

Abstract
Structures of all three poliovirus (PV) serotypes (PV1, PV2, and PV3) complexed with their cellular receptor, PV receptor (PVR or CD155), were determined by cryoelectron microscopy. Both glycosylated and fully deglycosylated CD155 exhibited similar binding sites and orientations in the viral canyon for all three PV serotypes, showing that all three serotypes use a common mechanism for cell entry. Difference maps between the glycosylated and deglycosylated CD155 complexes determined the sites of the carbohydrate moieties that, in turn, helped to verify the position of the receptor relative to the viral surface. The proximity of the CD155 carbohydrate site at Asn105 to the viral surface in the receptor-virus complex suggests that it might interfere with receptor docking, an observation consistent with the properties of mutant CD155. The footprints of CD155 on PV surfaces indicate that the south rim of the canyon dominates the virus-receptor interactions and may correspond to the initial CD155 binding state of the receptor-mediated viral uncoating. In contrast, the interaction of CD155 with the north rim of the canyon, especially the region immediately outside the viral hydrophobic pocket that normally binds a cellular “pocket factor,” may be critical for the release of the pocket factor, decreasing the virus stability and hence initiating uncoating. The large area of the CD155 footprint on the PV surface, in comparison with other picornavirus-receptor interactions, could be a potential limitation on the viability of PV escape mutants from antibody neutralization. Many of these are likely to have lost their ability to bind CD155, resulting in there being only three PV serotypes.
Poliovirus (PV), a human enterovirus (32) in the family of Picornaviridae, has three known serotypes: PV1, PV2, and PV3 (46). All three PV serotypes can cause poliomyelitis, a paralytic disease resulting from the destruction of motor neurons in the central nervous system (CNS) (38, 46). The three-dimensional atomic structures of PV1, PV2, and PV3 have been determined (12, 23, 28). They have very similar structural features among themselves, which are also similar to those of other human enteroviruses, coxsackieviruses (21, 35), echoviruses (13), and human rhinoviruses (HRVs) (43).
All three serotypes of PV recognize a common cellular receptor, CD155 (or poliovirus receptor previously abbreviated as PVR), for cell attachment and entry (33, 49). CD155 is a membrane-anchored glycoprotein with three immunoglobulin-like extracellular domains: D1, D2, and D3 (8). The fold of D1 (Fig. 1a) resembles that of an immunoglobulin variable domain, whereas the folds of D2 and D3 resemble immunoglobulin constant domains. Eight glycosylation sites are distributed among the three extracellular immunoglobulin domains of CD155. Four different isotypes (α, β, γ, and δ) of CD155 are produced by cells through alternate splicing of CD155 mRNA. The amino acid sequence of the extracellular parts is identical for these isotypes. CD155-α and -δ are membrane-bound forms and are used as PV receptors (25, 49). PV consists of 60 copies each of the capsid proteins VP1, VP2, VP3, and VP4 and one copy of the single-stranded RNA genome. Membrane-associated CD155 and even soluble CD155 are able to transform PV (160S) particles in vitro (1) to altered particles (A-particles, 135S) in which VP4 is absent, and to empty particles (80S) lacking both VP4 and the viral genome. The 135S and 80S particles are possible intermediates in the PV uncoating pathway (1, 10, 11).
FIG. 1.

(a) Ribbon diagram of CD155 domain D1. Residues in the virus-receptor binding interfaces are shown as colored spheres. Residues identified as being in the interface in all three serotypes are shown in red, residues identified in two serotypes are shown in blue, and residues identified in only one serotype are shown in black. Secondary structure elements are identified by A, B, C, C′, C", D, E, F, and G. Residues are numbered at strategic positions. (b) CD155 residues in the virus-receptor binding interface. Residues are colored as in Fig. 1a. The residues that are important for receptor binding, as indicated by mutagenesis studies, are marked by “x” (6, 17) and “+” (9, 30, 34).
Like wild-type CD155, the fully deglycosylated CD155 can also behave as a receptor for all three PV serotypes. Deglycosylated CD155 is significantly more efficient in mediating PV infection compared to the wild-type CD155 (5, 6). The glycosylation site at Asn105 of CD155 D1 has been identified to be responsible for the increased infectivity. This observation is somewhat unusual since wild-type receptors usually have higher activity than mutants. Although insect cells and mammalian cells normally produce different sugar moieties for glycoproteins, the soluble form of CD155 expressed in insect cells still retains binding activity for PV (1). This suggests that the sugar moiety at Asn105 is unlikely to be involved in the binding interface between CD155 and PV, and its influence on infectivity might be due to steric hindrance.
The normal physiological function of CD155 is slowly emerging. CD155 is a cell surface adhesion molecule with affinity to vitronectin (27) mediating cell-to-matrix contacts. It has been suggested that CD155 plays a role in the development of the CNS during embryogenesis (16). Moreover, recent evidence indicates that the cytoplasmic domain of CD155 can interact specifically with the light chain of a dynein motor complex, thus helping viral invasion of the CNS through the retrograde axonal pathway (36).
