

Abstract
Tegument proteins of herpes simplex virus type 1 (HSV-1) are hypothesized to contain the functional information required for the budding or envelopment process proposed to occur at cytoplasmic compartments of the host cell. One of the most abundant tegument proteins of HSV-1 is the UL49 gene product, VP22, a 38-kDa protein of unknown function. To study its subcellular localization, a VP22-green fluorescent protein chimera was expressed in transfected human melanoma (A7) cells. In the absence of other HSV-1 proteins, VP22 localizes to acidic compartments of the cell that may include the trans-Golgi network (TGN), suggesting that this protein is membrane associated. Membrane pelleting and membrane flotation assays confirmed that VP22 partitions with the cellular membrane fraction. Through truncation mutagenesis, we determined that the membrane association of VP22 is a property attributed to amino acids 120 to 225 of this 301-amino-acid protein. The above results demonstrate that VP22 contains specific information required for targeting to membranes of acidic compartments of the cell which may be derived from the TGN, suggesting a potential role for VP22 during tegumentation and/or final envelopment.
The virion proteins of herpes simplex virus type 1 (HSV-1) are arranged within three morphologically distinct components of the virus: the nucleocapsid, the host-derived lipid envelope containing glycoproteins and other transmembrane proteins, and the tegument, the proteinaceous region of the virus located between the capsid and the envelope (49). The herpesvirus assembly process involves a complex sequence of events occurring in numerous cellular compartments. It is well established that capsid assembly and DNA packaging occur within the nucleus (49). The assembled nucleocapsids are then shuttled into the cytoplasm by a budding-fusion process that takes place on the inner and outer nuclear membranes, respectively (reviewed in references 21, 39, and 40). Current evidence supports a model where final tegumentation and envelopment of herpesviruses occur when nucleocapsids bud into cytoplasmic vesicles, which may be derived from the trans-Golgi network (TGN) (9, 24-27, 31, 40, 50, 53, 56, 60, 63, 64, 66). Subsequently, these cytoplasmic vesicles containing enveloped particles are transported to the plasma membrane, where fusion results in the release of progeny virus (40).
The molecular mechanisms underlying the formation of the tegument region remain largely undefined for HSV-1 (40, 49). Historically, the tegument has been described as an amorphous proteinaceous region where the arrangement of composite proteins is largely random; however, recent evidence suggests that the tegument is an organized structure built through specific protein-protein interactions (reviewed in reference 40). Nevertheless, the nature of these interactions and the functions of most of the tegument proteins are unknown. Although certain tegument proteins tend to associate with the capsid while others may interact with glycoproteins or the lipid envelope, evidence indicates that collectively, tegument proteins contain all of the functions necessary to facilitate budding within cytoplasmic vesicles (38, 48, 58). Therefore, it is our goal to provide new insight regarding what tegument proteins are localized to cytoplasmic compartments as well as how they may interact to form stable structures as a prelude to the process of tegumentation and HSV-1 assembly. The studies described in this report are focused on HSV-1 VP22, a major component of the tegument region.
One of the most abundant tegument proteins in HSV-1 is the 301-amino-acid protein VP22, the product of the UL49 gene (20). According to studies performed by Heine et al., nearly 2,000 copies of VP22 were estimated to be present within each virion (29). Despite its abundance, very little is known regarding the role of VP22 during HSV-1 infection. Attempts to make a VP22-null virus have been unsuccessful, but the carboxy-terminal 89 amino acids do not appear to be necessary for production of infectious virus (46). VP22 is reported to be highly phosphorylated as well as capable of interacting with another major tegument protein, VP16 (13, 18, 19, 23). Several studies have focused on analyzing the localization of VP22 in both transfected and infected cells (1, 6, 14-17, 32, 37, 45). These studies have suggested dynamic trafficking properties of VP22, where the protein is capable of both cytoplasmic and nuclear accumulation (1, 6, 14-17, 32, 37, 45). In one study, by use of live-cell fluorescence during virus infection, VP22 demonstrated a punctate cytoplasmic localization reminiscent of the Golgi apparatus (16). This localization and its significance as an intrinsic property of VP22 have never been addressed.
In the present investigation, we examined the subcellular localization and membrane association of VP22. Using confocal microscopy, we observed that VP22 localized to acidic compartments of the cell, which may include the TGN. Given the punctate fluorescence exhibited by VP22, we addressed the ability of VP22 to associate with cellular membranes. Through truncation mutagenesis, we determined that membrane association of VP22 is a specific property that can be attributed to a particular region of the protein.
MATERIALS AND METHODS
Cells.
A7 (human melanoma) cells, a gift from Gary Thomas (The Oregon Health Sciences University, Portland), were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum, 2.25% sodium bicarbonate, 25 mM HEPES buffer, glutamine (0.3 μg/ml), penicillin (65 μg/ml), and streptomycin (131 μg/ml) (61).
Construction of VP22-GFP and VP22 truncation mutants.
The UL49 gene used for this study was PCR amplified from the HSV-1 KOS genome by using Platinum Taq polymerase (Invitrogen) (20, 54). PCR was performed with a forward primer (containing a BglII site) complementary to the sequence located 92 bp upstream of the VP22 start codon and a reverse primer (containing a HindIII site) complementary to the 3′ end of the UL49 gene but with a mutation to the stop codon of VP22 (TGA→TGC) to allow for read-through. This product was cut with BglII and HindIII and cloned into the multiple cloning site (MCS) of the pEGFP-N2 vector (Clontech), producing a plasmid, pVP22.GFP, that encodes a VP22-green fluorescent protein (GFP) chimera.
The VP22 C-terminal truncation mutants were made in a similar fashion, but pVP22.GFP was used as the source of the UL49 gene. A forward primer (containing a BglII site) complementary to the junction of the MCS and 5′ upstream sequence of UL49 and a reverse primer (containing a HindIII site) complementary to the region of the gene corresponding to the designated truncation were used to PCR amplify the truncated UL49 gene. This product was cut with BglII and HindIII and cloned into the MCS of the pEGFP-N2 vector, resulting in a vector that encodes a VP22.1−AA.GFP fusion protein.
