 390 also proved to be capable of binding to the full-length Tsg101 protein (Fig. 4B).
FIG. 4.

Tsg101 multimerization mediated by sequences in the leucine zipper and C-terminal domains. (A) 293T cells were transfected with a Myc-VPS28 expression plasmid alone (lanes −) or with both Tsg101-pA and Myc-VPS28 expression plasmids (lanes +). Cytoplasmic lysates (left panels) and IgG-bound protein complexes (right panels) were analyzed by SDS-polyacrylamide gel electrophoresis and Western blotting (WB) with anti-Tsg101 (α-Tsg101) (top and middle panels) or anti-Myc (lower panels) antibodies. (B) Yeast two-hybrid assay of Tsg101 multimerization. Y190 cells were transformed with GAL4-Tsg10(1-390) (full length) or GAL4-Tsg10(1-303) expression plasmids along with a plasmid expressing a full-length or deletion mutant VP16-Tsg101 expression plasmid, as indicated. The mean level of β-galactosidase expression (± standard deviation) in three pools of transformants is shown. (C) Coprecipitation of YFP-Tsg101 and Tsg101-pA. 293T cells were transfected with a YFP-Tsg101 (left panels) or a YFP-Tsg101(303-360) (right panels) expression plasmid in the presence (lanes +) or absence (lanes −) of a Tsg101-pA expression plasmid. Cell lysates (top panels) or IgG-bound protein complexes (bottom panels) were analyzed by Western blotting with an anti-GFP antibody. (D) Yeast two-hybrid analysis of mutant Tsg101 protein multimerization. Y190 cells were transformed with GAL4-Tsg101 along with a plasmid expressing a full-length or mutant VP16-Tsg101. The mean level of β-galactosidase expression (± standard deviation) in three pools of transformants is shown. (E) 293T cells were transfected with full-length YFP-Tsg101 (wild type [WT]) or YFP-Tsg101(EP4) and VPS28-pA expression plasmids. Cell lysates (top panel) or IgG-bound protein complexes (bottom panel) were analyzed by Western blotting with an anti-GFP antibody.
Thus, two nonoverlapping fragments of Tsg101 (residues 1 to 303 and 303 to 390) were able to bind to the full-length Tsg101 protein. To determine whether these two domains bound to each other or, alternatively, whether multimerization is mediated by binding of one domain to the same domain in a different polypeptide, we asked whether Tsg101(1-303) could bind to Tsg101(1-303) or to Tsg101(303-390). In fact, Tsg101(1-303) was capable of homomultimerization but did not bind to Tsg101(303-390) (Fig. 4B). Therefore, it appeared that independent homomultimerization of at least two Tsg101 domains was responsible for the overall multimerization properties of the full-length Tsg101 protein.
YFP-Tsg101 could also be coprecipitated by Tsg101-pA when the two proteins were coexpressed in mammalian cells, and a deletion analysis gave results similar to those obtained with the yeast two-hybrid assay (data not shown). Ultimately, we delineated a small protein domain derived from a region of Tsg101 that is responsible for multimerization that was capable of binding to the full-length protein in mammalian cells. Remarkably, this protein domain consisted of only 58 Tsg101 residues (303 to 360). Thus, this protein lacks a VPS28 binding site (Fig. 2) and also the leucine zipper domain. As is shown in Fig. 4C, YFP-Tsg101(303-360) could be coprecipitated with Tsg101-pA, albeit less efficiently than the full-length YFP-Tsg101 protein. Nevertheless, Tsg101(303-360) bound specifically to Tsg101, because it was not precipitated with IgG agarose beads when a full-length Tsg101 protein lacking the pA domain was coexpressed in place of Tsg101-pA.