A large number of enteroviruses and rhinoviruses use immunoglobulin-like molecules as their receptors for recognizing and entering host cells. Several of these picornavirus-receptor complexes have been studied by combining cryoelectron microscopy (cryoEM) and X-ray crystallography (45). Examples are major-group rhinoviruses and coxsackievirus A21, which use intracellular adhesion molecule-1 (ICAM-1) (26, 51); PVs, which use CD155 (4, 18, 52); and coxsackie B viruses (CVB), which use coxsackievirus-adenovirus receptor (CAR) (19). All of these receptors utilize the membrane-distal, N-terminal domain to bind into the viral canyon, a narrow depression around each of the icosahedral fivefold axes (43). It has been suggested that binding of the receptor into the canyon competes with the binding of a “pocket factor” into a hydrophobic pocket underneath the canyon. Release of the pocket factor could destabilize the virus and, thus, initiate uncoating (42). This property is utilized by a group of antiviral compounds (47) that stabilize the virus by binding into the pocket, thus inhibiting the capsid flexibility (“structural breathing”) and uncoating (29). Conservation of the receptor binding site among enteroviruses and major-group rhinoviruses confirms the significance of the canyon as a functionally important receptor-binding site and as a potential trigger for viral uncoating (42).
Two recent examples indicate that the canyon is not the only receptor-binding site on enterovirus and rhinovirus surfaces. In the case of human rhinovirus serotype 2 (HRV2), a member of the minor group of rhinoviruses, the very-low-density-lipoprotein receptor binds close to the icosahedral fivefold axes instead of the canyon (22). In addition, decay-accelerating factor, the receptor of many echoviruses and some coxsackieviruses (40, 41), binds close to the icosahedral twofold axes on the surface of echovirus 7 (20). Unlike the canyon-binding receptors, these non-canyon-binding receptors seem to be utilized only for recognition without initiating cell entry and uncoating. Although there is considerable diversity of virus-receptor interactions among different enteroviruses and rhinoviruses, the conservation of the receptor-binding mode is mostly retained among different serotypes that utilize a common cellular receptor. For instance, the structures of HRV14 and HRV16 each complexed with ICAM-1 (26) show that these different serotypes of major-group rhinoviruses bind receptor at the same location in similar orientations.
The PV1-CD155 complex has been studied by cryoEM (4, 18, 52) to ∼22-Å resolution. Although domain D1 of CD155 binds into the canyon, its tangential binding orientation relative to the viral surface is quite different from that of ICAM-1 (26, 51) and CAR (19). As a result, CD155 has a larger footprint than that of ICAM-1 because of an additional binding region on the east side of the canyon (18). Here, we report the structures of virus-receptor complexes of all three PV serotypes investigated by cryoEM image analysis. Comparison of glycosylated and fully deglycosylated CD155-virus complexes allowed the determination of the carbohydrate site positions. These sites provided restraints on the fitting of the CD155 homology model into the cryoEM density maps, thus increasing the accuracy of the atomic structure determination of the complex. This showed that CD155 utilized very similar binding modes on the surface of each of the PV serotypes.
MATERIALS AND METHODS
Purification of poliovirus.
PV1 Mahoney with the rhinovirus type 2 IRES (RIPO), a highly attenuated derivative of PV1 (Mahoney) [PV1(M)] from which the cognate internal ribosomal entry site was exchanged with that of HRV2 (15), was amplified in HeLa cells (in minimum essential medium modified for suspension cultures plus 10% bovine serum, 35°C) by infecting the cells with PV1 (RIPO) inoculum (multiplicity of infection of 10) for 9 h. The infected cells were collected and homogenized. Viral particles were purified by sedimentation through a 30% sucrose cushion and through a 15 to 45% sucrose gradient and then concentrated to ∼10 mg/ml in 0.1 M Tris buffer with 0.2 M NaCl at pH 8.3. The same procedure was applied for the amplification and purification of both PV2 and PV3 particles.
Expression of poliovirus receptor.
A soluble CD155 derivative was prepared by fusing the coding region of the 337 N-terminal codons of CD155 (including all three extracellular immunoglobulin-like domains) to the N-terminal coding region of human placental alkaline phosphatase (AP) (18) by using plasmid pAPtag2 to yield plasmid pCD155-AP (14). The secreted fusion protein, CD155-AP, was expressed with this vector in 293 cells and purified from cell supernatants through a size exclusion column. The fully deglycosylated CD155-AP fusion protein (in which the DX S/T motif had been changed to EX S/T) was expressed and purified similarly. Both purified CD155-AP samples were concentrated to ∼8 mg/ml in 0.1 M Tris buffer at pH 7.5.
cryoEM experiments.
Purified PV particles were mixed with CD155-AP samples at 4°C and incubated for ∼15 min. There were about five receptor molecules to every receptor-binding site. Small aliquots (∼3.5 μl) of this mixture were attached to carbon-coated electron microscope grids and vitrified in liquid ethane as described by Baker et al. (3). Electron micrographs were recorded on Kodak SO-163 film in a Philips CM300 FEG microscope at a nominal magnification of 45,000 and a dose level of ∼20 e−/Å2 (Table 1). Micrographs were then digitized on a Zeiss PHODIS microdensitometer at 14-μm intervals, which corresponds to 3.11 Å at the specimen.
TABLE 1.