The VP22 N-terminal truncations were constructed by overlap PCR mutagenesis. The forward primer (F1), which was also used for C-terminal truncation mutagenesis (containing a BglII site), and a reverse primer (R1) complementary to the start codon of VP22 and the sequence immediately upstream of the start codon were used to generate a product containing the upstream sequence and initiator codon of the UL49 gene. In a parallel PCR, a forward primer (F2) complementary to both the R1 primer and the site of truncation and a reverse primer (R2) (containing a HindIII site) complementary to the 3′ end of the UL49 gene were used to generate a product containing the N-terminal truncation of the UL49 gene. The upstream product and the UL49 truncation product were then combined along with primers F1 and R2 in a PCR to generate a truncated UL49 gene with the appropriate start codon and upstream sequence. This product was cut with BglII and HindIII and cloned into the MCS of the pEGFP-N2 vector, generating a vector that encodes a VP22.AA−301.GFP fusion protein. The UL49 gene that was cloned as a fusion construct with the gene encoding GFP and all of the subsequent UL49 truncation mutants were sequenced in their entirety to authenticate the identity of the PCR products.
Expression and metabolic labeling of VP22 chimeras.
A7 cells were transfected by the calcium phosphate method, as previously described (12). To measure the levels of expression and expected molecular masses of the VP22 chimeras, lysates of transfected cells were assayed by Western blotting using an antibody raised in rabbits against a peptide derived from the GFP protein (Clontech). Bound antibody was detected with a goat anti-rabbit antibody conjugated to peroxidase (Sigma) by chemiluminescence with ECL reagents (Amersham Pharmacia) and Kodak X-Omat AR5 film.
Metabolic labeling of A7 cells, grown in 35-mm-diameter plates, was performed at approximately 20 h posttransfection. Cells were starved for 15 min in methionine-free, cysteine-free DMEM (Invitrogen) and then metabolically labeled with 45 μCi (>1,000 Ci/mmol) of EXPRE35S35S Protein Labeling Mix (Perkin-Elmer) in 400 μl of methionine-free, cysteine-free DMEM for 2.5 h. Cells were harvested and mixed with radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate, 1% Triton X-100, 25 mM Tris [pH 8.0]) supplemented with protease inhibitors (Sigma). VP22 chimeras were then immunoprecipitated by using a polyclonal goat anti-GFP serum (Rockland) and protein A (65).
Immunoprecipitated proteins were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) on 10% polyacrylamide gels. The gels were both fixed and dried. Radiolabeled proteins were detected by autoradiography with Kodak X-Omat AR5 film. Gels were exposed to film for 1 to 2 days.
Confocal microscopy of VP22 chimeras.
A7 cells were transfected with the VP22 chimeras as described above. For transfection-infection experiments, A7 cells transfected for 24 h were infected with HSV-1 KOS (multiplicity of infection, 10) in 1% fetal bovine serum in phosphate-buffered saline (PBS). At various time points between 12 and 36 h posttransfection, cells were washed twice in Tris-buffered saline and immediately viewed by using a Zeiss laser-scanning microscope with a helium-argon laser.
In order to study the subcellular localization of VP22-GFP, immunofluorescence was performed on A7 cells. Approximately 24 h posttransfection, cells were fixed with 3% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100 in PBS. Subsequently, cell monolayers were incubated for 1 h with either a monoclonal antibody raised against AP-1 (Sigma) or a polyclonal sheep antibody raised against TGN-46 (Serotec). Cells were then incubated with either a goat anti-mouse immunoglobulin G antibody conjugated to tetramethyl rhodamine isothiocyanate (TRITC) (Sigma) or a rabbit anti-sheep antibody conjugated to Texas Red (Rockland). Intrinsic GFP fluorescence was observed as described above, while immunofluorescence was detected by using the helium-neon laser of the Zeiss laser-scanning microscope. Images were colored and digitally combined in Adobe Photoshop, version 7.0.
Live-cell fluorescence using LysoTracker Red (Molecular Probes) was performed at a concentration of 5 μl of LysoTracker/ml of DMEM. A7 cells were treated for 1.25 h at 37°C and were subsequently washed with Tris-buffered saline. GFP fluorescence and LysoTracker Red staining were observed by confocal microscopy as described above.
Membrane pelleting and quantitation of membrane association by fluorometry.
At 20 h posttransfection, A7 cells were washed twice and harvested in cold PBS. Intact cells were pelleted at 1,000 × g for 5 min (at 4°C) and resuspended by mild vortexing in 1 ml of hypotonic buffer (10 mM Tris, 0.2 mM MgCl2 [pH 7.4]) supplemented with Complete protease inhibitor cocktail (Roche). Following a 30-min incubation on ice, cells were lysed by Dounce homogenization (30 to 35 strokes). Cell lysates were centrifuged at 1,000 × g for 10 min (4°C) to remove unbroken cells and nuclei. Postnuclear supernatants were then centrifuged at 100,000 × g for 40 min (at 4°C) in a Beckman Optima TLX ultracentrifuge to pellet cellular membranes. As a control, replica samples were incubated with 0.5% Triton X-100 for 1 h (at 4°C) prior to centrifugation to solubilize cellular membranes. Soluble and pellet fractions were collected and adjusted to 0.5% Triton X-100 in hypotonic buffer (51). The amount of VP22 (fused to GFP) present in each fraction was quantitated by using an Aminco-Bowman series 2 fluorescence spectrophotometer with an excitation filter at 488 nm and an emission filter at 514 nm. The percentage of protein pelleted was calculated by dividing the amount in the pellet fraction (P) by the sum of the amounts in the soluble fraction (S) and the pellet fraction [P/(P + S)].
Membrane flotation gradient centrifugation.
Methods for analysis of membrane-associated proteins by sucrose density gradient flotation have been described previously (30, 42, 55). By using a modified form of this assay, a 100-mm-diameter plate of A7 cells was washed and harvested in cold PBS at 20 h posttransfection. Intact cells were pelleted at 1,000 × g for 5 min (at 4°C) and resuspended by mild vortexing in 0.8 ml of hypotonic buffer supplemented with Complete protease inhibitor cocktail (Roche). Following a 30-min incubation on ice, cells were lysed by Dounce homogenization (30 to 35 strokes). Cell lysates were centrifuged at 1,000 × g for 10 min (at 4°C) to remove unbroken cells and nuclei. Then 0.3 ml of postnuclear supernatant was mixed with 1.6 ml of 85% (wt/vol) sucrose in NTE buffer (100 mM NaCl, 10 mM Tris, 1 mM EDTA [pH 7.4]) (final sucrose concentration, 71.5%) and placed at the bottom of a centrifuge tube. Next, 7 ml of 65% sucrose in NTE and then 3.1 ml of 10% sucrose in NTE were layered into the centrifuge tube. This sucrose step gradient was then centrifuged at 100,000 × g for 18 h (at 4°C) in a Beckman SW41 rotor. Twelve 1-ml fractions were collected from the bottom of the tube for analysis by fluorometry. The amount of VP22 (fused to GFP) present in each fraction was determined by dividing the amount of fluorescence in a given fraction (Fx) by the sum of the fluorescences of all fractions [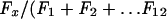 )].