To map amino acid residues within the domain from residue 303 to 390 that were important for Tsg101 multimerization, we performed a genetic screen in yeast. A library of random mutants of Tsg101(303-390) was constructed by error-prone PCR amplification and screened with the full-length Tsg101 protein as bait. Using this strategy, we isolated a single clone, termed EP4 (Fig. 1), that was well expressed in an intact form in yeast and contained a single amino acid substitution (Ile354-Lys) that abolished the ability of Tsg101(303-390) to bind the full-length Tsg101 protein (Fig. 4D). Because multiple Tsg101 domains apparently contributed to multimerization (Fig. 4B), we reasoned that the EP4 mutation might not abolish multimerization in the context of a full-length Tsg101 protein. This proved to be the case, and the full-length Tsg101(EP4) protein bound to Tsg101 as efficiently as the wild-type protein (Fig. 4D). Thus, because this single amino acid substitution has differential effects on multimerization in truncated [Tsg101(303-390)] versus full-length Tsg101 protein contexts, it seemed likely that the EP4 mutation affected only one of several contact sites that are responsible for Tsg101 multimerization.
Given the proximity of the EP4 mutation (residue 354) to residues that we had determined to be important for VPS28 binding (residues 360 to 363 [mutant A2] and 368 to 371 [mutant A3]), we examined whether the series of alanine scanning mutants were capable of multimerization in the context of a truncated Tsg101(303-390) protein. In fact, each of the A1 to A5 mutant Tsg101(303-390) proteins proved to be fully capable of binding to the full-length Tsg101 protein (Fig. 4D). We also determined whether the EP4 mutation affected the ability of Tsg101 to bind to VPS28, using the same coprecipitation approach that we had used to map the VPS28 binding site (Fig. 2). As shown in Fig. 4E, YFP-Tsg101(EP4) was coprecipitated with VPS28-pA as efficiently as the wild-type YFP-Tsg101 protein. Thus, even though Tsg101 residues that are important for multimerization or VPS28 binding lie in quite close proximity in the linear sequence, these two activities clearly segregate and therefore are independent.
Tsg101 multimerization, VPS28 binding, and leucine zipper domains are required for HIV-1 budding.
The above data indicate that the C-terminal domain of Tsg101 is capable of participating in at least two independent protein-protein interactions, namely, homomultimerization and VPS28 binding. We next determined whether either or both of these activities was required to mediate HIV-1 budding. In addition, to test whether the putative leucine zipper domain is important for HIV-1 budding, we constructed two additional Tsg101 mutants. Specifically, either a single central leucine residue within the putative leucine zipper domain (Fig. 1) was replaced by arginine (mutant L1) or three leucine residues were replaced by alanine (mutant L2). As shown in Fig. 5A, both Tsg101(L1) and Tsg101(L2) were fully capable of binding to Gag, Tsg101 and VPS28 in the yeast two-hybrid assay. Thus, the integrity of the putative leucine zipper is not required for any of these interactions.
FIG. 5.

Leucine zipper, multimerization, and VPS28 binding domains of Tsg101 are required to mediate HIV-1 virion budding. (A) Y190 yeast cells were transformed with plasmids expressing wild-type (WT) or leucine zipper mutant GAL4-Tsg101 proteins as well as VP16-HIV-1 Gag, VP16-Tsg101, or VP16-VPS28, as indicated. The mean level of β-galactosidase (β-Gal) expression (± standard deviation) in three pools of transformants is shown. (B) HOS cells were transfected with an L-domain-defective HIV-1 proviral plasmid, pNL(LTAL), and a complementing expression vector. The complementing vector expressed a Gagδp6 protein alone (lane −) or a Gagδp6 protein to which either p6 or wild-type or mutant forms of Tsg101 were fused at the C terminus. Cell lysates and extracellular particles were analyzed by Western blotting with an anti-HIV-1 CA antibody. (C) Culture supernatants from HOS cells transfected as for panel B were analyzed for the presence of infectious virions by inoculation of P4/R5 indicator cells. β-Galactosidase activity in cell lysates was measured 48 h after inoculation, and the mean and standard deviation are shown. The background β-galactosidase activity in lysates of uninfected P4/R5 cells (Un) was approximately 1,000 relative light units (R.L.U.).
To test whether each of the mutant Tsg101 proteins was competent to mediate viral budding, we used a previously described assay in which Tsg101 is expressed fused to the C terminus of an HIV-1 Gag protein that lacks p6 (22). This Gagδp6-Tsg101 fusion protein can functionally complement a PTAP-defective, but otherwise intact, proviral construct and thereby correct Gag processing and HIV-1 virion production defects that are induced by PTAP mutation. We examined cell-associated Gag processing as well as extracellular particle formation by using Western blot assays (Fig. 5B) and measured infectious virion production by using indicator target cells (P4/R5) that contain a β-galactosidase gene under transcriptional control of the HIV-1 long terminal repeat (Fig. 5C).