Statistics of the cryoEM reconstructions
| Sample | No. of micrographs | Incubation time (min) | Defocusa (μm) | Magnification | Pixel size (Å) | No. of particles (no. selected/total no.) | Correlation coefficientb | Resolutionc (Å) |
|---|---|---|---|---|---|---|---|---|
| PV1-glycoCD155 | 24 | 15 | 1.6-4.2 | 45,000 | 2.91 | 2,022/4,799 | 0.328 | 15 |
| PV1-deglycoCD155 | 15 | 15 | 1.5-3.4 | 45,000 | 2.88 | 1,109/2,404 | 0.326 | 16 |
| PV2-glycoCD155 | 13 | 15 | 1.2-4.6 | 45,000 | 2.93 | 512/1,159 | 0.308 | 21 |
| PV2-deglycoCD155 | 15 | 15 | 1.7-4.1 | 45,000 | 2.90 | 613/1,309 | 0.324 | 21 |
| PV3-glycoCD155 | 10 | 15 | 2.0-4.0 | 45,000 | 2.91 | 303/931 | 0.408 | 23 |
| PV3-deglycoCD155 | 10 | 15 | 2.0-5.0 | 45,000 | 2.88 | 842/1,759 | 0.359 | 19 |
Determined from the phase-contrast transfer function of the microscope.
Mean real-space correlation coefficient (CC), calculated as follows: 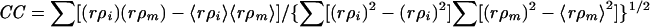 . In this formula, ρi, is the electron density of the boxed cryoEM image, ρm is the electron density of the model projection, and r is the radius of the corresponding density point used to assure proper weighting of the densities. The angle brackets indicate mean values.
. In this formula, ρi, is the electron density of the boxed cryoEM image, ρm is the electron density of the model projection, and r is the radius of the corresponding density point used to assure proper weighting of the densities. The angle brackets indicate mean values.
Resolution at which the correlation between two independent three-dimensional reconstructions falls below 0.5.
An earlier cryoEM image reconstruction of a PV1 (18) particle was used as the initial model for determining the orientation of each projected particle by means of the model-based polar-Fourier-transform method (2). Orientation refinement was monitored by correlation coefficients computed with real and reciprocal space data (3). The resolution of the resulting reconstruction (Table 1) was estimated by splitting the image data into two sets and comparing structure factors obtained in the separate reconstructions. The defocus level was calculated for each image and used to calculate the phase-contrast transfer function for the reconstructions (3). The hand of the resulting reconstruction was verified by comparing the asymmetric “V” shape of the canyon with that seen in the crystal structures of PVs and other picornaviruses. Reconstructions of PV1, PV2, and PV3, each complexed with both glycosylated CD155 and fully deglycosylated CD155, were calculated according to the same procedure (Fig. 2).
FIG. 2.

Stereoviews of cryoEM reconstructions of PV1, -2, and -3 complexed with glycosylated and fully deglycosylated CD155. Viral surfaces and CD155 are shown in gray and red, respectively. Resolutions of the different reconstructions are given in Table 1. Also shown (bottom left) is an enlarged, surface-shaded representation of the icosahedral asymmetric unit showing the complex of glycosylated CD155 with PV1. At the bottom right is an explanation of the north and south notation used to describe the canyon topology.
Difference map calculation.
The first step in the determination of the position of the carbohydrate sites was to make sure that all maps had the same magnification. This was achieved by identifying all map grid points (mask) that were associated with the capsid protein shell by comparison with the X-ray crystallographic atomic coordinates (PDB accession numbers: 2PLV for PV1, 1EAH for PV2, and 1PVC for PV3). The atomic coordinates were also used to compute structure factors representing the protein shell limited to 15-Å resolution data and using a temperature factor of 1,000 Å2. This map was then compared to the cryoEM map within the volume defined by the mask by computing correlation coefficients, assuming a series of pixel sizes for the electron microscopy (EM) map. The correct magnification of the EM map corresponded to the highest correlation. The scaled pixel sizes varied from 2.88 Å to 2.93 Å for the various cryoEM maps, whereas the independently EM-calibrated pixel size was 3.11 Å (Table 1). The second step was to determine a relative scale for the height of the densities. This scaling was done in reciprocal space by minimizing the squared difference between structure factors representing the X-ray and cryoEM maps within the volume of the mask. Having established both the radial and amplitude scale factors, vector difference maps were computed between receptor-virus complexes and the virus maps for each of the PV serotypes. In the third step, further difference maps were computed between the glycosylated receptor density and the deglycosylated receptor density, resulting in the identification of the carbohydrate sites (Table 2). This process compensated for possible unequal occupancy of the receptor on the viral surface. The final maps and difference maps were visualized using the program O (24).
TABLE 2.
Positions and significance of the glycosylation sites in difference maps
| Mapa | Coordinates and significanceb at:
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Asn105
|
Asn120
|
Asn188
|
Asn237
|
|||||||||||||
| x | y | z | H/N | x | y | z | H/N | x | y | z | H/N | x | y | z | H/N | |
| G-CD155-PV1 | −144.6 | 75.5 | 55.7 | 16/10 | −123.3 | 101.9 | 43.2 | 20/10 | −122.0 | 118.3 | 73.1 | 19/10 | −150.4 | 107.1 | 79.6 | 21/10 |
| G-CD155-PV2 | −121.5 | 101.1 | 38.5 | 23/11 | −120.6 | 114.4 | 62.1 | 15/11 | −149.1 | 97.5 | 83.1 | 18/11 | ||||
| G-CD155-PV3 | −120.5 | 99.6 | 43.9 | 30/10 | −116.6 | 116.0 | 67.3 | 15/10 | −157.5 | 109.1 | 76.6 | 18/10 | ||||
G-CD155-PV1 represents the fitted model in the density of glycosylated CD155 complexed with PV1. Similarly, D-CD155-PV1 refers to deglycosylated CD155.
H/N is the peak height in the carbohydrate difference map with respect to the noise. The mean density height of the difference map is 0. The noise is the biggest noncarbohydrate peak in the difference map.