)].
Radioimmunoprecipitation of membrane flotation gradient fractions required metabolic labeling of transfected A7 cells grown in 100-mm-diameter plates. Approximately 20 h posttransfection, cells were starved for 15 min in methionine-free, cysteine-free DMEM (Invitrogen) and then metabolically labeled with 135 μCi (>1,000 Ci/mmol) of EXPRE35S35S Protein Labeling Mix (Perkin-Elmer) in 2 ml of methionine-free, cysteine-free DMEM for 2.5 h. Cells were harvested and membranes were floated as described above. The 12 1-ml fractions were mixed in a 1:1 ratio with 2× RIPA buffer and then incubated overnight at 4°C with a polyclonal goat anti-GFP antibody (Rockland) (65). Fractions were immunoprecipitated with protein A (65). Immunoprecipitated proteins were resolved by SDS-PAGE on 10% polyacrylamide gels. The gels were both fixed and dried. Radiolabeled proteins were detected by autoradiography with Kodak X-Omat AR5 film. Gels were exposed to film for approximately 1 week. PhosphorImager analysis (Molecular Dynamics) was performed to determine the amount of VP22 that associated with cellular membranes. Quantitation was performed by dividing the amount of VP22 in one fraction (Fx) by the sum of the amounts of VP22 in all of the gradient fractions [ )].
)].
Differential extraction of cellular membranes was performed on the postnuclear supernatants of transfected, radiolabeled A7 cells. Membrane-associated proteins were extracted with either 1 M NaCl, 0.1 M Na2CO3 (pH 11), or 1% Triton X-100 for 1 h at 4°C and were subsequently processed by using the membrane flotation protocol described above (57). All experiments involving membrane flotation gradient centrifugation were repeated at least three times to ensure reproducibility.
RESULTS
Although VP22 is one of the most abundant HSV-1 tegument proteins, its role during the events of tegumentation and final envelopment remains poorly defined. The results presented in this report suggest that VP22 associates with cellular membranes and localizes to the acidic compartments of the cell, which may include the TGN, the proposed site for herpesvirus tegumentation and final envelopment.
Intracellular localization of VP22.
VP22 encoded in the HSV-1 KOS genome was cloned as a C-terminal fusion protein to GFP (Fig. 1A) so that its localization could be visualized in living cells. A7 human melanoma cells were used for all experiments because these cells are easily transfected, can be productively infected by HSV-1 (data not shown), and have been utilized to study membrane trafficking events at the TGN (44, 61).
FIG. 1.

Expression of VP22 within A7 cells. (A) Construction of VP22-GFP. VP22 from the HSV-1 KOS genome was cloned as a C-terminal fusion to the GFP protein. (B) Western blot analysis. A7 melanoma cells were transfected with the indicated plasmids, and cell lysates were separated by SDS-PAGE. Proteins transferred to nitrocellulose membranes were probed with a polyclonal antibody specific for GFP. (C) Subcellular localization of VP22. Plasmid DNA encoding VP22-GFP was transfected into A7 cells as in the experiment for which results are shown in panel B, and localization of the VP22-GFP chimera was observed in the absence or presence of HSV-1 infection. Fluorescence was visualized by live-cell confocal microscopy at approximately 32 h posttransfection (transfection only) or 32 h posttransfection-infection (24 h posttransfection plus 8 h postinfection).
Transfection of A7 cells with the plasmid encoding VP22-GFP followed by Western blot analysis (Fig. 1B) demonstrated that the chimeric protein was expressed and migrated at the expected molecular mass of approximately 64,000 Da. The intracellular localization of VP22 in transfected A7 cells was examined by live-cell fluorescence using confocal microscopy. VP22 exhibited a punctate perinuclear localization in A7 cells (Fig. 1C, left panel). This pattern was observed at various time points following transfection (data not shown). Transfection-infection experiments were conducted with A7 cells to determine whether expression of other HSV-1 proteins would alter the localization of VP22. Again, multiple time points were examined, and VP22 localization was visualized by live-cell fluorescence microscopy. At 8 h after infection of transfected cells with HSV-1, the localization of VP22 was similar to that in transfected A7 cells expressing only VP22 (Fig. 1C, right panel). Identical results were obtained for VP22-transfected Vero cells at early time points posttransfection (6 to 12 h), but in contrast to A7 cells, at later times after transfection (18 to 36 h), the punctate pattern was accompanied by a filamentous localization (data not shown). However, the filamentous pattern was not observed in Vero cells when the transfection-infection protocol was used; rather, only a punctate perinuclear fluorescence was observed, as in previous studies (data not shown) (16). These results suggest that the filamentous pattern of fluorescence exhibited in Vero cells by VP22 is cell type dependent, and therefore we chose to use A7 cells for most of our experiments.
To further define the subcellular localization of VP22, its position relative to those of cellular markers was assessed in fixed and permeabilized cells. Because herpesvirus budding is proposed to occur at the TGN, we hypothesized that this would be the compartment containing the tegument protein. The fluorescence pattern of VP22-GFP in A7 cells was compared to those of two cellular markers for the TGN: AP-1, a major clathrin adaptor protein of the TGN (27), and TGN-46, a resident glycoprotein of the TGN (27). Confocal microscopy showed that VP22-GFP (Fig. 2A and D) and the two cellular markers (Fig. 2B and E) appear to localize to similar regions of the cell (Fig. 2C and F). In these experiments, the expression levels of VP22-GFP were much higher than those of the endogenously expressed cellular proteins (AP-1 and TGN-46), and this may explain the failure of all of the VP22-GFP to colocalize with the two cellular proteins. Alternatively, VP22-GFP may be associated with other cytoplasmic vesicles within the cell.
FIG. 2.