As we have reported previously (22) and as shown in Fig. 5B and C, recruitment of Tsg101 to sites of HIV-1 assembly by direct fusion to Gag corrected processing defects and enhanced virion production by 10- to 15-fold. In contrast, the leucine zipper domain mutants (L1 and L2) and the mutants that had clear defects in VPS28 binding (A2 and A3) (Fig. 2) were completely unable to restore L-domain function by any criteria (Fig. 5B and C). Tsg101(EP4) was also defective, although a low residual level of restored Gag processing and virion production was observed. Surprisingly, the A1 and A4 Tsg101 mutants were also substantially defective (Fig. 5B and C), even though each of these mutants is fully competent to bind Tsg101 and VPS28 (Fig. 2 and 4). Finally, the A5 mutant retained significant activity in that it restored Gag processing and mediated HIV-1 virion formation at about 40% of the level of the wild-type Tsg101 protein. Overall these data suggest that the VPS28 binding activity of Tsg101 is required for viral particle production and that multimerization of Tsg101 may also play an important role. In addition, other mutations in the C-terminal portion of Tsg101, including those within the putative leucine zipper domain that did not affect VPS28 binding or multimerization (Fig. 2 and 5), also rendered Tsg101 unable to mediate HIV-1 budding. This last observation suggests that ESCRT-I has some function in addition to Tsg101 multimerization and Tsg101-VPS28 interaction that is important for HIV-1 particle release and that perhaps an intact ESCRT-I complex is required.
Minimal PTAP binding and multimerization domains of Tsg101 inhibit PTAP-type but not PPXY- or YXXL-type L-domain function.
If, as the above data suggest, an intact ESCRT-I complex is required for the activity of a PTAP L domain and HIV-1 particle budding, we reasoned that overexpression of a minimal domain of Tsg101 that is capable of multimerization might disrupt the stoichiometry of the complex and thereby block PTAP-type L-domain activity. Moreover, because Tsg101 is not required for MuLV budding (14), PPXY-type L-domain activity should not be affected by this manipulation. To address this, we used MuLV-based constructs which contain the natural PPXY L domain or, alternatively, in which the PPXY motif has been replaced with the PTAP L domain derived from HIV-1, termed MuLV(PY/p6) (40). Each proviral plasmid was cotransfected with an HIV-1 Tat-expressing MuLV-based retroviral vector in the presence of coexpressed YFP-Tsg101 fusion proteins or unfused YFP. Thereafter, particle formation was measured by Western blotting or by infectivity assays on P4/R5 indicator cells. Previously, it has been demonstrated that a PTAP binding fragment of Tsg101 can inhibit HIV-1 particle production by competing with endogenous Tsg101 for PTAP binding (12), and thus a similar Tsg101 fragment (residues 1 to 157) was used as a positive control for this experiment.
As expected, and as shown in Fig. 6A and B, overexpression of a minimal PTAP binding domain from Tsg101, fused to YFP, had little effect on MuLV virion production mediated by the PPXY L domain. Conversely, PTAP-dependent MuLV particle production was inhibited approximately fivefold. Notably, overexpression of the minimal Tsg101 multimerizing domain fused to YFP (residues 303 to 360) inhibited MuLV(PY/p6) particle egress to a similar extent (Fig. 6A and B). Like Tsg(101-157), however, the Tsg101(303-360) fragment had only a marginal effect on PPXY-dependent MuLV particle production. Thus, two independent fragments of Tsg101, one that binds PTAP and another that binds Tsg101 itself, both specifically inhibit Tsg101-dependent viral L-domain activity.
FIG. 6.