There are two (Asn105 and Asn120), three, and three potential glycosylation sites in domain D1, D2, and D3 of CD155, respectively (Table 3 and Fig. 3). Four carbohydrate density features, two on D1 and two on D2 (Asn188 and Asn237), were found in the cryoEM difference maps of the PV1-CD155 complexes. The volume of the carbohydrate sites at Asn120 and Asn237 are the largest and might indicate that these sites are associated with longer polysaccharide molecules. However, only three carbohydrate moieties (one on D1 and two on D2) were found in the PV2- and PV3-CD155 complexes. The missing site at Asn105 is close to the viral surface. The different densities between the glycosylated CD155 and the deglycosylated CD155 show some peaks that belong to the carbohydrate sites of D3. However, due to the low-density height of these peaks and the unavailability of the CD155 structure, it is difficult to be sure of the correct structural interpretation of the glycosylation sites for domain D3.
TABLE 3.
Fitting statistics of CD155 model into EM density
| Map |
sumfa
|
avgdsb (Å) | Distancec (Å)
|
|||||
|---|---|---|---|---|---|---|---|---|
| D1 | D2 | D3 | Asn105 | Asn120 | Asn188 | Asn237 | ||
| G-CD155-PV1 | 51.4 | 41.7 | 18.6 | 11.4 | 12.8 | 16.7 | 6.2 | 9.8 |
| G-CD155-PV1 (old) | 50.2 | 40.7 | 18.0 | 12.7 | 13.4 | 19.0 | 8.8 | 9.6 |
| D-CD155-PV1 | 57.8 | 38.8 | 14.8 | |||||
| G-CD155-PV2 | 56.4 | 42.3 | 23.7 | 14.3 | 19.8 | 9.1 | 14.0 | |
| D-CD155-PV2 | 55.7 | 40.4 | 20.4 | |||||
| G-CD155-PV3 | 55.0 | 44.0 | 22.6 | 14.0 | 14.3 | 10.0 | 17.6 | |
| D-CD155-PV3 | 54.4 | 43.6 | 22.3 | |||||
sumf is the average density at all Cα atoms when the maximum height of the map is set to 100. D1, D2, and D3 show the value of sumf for each separate domain.
avgds is the average distance between the observed carbohydrate sites to the presumed Cα atoms of the glycosylated residue.
The Asn105, Asn120, Asn188, and Asn237 columns list individual distances for each glycosylated site from the given residue.
FIG. 3.

Stereoviews of superposition of the carbohydrate site difference densities obtained by subtracting the deglycosylated from the glycosylated CD155 densities. The carbohydrate densities are colored in red, green, and blue for those derived from the PV1, PV2, and PV3 complexes, respectively. The Cα backbone of CD155 is shown in black.
Accession numbers.
The coordinates of the various CD155 complexes with PV1, PV2, and PV3 have been deposited with the Protein Data Bank (accession numbers 1NN8).
RESULTS AND DISCUSSION
Model fitting.
An earlier atomic model of CD155 had been generated based on homology of the three different domains to immunoglobulin-like structures and on adjustment with respect to the then-available ∼22-Å resolution cryoEM density of CD155 complexed with PV1 (18) (PDB accession no. 1DGI). This model fitted reasonably well into all of the six cryoEM maps presented here (Table 3). Nevertheless, attempts were made to improve the relative orientation of the individual domains within the CD155 model by using the program EMfit (44). To achieve this, adjustments were made to the model with respect to the new 15-Å resolution map for PV1 complexed with glycosylated CD155. This map had the highest resolution of all of the maps presented here and permitted restraining of the domain positions with respect to the four identified glycosylation sites. Since the highest density was associated with domain D1 and the weakest with domain D3 (Table 2), the domains were fitted separately starting with domain D1 and finishing with domain D3, while being restrained by the positions of the glycosylation sites. Fitting of domain D2 was further restrained by requiring a minimal distance between the carboxy end of the already-fitted domain D1 with the amino end of domain D3. The density of domain D3 was of insufficient quality and lacked observable glycosylation sites to permit accurate fitting. Hence, its position and orientation were taken from the earlier model. The new model retained the relative orientation and positions of domain D2 and D3 to each other, but domain D1 has a rotation of 41.9° about its long axis compared to the previous model. The biggest displacement was 6.8 Å for Cα atoms in residues that were in contact with the virus surface. This change in the model structure was based on a better fit of the D1 model to the density and carbohydrate sites.
The new model was used to fit into the cryoEM maps representing PV2 and PV3 complexed with glycosylated or deglycosylated CD155 (Table 2). The new model gave a higher average density (sumf) taken over all Cα atoms in all six complexes compared to the old model (Table 2). The average value of sumf systematically decreases from domain D1 to D3 on account of the progressive increase in flexibility of the receptor as its distance from the virus surface increases. The AP domain (53 kDa when unglycosylated), fused to the CD155 molecule at its carboxy terminus, appeared only as a diffuse, low-density feature at almost the level of the background noise. The slightly higher values (Table 3) of sumf for PV2 and PV3 compared to PV1 are probably the result of the lower resolution and, hence, blurring of these maps, thus presenting a more uniform density to the model. The average distance between the position of the glycosylation sites to the appropriate Cα atom varied from 11.4 to 14.3 Å, which is comparable to a similar measurement in the structural determination of Sindbis virus (53).