Subcellular localization of VP22 in A7 cells. (A to F) Subcellular localization of VP22 relative to those of cellular markers for the TGN. Immunofluorescence was performed on A7 cells that were transfected with the plasmid encoding VP22-GFP. After 20 h posttransfection, cells were fixed with paraformaldehyde and permeabilized with Triton X-100. In panels A to C, cells were labeled with a monoclonal antibody against AP-1, which was detected by a rabbit anti-mouse antibody conjugated to TRITC. Panels A and B are the same field viewed by confocal microscopy with the appropriate wavelength to excite GFP (A) or TRITC (B), and these images were digitally combined to produce the image in panel C. In panels D to F, cells were labeled with a polyclonal sheep antibody against TGN-46, which was detected by a rabbit anti-sheep antibody conjugated to Texas Red. Panels D and E are the same field viewed by confocal microscopy with the appropriate wavelength to excite GFP (D) or Texas Red (E), and these images were digitally combined to produce the image in panel F. (G to I) Subcellular localization of VP22 relative to the acidic compartments of the cell. Live-cell fluorescence was performed on A7 cells expressing VP22-GFP and stained with LysoTracker Red. Panels G and H are the same field viewed by confocal microscopy with the appropriate wavelength to excite GFP (G) or LysoTracker (H), and these images were digitally combined to produce the image in panel I.
Because fixation and permeabilization of cells can alter the localization of VP22 (6, 8), we also conducted localization experiments with living cells. For this, we used LysoTracker, a fluorescent pH-sensitive dye that stains the acidic compartments of cells, which include lysosomes, endosomes, and vesicles of the TGN (28). By use of confocal microscopy, the fluorescence pattern of VP22-GFP (Fig. 2G) and that of Lysotracker (Fig. 2H) in A7 cells were found to be similar (Fig. 2I). Thus, it appears that some but not all of the cytoplasmic VP22 localizes with cellular markers specific for acidic compartments of the cell that may include the TGN.
Association of VP22 with cellular membranes.
Based on the vesicular nature of VP22-GFP fluorescence and its putative localization to acidic compartments of the cell that may include the TGN, we hypothesized that VP22 is associated with cellular membranes. Initially, a standard cell fractionation technique involving membrane pelleting was used to determine whether VP22 associated with the membrane fraction (51). Since VP22 was fused to GFP, fluorometry could be used to measure the specific fluorescence associated with the soluble and pelleted fractions. As a positive control for membrane binding, we used a chimera consisting of UL11, an HSV-1 tegument protein that is both myristylated and palmitylated (5, 35, 36), fused to GFP. A fusion protein containing a myristate-negative mutant of UL11, Myr(−)UL11-GFP, was used as a negative control for membrane association (35). At 20 h posttransfection, cells were harvested and the membrane fraction was obtained as described in Materials and Methods. Approximately 70% of UL11 pellets with the cellular membrane fraction (Fig. 3). In contrast, only 30% of Myr(−)UL11 was associated with the membrane fraction. VP22 pelleted with the cellular membrane fraction in amounts similar to those observed with the membrane-associated UL11. To address the possibility that protein aggregation was responsible for the pelleting of VP22, we preincubated each of the cellular lysates with 0.5% Triton X-100 prior to centrifugation, which should solubilize cellular membranes but leave aggregates intact. The amounts of both UL11 and VP22 pelleted from the detergent-treated cell lysates were reduced following solubilization of membranes (Fig. 3). These results are supportive of the hypothesis that VP22 is associated with the cellular membrane fraction. However, previous reports have suggested that VP22 colocalizes with components of the cytoskeleton (15, 32), and these complexes might also pellet with membranes (42). Therefore, it is not possible to definitively evaluate the membrane association of VP22 by using this technique.
FIG. 3.

VP22 pellets with cellular membranes. A7 cells were transfected with the indicated plasmids, harvested at 20 h posttransfection, and incubated in hypotonic buffer. Swollen cells were disrupted by Dounce homogenization, and nuclei were removed by low-speed centrifugation. In order to separate membrane-bound entities from soluble forms, the supernatants underwent centrifugation at 100,000 × g. As a control, replica samples were incubated with 0.5% Triton X-100 prior to centrifugation to solubilize cellular membranes. Following centrifugation, soluble and pellet fractions were collected and analyzed for the presence of GFP chimeras by fluorometry. The fraction pelleted was calculated by dividing the amount pelleted by the total amount of protein in the soluble and membrane fractions (see Materials and Methods). Error bars, standard deviations for three independent experiments.
To distinguish between membrane-associated proteins and non-membrane-bound complexes, we also analyzed the membrane association of VP22 by membrane flotation gradient centrifugation (42). This assay has been successfully implemented to study the membrane association of the vesicular stomatitis virus M protein (11), the human immunodeficiency type 1 Gag polyprotein (42, 55), and the Ebola matrix protein VP40 (30). Postnuclear supernatants from A7 cells transfected with plasmids encoding either VP22-GFP, UL11-GFP, Myr(−)UL11-GFP, or GFP were harvested at 20 h posttransfection, adjusted to a concentration of 71.5% (wt/vol) sucrose, placed at the bottom of a centrifuge tube, overlaid with 65% (wt/vol) sucrose and 10% (wt/vol) sucrose, and centrifuged for 18 h at 100,000 × g. During centrifugation, membranes and their associated proteins will float to the 10%-65% sucrose interface due to the buoyant density of lipids while non-membrane-bound entities will remain trapped at the bottom of the gradient. Based on the refractometry data, the membrane-containing 10%-65% sucrose interface occurred between fractions 9 and 10 (data not shown). Each fraction was analyzed by fluorometry, and the percentage of total fluorescence was determined for each fraction (Fig. 4A). Nearly 40% of UL11 was found at the 10%-65% sucrose interface (fractions 9 and 10), and this could be eliminated by treatment of postnuclear supernatants with 1% Triton X-100 prior to analysis (data not shown). As expected, Myr(−)UL11 failed to float in these gradients. The flotation profile for VP22 mimicked the profile for UL11, with nearly 25% of VP22-GFP detected at the 10%-65% sucrose interface. GFP-expressing cells were included as an additional negative control for membrane flotation, and the flotation profile of GFP resembled that of Myr(−)UL11.
FIG. 4.