Selective inhibition of PTAP-type L-domain function by a minimal Tsg101 multimerizing domain. (A) 293T cells were transfected with an MuLV proviral plasmid and an MuLV-based HIV-1 Tat expression vector along with a YFP, YFP-Tsg101(1-157), or YFP-Tsg101(303-360) expression plasmid. The MuLV provirus harbored either the endogenous PPXY type L domain (left panel) or a PTAP motif derived from HIV-1 (right panel). Infectious MuLV virion production was measured by inoculation of P4/R5 indicator cells, and β-galactosidase (β-Gal) activity in lysates was measured 48 h later. The mean and standard deviation are shown, and the background β-galactosidase activity in lysates of uninfected P4/R5 cells was approximately 1,000 relative light units (R.L.U.). (B) Western blot (WB) analysis of extracellular MuLV virion production by 293T cells transfected with the same constructs as for panel A, using an anti-MuLV (α-MuLV) CA antibody. (C) 293T cells were transfected with an L-domain-defective HIV-1 proviral plasmid, (pNLδp6) and a complementing and Gagδp6 expression vector along with a YFP, YFP-Tsg101(1-157), or YFP-Tsg101(303-360) expression plasmid. The complementing Gagδp6 vector expressed either HIV-1 p6 (left panel) or EIAV p9 (right panel) fused at its C terminus. Infectious HIV-1 virion production was measured by inoculation of P4/R5 indicator cells, and β-galactosidase activity in lysates was measured 48 h later. The mean and standard deviation are shown. (D) Western blot analysis of cell lysates and extracellular HIV-1 virion production by 293T cells transfected with the same constructs as for panel C, using an anti-HIV-1 CA antibody.
We next tested whether these two independent dominant inhibitors of ESCRT-I-dependent L-domain function could inhibit the third class of viral L domain, namely, the YPDL-type L domain from EIAV. We used an experimental scheme in which a defective proviral plasmid that encodes a Gag protein lacking p6 (pNLδp6) is coexpressed with a Gag protein with p6 deleted to which is fused a heterologous L domain. We have previously used this strategy to demonstrate that the L domain of EIAV is able to functionally replace that of HIV-1, i.e., mediate budding of infectious HIV-1 particles (22). As was the case in the MuLV context, both PTAP binding and Tsg101 binding fragments of Tsg101, fused to YFP, inhibited p6-dependent HIV-1 virion egress, as measured by infectivity or Western blot assays (Fig. 6C and D). A reduced level of Gag processing, but not expression, was observed in each case, and a modest level of capsid-p2 late processing intermediate accumulated (Fig. 6D). Conversely, EIAV p9-dependent HIV-1 Gag processing and virion production were resistant to the effects of DN Tsg101 fragments (Fig. 6C and D). Thus, a Tsg101 fragment that binds to the C terminus of the full-length Tsg101 is a specific inhibitor of PTAP-type L-domain function, and these data are consistent with the notion that an intact ESCRT-I complex is important for PTAP-dependent viral budding. Moreover, they strongly suggest that the YPDL L domain contained within EIAV p9 functions independently of Tsg101.
The EIAV YPDL-type L domain requires a functional class E VPS pathway for activity.
The EIAV L domain interacts with a component of the clathrin adaptor complex (28), which has no obvious functional association with the class E VPS pathway. Moreover, it has been shown that EIAV L-domain function is not blocked by proteasome inhibitors (23, 25), which inhibit both PTAP- and PPXY-dependent viral budding (26, 32, 33, 36). Therefore, it seemed possible that the EIAV L domain might act by a mechanism entirely distinct from that of the PTAP- and PPXY-type L domains, i.e., independently of class E VPS factors. To address this, we tested whether p9-dependent HIV-1 particle egress could be inhibited by DN VPS4. As shown in Fig. 7, DN VPS4, but not wild-type VPS4, inhibited HIV-1 p6 and EIAV p9-dependent HIV-1 particle egress equivalently, by approximately 50-fold, as measured by infectivity (Fig. 7A) and Western blot (Fig. 7B) assays. Moreover, the characteristic cell-associated HIV-1 Gag processing defect that is indicative of a defective L-domain function was induced by DN VPS4 in the context of both p6 and p9 L domains (Fig. 7B). Thus, while neither PPXY- nor YXXL-type L domains require the ESCRT-I complex for activity, all three known classes of viral L domains require a functional class E VPS pathway to mediate particle budding.
FIG. 7.