To further validate the result of the EMfit program, the new model structure was used in conjunction with the SITUS program (50) for fitting into the various cryoEM maps. The algorithm used by SITUS for fitting is dependent on detecting the edges of density shape. In contrast, the EMfit program is dependent on maximizing the height of the density of all atoms in the cryoEM density. Furthermore, the EMfit results were restrained, when appropriate, by information on the position of the glycosylation sites, whereas the SITUS program fit is independent of the information derived for the glycosylation sites. In spite of the considerable difference in the algorithms used by these programs, the fits of the model into the densities were very similar, with root-mean-square (r.m.s.) deviation between equivalent Cα atoms being less than 2.3 Å in all cases (Table 4).
TABLE 4.
r.m.s. deviation (Å) between Cα atoms of the three-domain CD155 model when fitted into the various maps representing PV-CD155 complexesa
| Map | r.m.s. deviation (Å)
|
|||||
|---|---|---|---|---|---|---|
| G-CD155-PV1 | D-CD155-PV1 | G-CD155-PV2 | D-CD155-PV2 | G-CD155-PV3 | D-CD155-PV3 | |
| G-CD155-PV1 | 1.8 | 2.3 | 5.2 | 3.5 | 3.6 | 3.6 |
| D-CD155-PV1 | 2.3 | 5.5 | 3.6 | 4.7 | 4.8 | |
| G-CD155-PV2 | 2.1 | 4.1 | 4.3 | 2.5 | ||
| D-CD155-PV2 | 2.1 | 4.8 | 2.1 | |||
| G-CD155-PV3 | 2.2 | 3.2 | ||||
| D-CD155-PV3 | 1.2 | |||||
The values on the diagonal are the r.m.s. deviations between using the EMfit and SITUS programs for fitting the model into the cryoEM density.
The position and orientation of the new model in the glycosylated (restrained by the position of the carbohydrate sites) and deglycosylated cryoEM maps of PV1 differed by no more than the difference between using the EMfit and SITUS programs for fitting the model into the same density (Table 4). Thus, there was no detectable impact of the carbohydrate sites in the receptor-binding orientation or position in the complexes with PV1. The slightly larger difference between glycosylated and deglycosylated results for PV2 and PV3 are likely to be the consequence of the poorer resolution achieved for the cryoEM reconstructions involving PV2 and PV3 (Table 1).
The positions and orientations of the new model found for the different PV serotypes differed somewhat more than between the glycosylated and the deglycosylated structures for these serotypes. Whether this implies a real difference or merely an effect of the different qualities of the maps is uncertain. Nevertheless, at the current resolution, PV1, PV2, and PV3 have retained almost identical receptor-binding modes during their evolution, whereas their antigenic properties have changed. This finding is similar to the binding modes of ICAM-1 to HRV14 and HRV16 (26).
Virus-receptor interface.
The footprints of CD155 (7) onto the surface of PV1, PV2, and PV3 were determined in terms of the shortest distance any one atom of a specific viral residue made with any atom of CD155 (Fig. 4). There are two regions on CD155 that make contact with three regions on the viral surface. The C′-C"-D face of CD155 (Fig. 1a) makes contact with the south wall of the canyon, whereas the F-G face of CD155 interacts with the north wall of the canyon. The agreement between the positioning of mutations that alter virus infectivity and their proximity to the virus-receptor interface is a substantial improvement over the earlier structural studies (Fig. 1b) (18, 39). Even though some mutated residues (e.g., Asp117) are not in the virus-receptor interface, they may affect the CD155 binding affinity due to the change of the protein fold (6).
FIG. 4.

Footprints of CD155 on the surfaces of PV1 (top), PV2 (middle), and PV3 (bottom). Each figure is viewed in the same direction as those shown in Fig. 2, which is down an icosahedral twofold axis. The viral residues in the footprints are colored according to their nearest approach to a CD155 atom. The projections of the pocket factor to the CD155 footprints are outlined with a dashed line. The relative position of the CD155 footprint in the viral asymmetric unit is shown at the top of each panel. Shown at the top right is a compass to explain the notation used to describe the parts of the canyon.
The glycosylated Asn105 and Asn120 residues are on the E and F strands, respectively, of CD155. The carbohydrate difference densities show that Asn105 is close to the south rim and Asn120 is pointing toward the north rim of the canyon (Fig. 5). However, neither of these carbohydrate difference densities is in contact with the viral surface. Nevertheless, due to the proximity of Asn105 to the viral surface, the associated carbohydrate moiety, which is probably considerably bigger than the observed density, might cause steric hindrance with the south rim of the canyon during receptor binding. Hence, the absence of this carbohydrate moiety may increase binding affinity of the receptor, as was observed in mutagenesis studies (5, 6).
FIG. 5.

(a) Stereoviews of the carbohydrate difference density (red) of domain D1 and D2 of the PV1-CD155 complex. The deglycosylated CD155 density and the viral surface are shown in green and blue, respectively. (b) The Cα backbone of CD155 D1 is shown in black. The orientation of the canyon is marked as north, south, and east.