Membrane flotation of VP22. (A) Membrane flotation of VP22 assayed by fluorometry. A7 cells were transfected with the indicated plasmids, harvested, and incubated in hypotonic buffer. Swollen cells were disrupted by Dounce homogenization, and nuclei were removed by low-speed centrifugation. Membrane flotation gradient centrifugation was then performed on postnuclear supernatants as described in Materials and Methods. Gradient fractions were analyzed for the presence of GFP chimeras by fluorometry. The percentage of fluorescence for a given fraction was determined by dividing the fluorescence of a given fraction by the sum total fluorescence of all fractions (see Materials and Methods). Fluorometric results are depicted graphically. (B) Membrane flotation of VP22 assayed by immunoprecipitation. A7 cells were transfected with the indicated plasmids and were metabolically labeled with EXPRE35S35S protein labeling mix for 2.5 h. Membrane flotation gradient centrifugation was performed as described above. Gradient fractions were collected, diluted in RIPA buffer, and incubated with a polyclonal goat antibody specific for GFP.Immunoprecipitated fractions were separated by SDS-PAGE and visualized by autoradiography. (C) Quantification of membrane flotation. SDS-PAGE gels described above were analyzed by PhosphorImager analysis. The percentage of protein in a given fraction was determined by dividing the number of counts for a given fraction by the sum total number of counts of all fractions (see Materials and Methods). PhosphorImager results are displayed graphically.
As an alternative to fluorometry, we analyzed gradient fractions for the presence of VP22-GFP within membrane flotation gradient centrifugation fractions by immunoprecipitation with an anti-GFP antibody. For this, transfected cells were metabolically labeled with [35S]methionine and [35S]cysteine for 2.5 h at 20 h posttransfection. At 22.5 h posttransfection, cells were harvested and postnuclear supernatants were analyzed by membrane flotation gradient centrifugation. Immunoprecipitates were subjected to SDS-PAGE and analyzed by autoradiography (Fig. 4B). UL11 was found to float to the 10%-65% sucrose interface (fractions 9 and 10). VP22 also floated to the 10%-65% sucrose interface, while only a minimal amount of the negative control, GFP, was detected at the 10%-65% interface. The bands resolved on the polyacrylamide gels were quantitated by PhosphorImager analysis (Fig. 4C), and the resulting profiles were similar to those obtained by fluorometry (Fig. 4A). We conclude from these data that the HSV-1 tegument protein, VP22, is associated with cellular membranes. As demonstrated in Fig. 4, the relative amount of VP22 associated with the cellular membrane fraction is smaller than that of UL11. This could be due to the possibility that different phosphorylation species of VP22 may be localized to different cellular compartments. Alternatively, this observation may be due to the relative strength of the interaction of VP22 with membranes. This pattern of membrane association of VP22 was reproducible, as shown in the subsequent figures.
In an effort to further characterize the nature of the association of VP22 with cellular membranes, postnuclear supernatants from cells expressing VP22-GFP were treated with either 1 M NaCl, 0.1 M Na2CO3 (pH 11), or 1% Triton X-100 for 1 h prior to their analysis by membrane flotation gradient centrifugation. Fractions from the gradients were immunoprecipitated with an anti-GFP antibody, analyzed by SDS-PAGE, visualized by autoradiography, and quantitated by PhosphorImager analysis (Fig. 5). Mock-treated VP22 floated to the 10%-65% sucrose interface, as observed previously. Pretreatment of postnuclear supernatants with either 1 M NaCl or 1% Triton X-100 resulted in the failure of VP22 to float to the 10%-65% interface, indicating that VP22 can be extracted from cellular membranes by either altering the electrostatic conditions (1 M NaCl) or solubilizing the membrane (1% Triton X-100). These two results were anticipated given the extremely basic nature of VP22 and the hypothesis that it is a peripheral membrane protein. In contrast, we found that pretreatment with 0.1 M Na2CO3 had little effect on the ability of VP22 to float in the gradient. Resistance to extraction by 0.1 M Na2CO3 may suggest two different possibilities: either insufficient electrostatic conditions to extract VP22 or actual resistance to alkaline conditions, indicating that VP22 may possess integral membrane protein-like characteristics (57). Given the 1 M NaCl extraction results, the latter possibility seems unlikely.
FIG. 5.

Differential extraction of VP22 from cellular membranes. A7 cells were transfected with the plasmid encoding VP22-GFP and were metabolically labeled with EXPRE35S35S protein labeling mix for 2.5 h, prior to harvesting at 22.5 h posttransfection. Following incubation in hypotonic buffer, the swollen cells were disrupted by Dounce homogenization, and nuclei were removed by low-speed centrifugation. Postnuclear supernatants were either treated with 1 M NaCl, 0.1 M Na2CO3, or 1% Triton X-100 or mock treated for 1 h prior to membrane flotation gradient centrifugation. Membrane flotation gradient centrifugation was then performed as described in Materials and Methods. PhosphorImager quantification was performed as described in the legend to Fig. 4C, and results are displayed graphically.
Is a specific domain of VP22 responsible for membrane association?
To further characterize the interaction of VP22 with cellular membranes, we attempted to map the specific region of VP22 responsible for its membrane association. A series of C-terminal truncation mutants (Fig. 6A) and a series of reciprocal N-terminal truncation mutants (Fig. 7A) were constructed as described in Materials and Methods. A Kyte-Doolittle hydrophobicity plot (33) of VP22 was used to strategically select the sites for truncation mutagenesis. This approach was used in an attempt to avoid major disruptions in protein folding that could result from cutting the protein in hydrophobic regions. Both the C-terminal (Fig. 6B) and the N-terminal (Fig. 7B) VP22 truncation mutants were expressed in A7 cells, immunoprecipitated with an anti-GFP antibody, and assayed by SDS-PAGE, which revealed that all constructs were expressed and had the expected molecular masses. Using both live-cell fluorescence and membrane flotation gradient centrifugation, we investigated the abilities of the C- and N-terminal truncation mutants of VP22 to associate with cellular membranes.
FIG. 6.

Membrane association of VP22 C-terminal truncation mutants. (A) VP22 C-terminal truncations. Shown is a schematic representation of full-length and C-terminal truncated forms of VP22. Sites for truncation were based on the Kyte-Doolittle hydrophobicity plot of VP22. (B) Expression of C-terminal truncations of VP22. A7 cells were transfected with the indicated plasmids and at 20 h posttransfection were metabolically labeled with EXPRE35S35S protein labeling mix for 2.5 h. Cells were harvested, mixed in RIPA buffer, and incubated with a polyclonal goat antibody against GFP. Proteins were immunoprecipitated, separated by SDS-PAGE, and visualized by autoradiography. (C) Subcellular localization of VP22 C-terminal truncation mutants. A7 cells were transfected with the designated plasmids, and the fluorescence of these C-terminal truncation mutants was observed by confocal microscopy at 20 h posttransfection. (D) Membrane flotation analysis of C-terminal truncation mutants. A7 cells were transfected with the appropriate plasmids and at 20 h posttransfection were metabolically labeled with EXPRE35S35S protein labeling mix for 2.5 h. Cells were harvested and incubated in hypotonic buffer. Swollen cells were disrupted by Dounce homogenization, and nuclei were removed by low-speed centrifugation. Membrane flotation gradient centrifugation was performed as described in Materials and Methods. PhosphorImager quantification was performed as described in the legend to Fig. 4C, and results are displayed graphically.