EIAV YPDL-type L-domain activity is inhibited by catalytically inactive VPS4. (A) 293T cells were transfected with an L-domain-defective HIV-1 proviral plasmid, (pNLδp6) and a complementing Gagδp6 expression vector that expressed either no L domain (None), HIV-1 p6, or EIAV p9 fused at its C terminus, as indicated. Either no VPS4 or wild-type (WT) or DN mutant forms of VPS4 were coexpressed. Infectious HIV-1 virion production was measured by inoculation of P4/R5 indicator cells, and β-galactosidase (β-Gal) activity in lysates was measured 48 h later. The mean and standard deviation are shown. R.L.U., relative light units. (B) Western blot analysis of cell lysates and extracellular HIV-1 virion production by 293T cells transfected with the same constructs as for panel A, using an anti-HIV-1 CA antibody.
DISCUSSION
There is compelling evidence that an interaction between Tsg101 and a PTAP motif is required for the budding of viruses that encode this tetrapeptide as part of their L domain (12, 14, 22). Subsequent events that ultimately result in the budding of a nascent virion are poorly defined. In this study we demonstrate the Tsg101 C-terminal domain plays a central role in budding of HIV-1 particles. In contrast, retroviruses that encode other known classes of L domains apparently do not require Tsg101.
The only other mammalian class E VPS protein that is known to bind Tsg101 is VPS28 (8). This protein binds sequences close to the Tsg101 C terminus and, like Tsg101, is relocalized to the plasma membrane upon coexpression with HIV-1 Gag or EbVp40, in a PTAP-dependent manner. Moreover, mutational inactivation of the VPS28 binding site on Tsg101 prevents recruitment of VPS28 to sites of particle assembly at the plasma membrane and abolishes the ability of Tsg101 to mediate HIV-1 budding. These data indicate that Tsg101 and VPS28 are recruited as a complex to sites of virus assembly and strongly suggest that VPS28 is an essential component of the machinery that mediates the activity of PTAP-type viral L domains.
While recruitment of Tsg101 and VPS28 appears to be essential for the activity of PTAP-type L domains, it is not in itself sufficient. Tsg101 is also capable of homomultimerization, and at least two distinct domains within Tsg101 contribute to this activity, suggesting that there are multiple protein-protein contact sites. Most, perhaps all, of the Tsg101 and VPS28 content of the cell is present in the form of a large (approximately 350-kDa) protein complex termed ESCRT-I (3). While the precise composition of this complex in mammalian cells is not defined, the analogous ESCRT-I complex in yeast is of similar size yet contains only three polypeptides, Vps23, Vps28, and Vps37 (19). Thus, it might be that Tsg101 multimerization contributes to the formation of the intact ESCRT-I complex. However, experiments very similar to those described here did not reveal any tendency for the yeast ortholog of Tsg101 (Vps23) to multimerize (3). At this point, it is not clear whether this apparent discrepancy is the result of methodological differences in the two studies or whether this reflects a genuine biological difference in the composition of the mammalian and yeast ESCRT-I complexes. Nonetheless, a mutation in Tsg101 that selectively eliminates one of the Tsg101-Tsg101 contacts, but does not affect VPS28 binding, attenuates the ability of Tsg101 to mediate HIV-1 particle budding. While we cannot exclude the possibility that this mutation additionally affects interactions between Tsg101 and as-yet-unidentified ESCRT-I components, these data do suggest that the Tsg101-Tsg101 interaction plays an important role in viral budding that is mediated by PTAP-type L domains.
Other sequences in the C-terminal domain of Tsg101 that are not required either for VPS28 binding or for homomultimerization also appear to be important in Tsg101-dependent viral budding. Most notably, alteration of leucine residues within the putative leucine zipper domain of Tsg101 completely abolished budding activity. The leucine zipper is conserved in Tsg101 and Vps23 proteins from all eukaryotes examined (8), but what role it plays in Tsg101 function is unknown. However, in yeast, Vps23 mutants that lack this domain participate in the formation of ESCRT-I complexes of reduced size that lack Vps37 (19). The mammalian ortholog of Vps37, if it exists, is not yet identified, but a plausible explanation for the lack of activity of Tsg101 leucine zipper mutants is that they fail to bind this protein. Alternatively, it is likely that the leucine zipper domain contains Tsg101-Tsg101 contact sites. Consistent with this, a Tsg101 fragment containing the leucine zipper domain (residues 1 to 303) multimerizes almost as efficiently as the full-length protein, while a fragment (residues 1 to 250) lacking the 53 residues that encode the leucine zipper domain multimerizes only poorly. Whatever contribution the leucine zipper domain makes to the anatomy of the ESCRT-I complex, its integrity appears to be essential for the budding of HIV-1 particles.