The closest contacts between CD155 and the virus are with the south rim and east end of the canyon, whereas the residues on the north wall of the canyon make relatively distant contacts (Fig. 4). The contact region with the north wall of the canyon, formed by residues Val1107 and Lys1109 in PV1 and PV2 or by Met1107 and Arg1109 in PV3, respectively, is close to the hydrophobic pocket in VP1. (Poliovirus residues are numbered sequentially from 1001, 2001, 3001, and 4001 for the viral proteins VP1, VP2, VP3, and VP4, respectively. Residues of CD155 are identified with only two or three digits.) Thus, the receptor interaction with the north side of the canyon is likely to be important for the release of the pocket factor and therefore the destabilization of the virus, whereas the contacts with the south wall and the east end of the canyon are likely to be more important for receptor recognition. This hypothesis finds support in the properties of a CD155 mutant (Q130G, G131D) that is unable to bind to PV1 and PV2 but can bind to PV3; however, the binding of this CD155 mutant to PV3 does not lead to viral infection, presumably because it cannot initiate viral uncoating (17). These two mutated residues in CD155 are close to the north wall of the canyon in the virus-receptor complexes, with residue Gln130 close to the conserved viral residues 1107 and 1109 and residue Gly131 in the proximity of residue Glu1168 in PV1, Gly1168 in PV2, and Lys1168 in PV3. The phenotype of the mutant CD155 can be rationalized in that binding to PV1 and PV2 is destroyed by the extra charge on CD155 residue Asp131, whereas in PV3 there may be a formation of a salt bridge between Asp131 and Lys1168. However, due to the glycine at position 130, the CD155 mutant would no longer have the capacity to affect the binding of the pocket factor and therefore would be unable to alter the viral stability of PV3 as required for uncoating.
Receptor-mediated viral uncoating.
It has been shown that PV interacts with the cellular receptor in two distinct steps (31, 48). The first step, probably electrostatic in nature, can be isolated at 4°C. The second step, which dominates at higher temperatures, leads to irreversible structural changes of the virion, resulting in the formation of A-particles and 130S particles (49). Recently, these steps have been correlated with two distinct binding affinities of CD155 when it binds to PV (31). The relative abundance of the high binding affinity site decreases at low temperature, whereas the abundance of the low binding affinity site remains almost constant at different temperatures (31). Therefore, the cryoEM structures of the PV-CD155 complexes, which were formed at 4°C, would represent the dominant “low-affinity” binding site. This correlates well with data suggesting that PV and CD155 may form an initial binding complex, which later is transformed to an activated complex (48). Furthermore, cryoEM investigations (26, 37) showed that complexes of HRVs with their ICAM-1 receptor are more stable at 4°C. Therefore, the dominant low-temperature CD155 binding site of McDermott et al. (31) and the initial binding complex of Tsang et al. (48) probably correspond to the structures observed by cryoEM. Hence, the tighter receptor-virus contacts with the south rim and the east end of the canyon define the initial binding orientation of CD155, whereas the north rim of the canyon may be responsible for forming the transient activated complex, which favors pocket factor release and uncoating. Increase of temperature could increase the “structural breathing” of the virion (29), allowing the receptor to make better contacts with the north rim of the canyon (Fig. 6).
FIG. 6.

Model of receptor-mediated PV uncoating. The south wall of the canyon is probably responsible for forming the initial binding state of the PV-CD155 complex. The interaction between the north wall of the canyon and CD155 immediately above the hydrophobic binding pocket helps the release of the pocket factor, thus destabilizing the virus. Heating increases the structural “breathing” of the virus, thus providing the conformational change required for forming the “activated state” of the PV-CD155 complex.
Although the contact regions of ICAM-1 with HRVs and of CAR with CVB3 (19) lack the additional region at the east end of the canyon, nevertheless the more extensive contact regions between receptor and virus are also on the south wall of the canyon (26). Thus, the proposed functional division between different parts of the receptor footprint is likely to be equally valid for other rhinoviruses and enteroviruses. Moreover, the larger surface area utilized by CD155 in making contacts with the virus would require that more surface residues need to be conserved to retain the receptor-binding activity. This suggests that PV escape mutants from antibody neutralization may also lose their ability to bind to CD155. Thus, the large interface of CD155 with PVs, as opposed to ICAM-1 with HRVs or CAR with CVBs, would limit the number of possible PV serotypes, a hypothesis entertained previously (17).
Acknowledgments
We thank Wei Zhang and Chuan Xiao for the use of their programs regarding the cryoEM reconstructions. We are grateful to Rob Ashmore for the use of his RobEM suite of programs. We thank Timothy S. Baker for his support. We thank Sharon Wilder and Cheryl Towell for help in the preparation of the manuscript.
This research was supported by NIH grants to M.G.R. (AI11219), to E.W. (AI39485), and to T. S. Baker (GM33050), as well as by facility grants from the Keck Foundation and Purdue University.