FIG. 7.

Membrane association of VP22 N-terminal truncation mutants. N-terminal truncation mutants of VP22 were constructed, expressed, and analyzed as described for C-terminal truncation mutants in the legend to Fig. 6. (A) VP22 N-terminal truncations. (B) Expression of N-terminal truncations of VP22. (C) Subcellular localization of VP22 N-terminal truncation mutants. (D) Membrane flotation analysis of N-terminal truncation mutants.
As shown in Fig. 6C, deletion of the C-terminal 258, 215, 181, or 137 amino acids resulted in a fluorescence pattern similar to that for GFP expressed alone. In contrast, the wild-type pattern of VP22-GFP fluorescence was retained when only the C-terminal 76 and 31 amino acids were deleted. These results suggest that the region of VP22 that is required for membrane association resides somewhere in the first 225 amino acids. As predicted, only mutants VP22.1-225 and VP22.1-270 associate with cellular membranes as determined by membrane flotation gradient centrifugation (Fig. 6D)
To further map the membrane association regions of VP22, we also characterized the N-terminal truncation mutants by live-cell fluorescence and membrane flotation assays (Fig. 7C and D). Deletion of the N-terminal 43, 86, or 120 amino acids resulted in various forms of the punctate cytoplasmic and perinuclear fluorescence, whereas removal of the N-terminal 164 amino acids resulted in a primarily nuclear fluorescence with some punctate perinuclear fluorescence. In contrast, still larger deletions of 225 or 270 amino acids resulted in a fluorescence pattern that resembled that of GFP expressed alone. These results suggest that only mutants VP22.43-301, VP22.86-301, and VP22.120-301 retain the membrane-binding domain and should associate with cellular membranes. Flotation gradient analysis confirmed this prediction (Fig. 7D) and revealed that two of these mutants actually appear to bind membranes better than wild-type VP22. Collectively, all of the truncation mutagenesis experiments indicate that amino acids 120 to 225 are responsible for the membrane association of VP22.
DISCUSSION
Although several studies have addressed the intracellular localization of various tegument proteins, the molecular events associated with the process of tegumentation and final envelopment of HSV-1 nucleocapsids are not well defined. In this report, we provide evidence that an abundant tegument protein, VP22, localizes to acidic compartments of the cell that may include the TGN, the proposed site of herpesvirus final envelopment. Using membrane flotation gradient analysis, we also have shown that VP22 associates with cellular membranes by means of a functional domain located between residues 120 and 225. To our knowledge, this is the first report that the VP22 tegument protein is both membrane associated and localized to the acidic compartments of the cell. The association of VP22 with cytoplasmic membranes may prove instrumental for the proper tegumentation and envelopment of virions.
Subcellular localization of VP22.
A significant array of viral proteins, including tegument proteins, glycoproteins, and assembled nucleocapsids, converge at cytoplasmic vesicles to become incorporated into virions. In fact, within neuronal cultures, it has been demonstrated that tegument proteins, glycoproteins, and capsids are transported down axons independently of one another to the putative site of virion assembly (43, 59). Therefore, each virion protein must have a mechanism for localizing to membrane vesicles—either an intrinsic sorting signal or an interaction with a protein that possesses such a signal. For instance, incorporation of herpesvirus glycoproteins gB, gD, gE, and gI into virions and trafficking through the TGN are facilitated by specific amino acid sequences (2-4, 47, 62, 64, 66, 67). The cooperative role of an acidic cluster of amino acids and palmitylation of the UL11 tegument protein, which facilitate its localization to the Golgi apparatus, have been described previously (7, 35). With the exception of UL11, it is not clear which tegument proteins localize to the site of final tegumentation and envelopment, and what molecular mechanisms are responsible for their localization. Since VP22 localizes to acidic compartments of the cell, which may include the TGN within transfected cells, these data indicate that VP22 contains specific intracellular trafficking sequences necessary for directing and retaining the protein at this site. The findings presented in this report provide new insight as well as the groundwork for further studies on the localization of VP22 as it pertains to vesicular trafficking. Perhaps VP22 possesses trafficking signals, analogous to those of UL11 and the glycoproteins, that are required for proper localization of the protein to the site of virion envelopment.
Membrane association of VP22.
One of the unexpected findings from this study was that VP22 associates with cellular membranes. Truncation mutagenesis of both the C-terminal and N-terminal regions of VP22 revealed that membrane association is a property attributable to a specific region of the protein. Collectively, the VP22 truncation mutagenesis data suggest that amino acids 120 to 225 contribute to the membrane association of VP22. Although the truncation mutagenesis results indicate that a particular region of VP22 is responsible for its membrane association, we cannot rule out the possibility that the membrane association domain of VP22 may extend beyond this region, due to the potential disruption of tertiary structure. Interestingly, primary structure alignment reveals that the region of HSV-1 VP22 within amino acids 120 through 225 is highly conserved among VP22 homologues of herpesviruses, opening the possibility that membrane association is a conserved attribute of VP22. Studies are now in progress to further dissect this membrane association domain of VP22.
With regard to the mechanism of membrane association, VP22 may bind to membranes either directly, by interacting with phospholipids, or indirectly, by binding to a cellular membrane-associated protein. Given the basic nature of VP22 and its susceptibility to extraction under high electrostatic conditions, we hypothesize that the positively charged basic amino acid residues of VP22 could interact with acidic phospholipids of cellular membranes. Intriguingly, the truncation mutants that float best by gradient analysis also exhibit the greatest net positive charge (number of basic amino acids minus number of acidic amino acids) (data not shown). Similarly, the membrane-binding domain of the Gag polyprotein of Rous sarcoma virus is composed exclusively of basic residues and is extremely sensitive to alterations of the charge, which directly impact the functionality of this protein (10).