In fact, the majority of mutations that we introduced into the C-terminal 140 residues of Tsg101 had clearly deleterious effects on its ability to support HIV-1 budding. In contrast, we have previously shown that deletion of the N-terminal 249 residues does not markedly affect viral budding activity, provided that the remaining portion of the protein is recruited to sites of particle assembly (22). Thus, it appears that Tsg101 has two domains that are important for PTAP-type L-domain function: an N-terminal ubiquitin conjugase (UBC) homology region of Tsg101 (residues 1 to 157) that targets ESCRT-I to viral particle assembly sites and a C-terminal domain (residues 250 to 390) that participates in multiple protein-protein interactions. Because Tsg101-dependent HIV-1 budding appears to be sensitive to a variety of mutations in the C-terminal Tsg101 domain, it seems likely that an intact ESCRT-I complex is required for the budding of HIV-1 particles.
As might be expected, an N-terminal PTAP binding fragment of Tsg101 (residues 1 to 157) acts as a specific inhibitor of PTAP-dependent viral budding, presumably by competing with endogenous ESCRT-I for binding to PTAP (12). More surprisingly, we found that expression of a small, 58-amino-acid fragment of Tsg101 (residues 303 to 360) inhibited HIV-1 L-domain function. This protein lacks PTAP or VPS28 binding sites but does bind to the full-length Tsg101 protein in mammalian cells. Most likely, it inhibits PTAP L-domain function by disrupting the endogenous ESCRT-I complex, by blocking Tsg101-Tsg101 interactions or, possibly, by blocking interactions between Tsg101 and other ESCRT-I components. These observations are consistent with the notion that an intact ESCRT-I complex is required for PTAP-type L-domain function. Importantly, inhibition of viral budding by Tsg101(1-157) and Tsg101(303-360) was specific; MuLV budding was insensitive to either fragment but became sensitive when the native PPXY L domain was replaced with the PTAP L domain of HIV-1. Moreover, the budding of HIV-1 itself was inhibited by both Tsg101(1-157) and Tsg101(303-360), but replacement of the endogenous PTAP-type L domain with the YPDL-type L domain conferred resistance. Thus, in two entirely different retroviral contexts, the requirement for ESCRT-I in virion budding is governed by the origin of the L-domain sequence and not by any other viral determinant.
It has previously been shown that the activities of both PTAP- and PPXY-type viral L domains are inhibited by DN VPS4 (14), suggesting that each has a related mechanism of action. DN VPS4 could act by inducing the sequestration of class E VPS factors at the surface of aberrant endosomes or, alternatively, by preventing the disassembly of ESCRT complexes at the sites of particle assembly, thereby blocking virus release. In either case, it appears that at least a subset of the class E VPS factors are required for the activity of both PTAP- and PPXY-mediated viral budding. In this study, we found that a distinct class of L domain, namely, the YPDL type encoded by EIAV, is similarly inhibited by DN VPS4. Thus, it is apparent that each of the three known classes of L domains requires a functional class E pathway in order to effect viral budding.
PTAP-type L domains appear to be unique among the known L-domain types in their requirement for Tsg101 and an intact ESCRT-I complex. Moreover, the requirement for ESCRT-I in HIV-1 budding can be alleviated by replacing the native PTAP-type L domain with a YPDL-type L domain. Therefore, it may be that the sole role of ESCRT-I in PTAP-dependent viral budding is to act as an interface between PTAP-type L domains and the remaining components of the class E pathway. One model for vesicle budding into endosomes involves the sequential action of ESCRT-I and ESCRT-II followed by the assembly of the ESCRT-III complexes, which play the central role in vesicle formation (1, 2). It might be that PPXY- and YPDL-type L domains simply bypass ESCRT-I and interact directly or indirectly with downstream factors. Nevertheless, how they and their reported cellular binding partners (ubiquitin ligases and AP2, respectively) access the class E VPS machinery is presently unclear. It is also formally possible that, rather than simply acting as an ‘adaptor' to link PTAP to the class E VPS machinery, ESCRT-I participates in an active way in the viral budding process and that PPXY and YPDL motifs recruit an alternative activity that can functionally replace that of ESCRT-I.