REFERENCES
- 1.Arita, M., S. Koike, J. Aoki, H. Horie, and A. Nomoto. 1998. Interaction of poliovirus with its purified receptor and conformational alteration in the virion. J. Virol. 72:3578-3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker, T. S., and R. H. Cheng. 1996. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J. Struct. Biol. 116:120-130. [DOI] [PubMed] [Google Scholar]
- 3.Baker, T. S., N. H. Olson, and S. D. Fuller. 1999. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol. Mol. Biol. Rev. 63:862-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belnap, D. M., B. M. McDermott, Jr., D. J. Filman, N. Cheng, B. L. Trus, H. J. Zuccola, V. R. Racaniello, J. M. Hogle, and A. C. Steven. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA 97:73-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernhardt, G., J. A. Bibb, J. Bradley, and E. Wimmer. 1994. Molecular characterization of the cellular receptor for poliovirus. Virology 199:105-113. [DOI] [PubMed] [Google Scholar]
- 6.Bernhardt, G., J. Harber, A. Zibert, M. deCrombrugghe, and E. Wimmer. 1994. The poliovirus receptor: identification of domains and amino acid residues critical for virus binding. Virology 203:344-356. [DOI] [PubMed] [Google Scholar]
- 7.Chapman, M. S. 1993. Mapping the surface properties of macromolecules. Protein Sci. 2:459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chothia, C., and E. Y. Jones. 1997. The molecular structure of cell adhesion molecules. Annu. Rev. Biochem. 66:823-862. [DOI] [PubMed] [Google Scholar]
- 9.Colston, E., and V. R. Racaniello. 1994. Soluble receptor-resistant poliovirus mutants identify surface and internal capsid residues that control interaction with the cell receptor. EMBO J. 13:5855-5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curry, S., M. Chow, and J. M. Hogle. 1996. The poliovirus 135S particle is infectious. J. Virol. 70:7125-7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dove, A. W., and V. R. Racaniello. 1997. Cold-adapted poliovirus mutants bypass a postentry replication block. J. Virol. 71:4728-4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filman, D. J., R. Syed, M. Chow, A. J. Macadam, P. D. Minor, and J. M. Hogle. 1989. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 8:1567-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filman, D. J., M. W. Wien, J. A. Cunningham, J. M. Bergelson, and J. M. Hogle. 1998. Structure determination of echovirus 1. Acta Crystallogr. D54:1261-1272. [DOI] [PubMed]
- 14.Flanagan, J. G., and P. Leder. 1990. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell 63:185-194. [DOI] [PubMed] [Google Scholar]
- 15.Gromeier, M., L. Alexander, and E. Wimmer. 1996. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl. Acad. Sci. USA 93:2370-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gromeier, M., D. Solecki, D. D. Patel, and E. Wimmer. 2000. Expression of the human poliovirus receptor/CD155 gene during development of the central nervous system: implications for the pathogenesis of poliomyelitis. Virology 273:248-257. [DOI] [PubMed] [Google Scholar]
- 17.Harber, J., G. Bernhardt, H.-H. Lu, J. Y. Sgro, and E. Wimmer. 1995. Canyon rim residues, including antigenic determinants, modulate serotype-specific binding of polioviruses to mutants of the poliovirus receptor. Virology 214:559-570. [DOI] [PubMed] [Google Scholar]
- 18.He, Y., V. D. Bowman, S. Mueller, C. M. Bator, J. Bella, X. Peng, T. S. Baker, E. Wimmer, R. J. Kuhn, and M. G. Rossmann. 2000. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA 97:79-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He, Y., P. R. Chipman, J. Howitt, C. M. Bator, M. A. Whitt, T. S. Baker, R. J. Kuhn, C. W. Anderson, P. Freimuth, and M. G. Rossmann. 2001. Interaction of coxsackievirus B3 with the full-length coxsackievirus-adenovirus receptor. Nat. Struct. Biol. 8:874-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He, Y., F. Lin, P. R. Chipman, C. M. Bator, T. S. Baker, M. Shoham, R. J. Kuhn, M. E. Medof, and M. G. Rossmann. 2002. Structure of decay-accelerating factor bound to echovirus 7: a virus-receptor complex. Proc. Natl. Acad. Sci. USA 99:10325-10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hendry, E., H. Hatanaka, E. Fry, M. Smyth, J. Tate, G. Stanway, J. Santti, M. Maaronen, T. Hyypiä, and D. Stuart. 1999. The crystal structure of coxsackievirus A9: new insights into the uncoating mechanisms of enteroviruses. Structure 7:1527-1538. [DOI] [PubMed] [Google Scholar]
- 22.Hewat, E. A., E. Neumann, J. F. Conway, R. Moser, B. Ronacher, T. C. Marlovits, and D. Blaas. 2000. The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: a structural view. EMBO J. 19:6317-6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogle, J. M., M. Chow, and D. J. Filman. 1985. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science 229:1358-1365. [DOI] [PubMed] [Google Scholar]
- 24.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47:110-119. [DOI] [PubMed] [Google Scholar]
- 25.Koike, S., H. Horie, I. Ise, A. Okitsu, M. Yoshida, N. Iizuka, K. Takeuchi, T. Takegami, and A. Nomoto. 1990. The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 9:3217-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolatkar, P. R., J. Bella, N. H. Olson, C. M. Bator, T. S. Baker, and M. G. Rossmann. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18:6249-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lange, R., X. Peng, E. Wimmer, M. Lipp, and G. Bernhardt. 2001. The poliovirus receptor CD155 mediates cell-to-matrix contacts by specifically binding to vitronectin. Virology 285:218-227. [DOI] [PubMed] [Google Scholar]