As with the well-characterized retrovirus system, certain tegument proteins of herpesviruses could collectively be considered both structurally and functionally equivalent to the matrix proteins of RNA viruses. Drawing such a parallel, one could hypothesize that certain tegument proteins may contain all of the functional information required to drive HSV-1 final envelopment. In fact, HSV-1 produces L particles, which contain a full complement of tegument and glycoproteins but are devoid of capsids (38, 48, 58). Increasing experimental evidence supports the idea that the tegument protein, VP22, is a critical structural component of the virion with potential implications for virus assembly. VP22 is capable of interacting with VP16 (13), and yeast two-hybrid analysis has revealed that pseudorabies virus VP22 may interact with both gM and gE (22). Adding further complexity to the protein-protein interactions that can occur within the virion, VP22 has recently been shown to bind RNA in vitro (52). With regard to assembly, overexpression of VP22 during viral infection dramatically increases L particle production (34). Recently, quantitative transmission immunoelectron microscopy analysis of VP22 during HSV-1 infection revealed that approximately 90% of unenveloped capsids within the cytoplasm lack VP22, suggesting that VP22 is acquired as capsids bud into a cytoplasmic compartment (41). The latter result is particularly interesting if considered with the data presented in this report, which show that VP22 is localized to vesicular membranes. Future research will address the potential functional role of VP22 during tegumentation and final envelopment.
Acknowledgments
We thank Michael G. Fried (Department of Biochemistry and Molecular Biology, The Pennsylvania State University College of Medicine) for the use of the fluorometer and for assistance with the fluorometric analyses. We also thank Sandra Allen, Eric Callahan, and Kevin O'Regan for discussions and careful review of the manuscript.
This work was supported by National Institutes of Health (NIH) grant CA42460. M.J.B. and J.S.L. were partially supported by NIH training grant CA60395.
REFERENCES
- 1.Aints, A., H. Guven, G. Gahrton, C. I. Smith, and M. S. Dilber. 2001. Mapping of herpes simplex virus-1 VP22 functional domains for inter- and subcellular protein targeting. Gene Ther. 8:1051-1056. [DOI] [PubMed] [Google Scholar]
- 2.Alconada, A., U. Bauer, L. Baudoux, J. Piette, and B. Hoflack. 1998. Intracellular transport of the glycoproteins gE and gI of the varicella-zoster virus. gE accelerates the maturation of gI and determines its accumulation in the trans-Golgi network. J. Biol. Chem. 273:13430-13436. [DOI] [PubMed] [Google Scholar]
- 3.Alconada, A., U. Bauer, and B. Hoflack. 1996. A tyrosine-based motif and a casein kinase II phosphorylation site regulate the intracellular trafficking of the varicella-zoster virus glycoprotein I, a protein localized in the trans-Golgi network. EMBO J. 15:6096-6110. [PMC free article] [PubMed] [Google Scholar]
- 4.Alconada, A., U. Bauer, B. Sodeik, and B. Hoflack. 1999. Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 73:377-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baines, J. D., R. J. Jacob, L. Simmerman, and B. Roizman. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 69:825-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blouin, A., and J. A. Blaho. 2001. Assessment of the subcellular localization of the herpes simplex virus structural protein VP22 in the absence of other viral gene products. Virus Res. 81:57-68. [DOI] [PubMed] [Google Scholar]
- 7.Bowzard, J. B., R. J. Visalli, C. B. Wilson, J. S. Loomis, E. M. Callahan, R. J. Courtney, and J. W. Wills. 2000. Membrane targeting properties of a herpesvirus tegument protein-retrovirus Gag chimera. J. Virol. 74:8692-8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brewis, N., A. Phelan, J. Webb, J. Drew, G. Elliott, and P. O'Hare. 2000. Evaluation of VP22 spread in tissue culture. J. Virol. 74:1051-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Browne, H., S. Bell, T. Minson, and D. W. Wilson. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 70:4311-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callahan, E. M., and J. W. Wills. 2000. Repositioning basic residues in the M domain of the Rous sarcoma virus Gag protein. J. Virol. 74:11222-11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong, L. D., and J. K. Rose. 1993. Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J. Virol. 67:407-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craven, R. C., A. E. Leure-duPree, R. A. Weldon, Jr., and J. W. Wills. 1995. Genetic analysis of the major homology region of the Rous sarcoma virus Gag protein. J. Virol. 69:4213-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott, G., G. Mouzakitis, and P. O'Hare. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932-7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott, G., and P. O'Hare. 1997. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 88:223-233. [DOI] [PubMed] [Google Scholar]
- 15.Elliott, G., and P. O'Hare. 1998. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 72:6448-6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elliott, G., and P. O'Hare. 1999. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J. Virol. 73:4110-4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott, G., and P. O'Hare. 2000. Cytoplasm-to-nucleus translocation of a herpesvirus tegument protein during cell division. J. Virol. 74:2131-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elliott, G., D. O'Reilly, and P. O'Hare. 1996. Phosphorylation of the herpes simplex virus type 1 tegument protein VP22. Virology 226:140-145. [DOI] [PubMed] [Google Scholar]
- 19.Elliott, G., D. O'Reilly, and P. O'Hare. 1999. Identification of phosphorylation sites within the herpes simplex virus tegument protein VP22. J. Virol. 73:6203-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott, G. D., and D. M. Meredith. 1992. The herpes simplex virus type 1 tegument protein VP22 is encoded by gene UL49. J. Gen. Virol. 73:723-726. [DOI] [PubMed] [Google Scholar]
- 21.Enquist, L. W., P. J. Husak, B. W. Banfield, and G. A. Smith. 1998. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 51:237-347. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs, W., B. G. Klupp, H. Granzow, C. Hengartner, A. Brack, A. Mundt, L. W. Enquist, and T. C. Mettenleiter. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 76:8208-8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geiss, B. J., J. E. Tavis, L. M. Metzger, D. A. Leib, and L. A. Morrison. 2001. Temporal regulation of herpes simplex virus type 2 VP22 expression and phosphorylation. J. Virol. 75:10721-10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gershon, A. A., D. L. Sherman, Z. Zhu, C. A. Gabel, R. T. Ambron, and M. D. Gershon. 1994. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J. Virol. 68:6372-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granzow, H., B. G. Klupp, W. Fuchs, J. Veits, N. Osterrieder, and T. C. Mettenleiter. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Granzow, H., F. Weiland, A. Jons, B. G. Klupp, A. Karger, and T. C. Mettenleiter. 1997. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J. Virol. 71:2072-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harley, C. A., A. Dasgupta, and D. W. Wilson. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 75:1236-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haugland, R. P. 1996. Probes for organelles, p. 265-286. In M. T. Z. Spence (ed.), Handbook of fluorescent probes and research chemicals. Molecular Probes, Eugene, Oreg.