While it is apparent that class E VPS factors are important for both viral budding at the plasma membrane and vesicle budding at the endosomal limiting membrane, the two processes may not be mechanistically identical. Endosomal vesicle budding, at least in yeast, requires each of the ESCRT-I components (3, 19, 30), but viral budding in mammalian cells is not strictly ESCRT-I dependent, except when the L domain is of the PTAP type. Nevertheless, given that vesicle budding at endosomes and retroviral budding at plasma membranes share a common topology and require at least some of the same cellular factors, it seems likely that certain steps in the two processes will be mechanistically similar. It will be interesting to determine whether ESCRT-II, -III, and each of the remaining class E VPS proteins that are required for the formation of endosomal vesicles are also required for viral budding, in a general or L-domain type-specific manner.
Acknowledgments
We thank Kelvin Li for technical assistance; N. Landau, P. Charneau, B. Chesebro, and H. Chen for providing P4/R5 and the 183-H12-5C hybridoma cell lines through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; and Stephen Goff for MuLV proviral plasmids and the anti-MuLV capsid antibody.
This work was supported by grants from the NIH (RO1 AI52774) and AMFAR (02865-31) (to P.D.B). J.M.-S. is a recipient of a postdoctoral fellowship from the Spanish Ministerio de Educación, Cultura y Deporte. P.D.B. is an Elizabeth Glaser scientist of the Elizabeth Glaser Pediatric AIDS Foundation.
REFERENCES
- 1.Babst, M., D. Katzmann, E. Estepa-Sabal, T. Meerloo, and S. Emr. 2002. Escrt-III. An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell 3:271.. [DOI] [PubMed] [Google Scholar]
- 2.Babst, M., D. Katzmann, W. Snyder, B. Wendland, and S. Emr. 2002. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev. Cell 3:283.. [DOI] [PubMed] [Google Scholar]
- 3.Babst, M., G. Odorizzi, E. J. Estepa, and S. D. Emr. 2000. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 1:248-258. [DOI] [PubMed] [Google Scholar]
- 4.Babst, M., T. K. Sato, L. M. Banta, and S. D. Emr. 1997. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO J. 16:1820-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babst, M., B. Wendland, E. J. Estepa, and S. D. Emr. 1998. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17:2982-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bieniasz, P. D., T. A. Grdina, H. P. Bogerd, and B. R. Cullen. 1998. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 17:7056-7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop, N., A. Horman, and P. Woodman. 2002. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 157:91-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bishop, N., and P. Woodman. 2001. TSG101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem. 276:11735-11742. [DOI] [PubMed] [Google Scholar]
- 9.Bogerd, H. P., R. A. Fridell, W. S. Blair, and B. R. Cullen. 1993. Genetic evidence that the Tat proteins of human immunodeficiency virus types 1 and 2 can multimerize in the eukaryotic cell nucleus. J. Virol. 67:5030-5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins, B. M., A. J. McCoy, H. M. Kent, P. R. Evans, and D. J. Owen. 2002. Molecular architecture and functional model of the endocytic AP2 complex. Cell 109:523-535. [DOI] [PubMed] [Google Scholar]
- 11.Craven, R. C., R. N. Harty, J. Paragas, P. Palese, and J. W. Wills. 1999. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol. 73:3359-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demirov, D. G., A. Ono, J. M. Orenstein, and E. O. Freed. 2002. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 99:955-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Einbond, A., and M. Sudol. 1996. Towards prediction of cognate complexes between the WW domain and proline-rich ligands. FEBS Lett. 384:1-8. [DOI] [PubMed] [Google Scholar]
- 14.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55-65. [DOI] [PubMed] [Google Scholar]
- 15.Gottlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 88:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harty, R. N., M. E. Brown, G. Wang, J. Huibregtse, and F. P. Hayes. 2000. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. USA 97:13871-13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harty, R. N., J. Paragas, M. Sudol, and P. Palese. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, M., J. M. Orenstein, M. A. Martin, and E. O. Freed. 1995. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69:6810-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katzmann, D. J., M. Babst, and S. D. Emr. 2001. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106:145-155. [DOI] [PubMed] [Google Scholar]