- 28.Lentz, K. N., A. D. Smith, S. C. Geisler, S. Cox, P. Buontempo, A. Skelton, J. DeMartino, E. Rozhon, J. Schwartz, V. Girijavallabhan, J. O'Connell, and E. Arnold. 1997. Structure of poliovirus type 2 Lansing complexed with antiviral agent SCH48973: comparison of the structural and biological properties of the three poliovirus serotypes. Structure 5:961-978. [DOI] [PubMed] [Google Scholar]
- 29.Lewis, J. K., B. Bothner, T. J. Smith, and G. Siuzdak. 1998. Antiviral agent blocks breathing of the common cold virus. Proc. Natl. Acad. Sci. USA 95:6774-6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao, S., and V. R. Racaniello. 1997. Allele-specific adaptation of poliovirus VP1 B-C loop variants to mutant cell receptors. J. Virol. 71:9770-9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDermott, Jr., B. M., A. H. Rux, R. J. Eisenberg, G. H. Cohen, and V. R. Racaniello. 2000. Two distinct binding affinities of poliovirus for its cellular receptor. J. Biol. Chem. 275:23089-23096. [DOI] [PubMed] [Google Scholar]
- 32.Melnick, J. L. 1996. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p. 655-712. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 33.Mendelsohn, C. L., E. Wimmer, and V. R. Racaniello. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56:855-865. [DOI] [PubMed] [Google Scholar]
- 34.Morrison, M. E., Y.-J. He, M. W. Wien, J. M. Hogle, and V. R. Racaniello. 1994. Homolog-scanning mutagenesis reveals poliovirus receptor residues important for virus binding and replication. J. Virol. 68:2578-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muckelbauer, J. K., M. Kremer, I. Minor, G. Diana, F. J. Dutko, J. Groarke, D. C. Pevear, and M. G. Rossmann. 1995. The structure of coxsackievirus B3 at 3.5 Å resolution. Structure 3:653-667. [DOI] [PubMed] [Google Scholar]
- 36.Mueller, S., X. Cao, R. Welker, and E. Wimmer. 2002. Interaction of the poliovirus receptor CD155 with the dynein light chain Tctex-1 and its implication for poliovirus pathogenesis. J. Biol. Chem. 277:7897-7904. [DOI] [PubMed] [Google Scholar]
- 37.Olson, N. H., P. R. Kolatkar, M. A. Oliveira, R. H. Cheng, J. M. Greve, A. McClelland, T. S. Baker, and M. G. Rossmann. 1993. Structure of a human rhinovirus complexed with its receptor molecule. Proc. Natl. Acad. Sci. USA 90:507-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pallansch, M. A., and R. P. Roos. 2001. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p. 723-775. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 1. Lippincott/The Williams & Wilkins Co., Philadelphia, Pa.
- 39.Racaniello, V. R. 1996. Early events in poliovirus infection: virus-receptor interactions. Proc. Natl. Acad. Sci. USA 93:11378-11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Racaniello, V. R. 2001. Picornaviridae: the viruses and their replication, p. 685-722. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 1. Lippincott/The Williams & Wilkins Co., Philadelphia, Pa.
- 41.Rieder, E., and E. Wimmer. 2002. Cellular receptors of picornaviruses: an overview, p. 61-70. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. American Society for Microbiology, Washington, D.C.
- 42.Rossmann, M. G. 1994. Viral cell recognition and entry. Protein Sci. 3:1712-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossmann, M. G., E. Arnold, J. W. Erickson, E. A. Frankenberger, J. P. Griffith, H. J. Hecht, J. E. Johnson, G. Kamer, M. Luo, A. G. Mosser, R. R. Rueckert, B. Sherry, and G. Vriend. 1985. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 317:145-153. [DOI] [PubMed] [Google Scholar]
- 44.Rossmann, M. G., R. Bernal, and S. V. Pletnev. 2001. Combining electron microscopic with X-ray crystallographic structures. J. Struct. Biol. 136:190-200. [DOI] [PubMed] [Google Scholar]
- 45.Rossmann, M. G., Y. He, and R. J. Kuhn. 2002. Picornavirus-receptor interactions. Trends Microbiol. 10:324-331. [DOI] [PubMed] [Google Scholar]
- 46.Rueckert, R. R. 1996. Picornaviridae: the viruses and their replication, p. 609-654. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 47.Smith, T. J., M. J. Kremer, M. Luo, G. Vriend, E. Arnold, G. Kamer, M. G. Rossmann, M. A. McKinlay, G. D. Diana, and M. J. Otto. 1986. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science 233:1286-1293. [DOI] [PubMed] [Google Scholar]
- 48.Tsang, S. K., B. M. McDermott, V. R. Racaniello, and J. M. Hogle. 2001. Kinetic analysis of the effect of poliovirus receptor on the viral uncoating: the receptor as a catalyst. J. Virol. 75:4984-4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wimmer, E., J. J. Harber, J. A. Bibb, M. Gromeier, H.-H. Lu, and G. Bernhardt. 1994. Poliovirus receptors, p. 101-127. In E. Wimmer (ed.), Cellular receptors for animal viruses. Cold Spring Harbor Press, Cold Spring Harbor, N.Y.
- 50.Wriggers, W., R. A. Milligan, and J. A. McCammon. 1999. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol. 125:185-189. [DOI] [PubMed] [Google Scholar]
- 51.Xiao, C., C. M. Bator, V. D. Bowman, E. Rieder, Y. He, B. Hébert, J. Bella, T. S. Baker, E. Wimmer, R. J. Kuhn, and M. G. Rossmann. 2001. Interaction of coxsackievirus A21 with its cellular receptor, ICAM-1. J. Virol. 75:2444-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xing, L., K. Tjarnlund, B. Lindqvist, G. G. Kaplan, D. Feigelstock, R. H. Cheng, and J. M. Casasnovas. 2000. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J. 19:1207-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang, W., S. Mukhopadhyay, S. V. Pletnev, T. S. Baker, R. J. Kuhn, and M. G. Rossmann. 2002. Placement of the structural proteins in Sindbis virus. J. Virol. 76:11645-11658. [DOI] [PMC free article] [PubMed]