- 29.Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jasenosky, L. D., G. Neumann, I. Lukashevich, and Y. Kawaoka. 2001. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 75:5205-5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones, F., and C. Grose. 1988. Role of cytoplasmic vacuoles in varicella-zoster virus glycoprotein trafficking and virion envelopment. J. Virol. 62:2701-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotsakis, A., L. E. Pomeranz, A. Blouin, and J. A. Blaho. 2001. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 75:8697-8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 34.Leslie, J., F. J. Rixon, and J. McLauchlan. 1996. Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology 220:60-68. [DOI] [PubMed] [Google Scholar]
- 35.Loomis, J. S., J. B. Bowzard, R. J. Courtney, and J. W. Wills. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209-12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacLean, C. A., B. Clark, and D. J. McGeoch. 1989. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J. Gen. Virol. 70:3147-3157. [DOI] [PubMed] [Google Scholar]
- 37.Martin, A., P. O'Hare, J. McLauchlan, and G. Elliott. 2002. Herpes simplex virus tegument protein VP22 contains overlapping domains for cytoplasmic localization, microtubule interaction, and chromatin binding. J. Virol. 76:4961-4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLauchlan, J., and F. J. Rixon. 1992. Characterization of enveloped tegument structures (L particles) produced by alphaherpesviruses: integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 73:269-276. [DOI] [PubMed] [Google Scholar]
- 39.Mettenleiter, T. C. 2000. Aujeszky's disease (pseudorabies) virus: the virus and molecular pathogenesis—state of the art, June 1999. Vet. Res. 31:99-115. [DOI] [PubMed] [Google Scholar]
- 40.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miranda-Saksena, M., R. A. Boadle, P. Armati, and A. L. Cunningham. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J. Virol. 76:9934-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ono, A., and E. O. Freed. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136-4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penfold, M. E., P. Armati, and A. L. Cunningham. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA 91:6529-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piguet, V., L. Wan, C. Borel, A. Mangasarian, N. Demaurex, G. Thomas, and D. Trono. 2000. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2:163-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pomeranz, L. E., and J. A. Blaho. 1999. Modified VP22 localizes to the cell nucleus during synchronized herpes simplex virus type 1 infection. J. Virol. 73:6769-6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pomeranz, L. E., and J. A. Blaho. 2000. Assembly of infectious herpes simplex virus type 1 virions in the absence of full-length VP22. J. Virol. 74:10041-10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radsak, K., M. Eickmann, T. Mockenhaupt, E. Bogner, H. Kern, A. Eis-Hubinger, and M. Reschke. 1996. Retrieval of human cytomegalovirus glycoprotein B from the infected cell surface for virus envelopment. Arch. Virol. 141:557-572. [DOI] [PubMed] [Google Scholar]
- 48.Rixon, F. J., C. Addison, and J. McLauchlan. 1992. Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J. Gen. Virol. 73:277-284. [DOI] [PubMed] [Google Scholar]
- 49.Roizman, B., and D. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 50.Sanchez, V., K. D. Greis, E. Sztul, and W. J. Britt. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandefur, S., V. Varthakavi, and P. Spearman. 1998. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 72:2723-2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sciortino, M. T., B. Taddeo, A. P. Poon, A. Mastino, and B. Roizman. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. USA 99:8318-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment→deenvelopment→reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith, K. O. 1964. Relationship between the envelope and infectivity of the herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814-816. [DOI] [PubMed] [Google Scholar]
- 55.Spearman, P., R. Horton, L. Ratner, and I. Kuli-Zade. 1997. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 71:6582-6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stackpole, C. W. 1969. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J. Virol. 4:75-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steck, T. L., and C. F. Fox. 1972. Membrane proteins, p. 27-75. In C. F. Fox and A. D. Keith (ed.), Membrane molecular biology. Sinauer Associates, Stamford, Conn.
- 58.Szilagyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72:661-668. [DOI] [PubMed] [Google Scholar]
- 59.Tomishima, M. J., G. A. Smith, and L. W. Enquist. 2001. Sorting and transport of alpha herpesviruses in axons. Traffic 2:429-436. [DOI] [PubMed] [Google Scholar]
- 60.van Genderen, I. L., R. Brandimarti, M. R. Torrisi, G. Campadelli, and G. van Meer. 1994. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology 200:831-836. [DOI] [PubMed] [Google Scholar]
- 61.Wan, L., S. S. Molloy, L. Thomas, G. Liu, Y. Xiang, S. L. Rybak, and G. Thomas. 1998. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94:205-216. [DOI] [PubMed] [Google Scholar]
- 62.Wang, Z. H., M. D. Gershon, O. Lungu, Z. Zhu, and A. A. Gershon. 2000. Trafficking of varicella-zoster virus glycoprotein gI: T(338)-dependent retention in the trans-Golgi network, secretion, and mannose 6-phosphate-inhibitable uptake of the ectodomain. J. Virol. 74:6600-6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whealy, M. E., J. P. Card, R. P. Meade, A. K. Robbins, and L. W. Enquist. 1991. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J. Virol. 65:1066-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whiteley, A., B. Bruun, T. Minson, and H. Browne. 1999. Effects of targeting herpes simplex virus type 1 gD to the endoplasmic reticulum and trans-Golgi network. J. Virol. 73:9515-9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wills, J. W., R. C. Craven, and J. A. Achacoso. 1989. Creation and expression of myristylated forms of Rous sarcoma virus Gag protein in mammalian cells. J. Virol. 63:4331-4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu, Z., M. D. Gershon, Y. Hao, R. T. Ambron, C. A. Gabel, and A. A. Gershon. 1995. Envelopment of varicella-zoster virus: targeting of viral glycoproteins to the trans-Golgi network. J. Virol. 69:7951-7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu, Z., Y. Hao, M. D. Gershon, R. T. Ambron, and A. A. Gershon. 1996. Targeting of glycoprotein I (gE) of varicella-zoster virus to the trans-Golgi network by an AYRV sequence and an acidic amino acid-rich patch in the cytosolic domain of the molecule. J. Virol. 70:6563-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]