- 20.Kikonyogo, A., F. Bouamr, M. L. Vana, Y. Xiang, A. Aiyar, C. Carter, and J. Leis. 2001. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 98:11199-11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotsopoulou, E., V. N. Kim, A. J. Kingsman, S. M. Kingsman, and K. A. Mitrophanous. 2000. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J. Virol. 74:4839-4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313-1319. [DOI] [PubMed] [Google Scholar]
- 23.Ott, D. E., L. V. Coren, R. C. Sowder II, J. Adams, K. Nagashima, and U. Schubert. 2002. Equine infectious anemia virus and the ubiquitin-proteasome system. J. Virol. 76:3038-3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parent, L. J., R. P. Bennett, R. C. Craven, T. D. Nelle, N. K. Krishna, J. B. Bowzard, C. B. Wilson, B. A. Puffer, R. C. Montelaro, and J. W. Wills. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 69:5455-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patnaik, A., V. Chau, F. Li, R. C. Montelaro, and J. W. Wills. 2002. Budding of equine infectious anemia virus is insensitive to proteasome inhibitors. J. Virol. 76:2641-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patnaik, A., V. Chau, and J. W. Wills. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 97:13069-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puffer, B. A., L. J. Parent, J. W. Wills, and R. C. Montelaro. 1997. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J. Virol. 71:6541-6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puffer, B. A., S. C. Watkins, and R. C. Montelaro. 1998. Equine infectious anemia virus Gag polyprotein late domain specifically recruits cellular AP-2 adapter protein complexes during virion assembly. J. Virol. 72:10218-10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raymond, C. K., I. Howald-Stevenson, C. A. Vater, and T. H. Stevens. 1992. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell 3:1389-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieder, S. E., L. M. Banta, K. Kohrer, J. M. McCaffery, and S. D. Emr. 1996. Multilamellar endosome-like compartment accumulates in the yeast vps28 vacuolar protein sorting mutant. Mol. Biol. Cell 7:985-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030-1032. [DOI] [PubMed] [Google Scholar]
- 32.Schubert, U., D. E. Ott, E. N. Chertova, R. Welker, U. Tessmer, M. F. Princiotta, J. R. Bennink, H. G. Krausslich, and J. W. Yewdell. 2000. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 97:13057-13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strack, B., A. Calistri, M. A. Accola, G. Palu, and H. G. Gottlinger. 2000. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 97:13063-13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Timmins, J., S. Scianimanico, G. Schoehn, and W. Weissenhorn. 2001. Vesicular release of Ebola virus matrix protein VP40. Virology 283:1-6. [DOI] [PubMed] [Google Scholar]
- 35.VerPlank, L., F. Bouamr, T. J. LaGrassa, B. Agresta, A. Kikonyogo, J. Leis, and C. A. Carter. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 98:7724-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogt, V. M. 2000. Ubiquitin in retrovirus assembly: actor or bystander? Proc. Natl. Acad. Sci. USA 97:12945-12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wills, J. W., C. E. Cameron, C. B. Wilson, Y. Xiang, R. P. Bennett, and J. Leis. 1994. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol. 68:6605-6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang, Y., C. E. Cameron, J. W. Wills, and J. Leis. 1996. Fine mapping and characterization of the Rous sarcoma virus Pr76gag late assembly domain. J. Virol. 70:5695-5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasuda, J., and E. Hunter. 1998. A proline-rich motif (PPPY) in the Gag polyprotein of Mason-Pfizer monkey virus plays a maturation-independent role in virion release. J. Virol. 72:4095-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan, B., S. Campbell, E. Bacharach, A. Rein, and S. P. Goff. 2000. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 74:7250-7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan, B., X. Li, and S. P. Goff. 1999. Mutations altering the Moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J. 18:4700-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]