

Abstract
Urothelial barrier function is maintained by apical membrane plaques and intercellular tight junctions (TJ). Little is known about the composition and regulation of TJ expression in human urothelium. In this study, we have characterised the expression of TJ components in situ and their regulation in an in vitro model of differentiating normal human urothelial (NHU) cells. In normal ureteric urothelium in situ, there was a differentiation-associated profile of claudins 3, 4, 5, 7, ZO1 and occludin proteins. Proliferating NHU cells in vitro expressed predominantly claudin 1 protein and transcripts for claudins 1–5 and 7. Following induction of differentiation by pharmacological activation of PPARγand blockade of EGFR, there was de novo expression of claudin 3 mRNA and protein and downregulation of claudin 2 transcription. There was also a massive increase in expression of claudin 4 and 5 proteins which was due to inhibition of proteasomal degradation of claudin 4 and consequential stabilisation of the claudin 5 heterodimerisation partner. NHU cell differentiation was accompanied by relocalisation of TJ proteins to intercellular junctions. The differentiation-associated development of TJ formation in vitro reflected the stage-related TJ expression seen in situ. This was distinct from changes in TJ composition of NHU cells mediated by increasing the calcium concentration of the medium. Our results imply a role for PPARγ and EGFR signalling pathways in regulating TJ formation in NHU cells and support the hypothesis that TJ development is an integral part of the urothelial differentiation programme.
The maintenance of epithelial barrier function requires that the transepithelial passage of water and solutes be tightly regulated. Ion channels and membrane pumps located in the apical and basolateral membrane compartments control transcellular ion transport, whereas tight junctions (TJ), located at the superior aspect of the intercellular junctional complex, control paracellular diffusion (Schneeberger and Lynch, 2004). TJ are composed of cytoplasmic plaque proteins, such as the zonular occludens (ZO) proteins that link the TJ to the cytoskeleton, and integral transmembrane proteins, such as occludin, junctional adhesion molecule (JAM) and claudins that define the properties of the paracellular pore (Tsukita and Furuse, 2002; Gonzalez-Mariscal et al., 2003). The TJ not only limits paracellular movement, but maintain polarity by restricting the movement of proteins and lipids between apical and basolateral membrane compartments.
The claudins, which represent a multigene family of some 24 members ranging from 20,000 to 27,000 Mr, are considered to represent the primary seal-forming fibrils of the TJ. Claudins are tetra-spanning proteins, comprising two extracellular loops and short amino and carboxy termini (Schneeberger and Lynch, 2004). The expression of different claudin proteins and the pairing of claudins to form homotypic or heterotypic fibrils affords a large number of potential combinations, thus providing a mechanism for determining differential paracellular functions by different epithelia (Tsukita and Furuse, 2002).
The barrier function of the urinary bladder is served by the epithelial lining, termed the urothelium. Urothelium is organised into basal, intermediate and terminally-differentiated superficial cell layers, the latter having a number of specialised molecular features supporting urinary barrier function. Best characterised are the plaques of asymmetric unit membrane (AUM), which are uniquely present in the apical membrane of the superficial cell. These AUM plaques, formed as a result of interactions between specific uroplakin (UP) proteins, constitute a barrier to the free movement of water and solutes across the apical membrane (Hu et al., 2002). However, very little is known of the composition of the urothelial TJ, which is responsible for maintaining the paracellular barrier. Although urothelium is regarded as one of the ‘tightest’ of all epithelia, there has been very little work on TJ composition in man (Slobodov et al., 2004) or other species (Acharya et al., 2004).
The morphology of the urothelium and the expression of AUM plaques by the superficial cells imply a differentiation-associated acquisition of barrier function. In order to address this issue, we have exploited our well established in vitro model of normal human urothelial (NHU) cells (Southgate et al., 1994, 2002), using pharmacological activation of peroxisome proliferator activated receptor γ (PPARγ) to induce urothelial cytodifferentiation (Varley et al., 2004a,b). PPARγ is a nuclear hormone receptor and a ligand-activated transcription factor that requires heterodimerisation with the retinoid X receptor α (RXRα) to bind specific PPAR response elements (PPRE) in the promoter region of target genes (Kliewer et al., 1992; Issemann et al., 1993). PPARγ is expressed by urothelium during murine development and in the mature urothelium of mice, rabbits and man (Jain et al., 1998; Guan et al., 1999; Kawakami et al., 2002). We have shown that in cultures of NHU cells, PPARγ agonists activate the urothelial differentiation programme, as shown by induction of expression of uroplakins and other urothelial differentiation-associated proteins (Varley et al., 2004a,b). Activation of the PPARγ-dependent urothelial cytodifferentiation programme requires inhibition of the autocrine-activated epidermal growth factor receptor (EGFR) pathway, which in NHU cell cultures, mediates a highly proliferative regenerative phenotype (Varley et al., 2004a, 2005).
In this report, we have studied the expression of claudins by human ureteric urothelium to identify which claudin family members are expressed and hence implicated in urothelial barrier function. Using our NHU cell culture systems, we have characterised the claudin expression patterns associated with a proliferative phenotype in low exogenous calcium (0.09 mM) serum-free medium and following induction of stratification in near physiological calcium (2 mM) concentrations. We have further investigated changes and differential regulation of claudin expression in response to PPARγ activation and how this is modulated by epidermal growth factor (EGF) signalling, during the progression to terminal differentiation.
MATERIALS AND METHODS
Materials
The EGF receptor tyrosine kinase inhibitor, PD153035, was obtained from Calbiochem-Novabiochem Biosciences Ltd (Nottingham, UK). Troglitazone (TZ) was a kind gift from Parke-Davis Pharmaceutical Research (Ann Arbor); rosiglitazone (RZ) and GW9662 were provided as gifts by GlaxoSmithKline (Worthing, UK). The PPARγ-specific antagonist, T0070907, was obtained from Cayman Chemical Co. (supplied by AXXORA UK, Ltd., Bingham, Nottingham).
Tissues
Surgical specimens of normal human ureter were collected with patient consent and approval of the relevant Local Research Ethics Committees from patients with no history of urothelial dysplasia or malignancy. Tissues were collected into Transport Medium consisting of Hanks’ balanced salt solution (HBSS) containing 10 mM HEPES pH 7.6 and 20 KIU aprotinin (Trasylol, Bayer plc, Newbury, UK), as described (Southgate et al., 1994, 2002). Representative pieces of each tissue sample were fixed in 10% formalin and processed into paraffin wax for histology to confirm normal histioarchitecture and absence of atypia or inflammation, and for immunohistochemistry. The remaining tissue was used to isolate intact sheets of urothelium by incubation at 4°C in Transport Medium (Ca2+ and Mg2+-free) containing 0.1% (w/v) EDTA (Southgate et al., 1994, 2002). The isolated urothelial cell sheets were used for cell culture or to extract RNA.
Cell culture
NHU cell cultures were established and maintained as finite cell lines in keratinocyte serum-free medium (KSFM), containing bovine pituitary extract and EGF at the manufacturer’s recommended concentrations (Invitrogen Ltd., Paisley, UK) and supplemented with 30 ng/ml cholera toxin (Sigma-Aldrich Ltd., Poole, UK), as previously described (Southgate et al., 1994, 2002) and referred to as KSFMc. NHU cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2 in air and passaged at just-confluence, as detailed (Southgate et al., 1994, 2002). For the experiments described here, NHU cell lines were used between passages 3 and 5, inclusive, and were seeded at 1.8 × 104 cells per cm2 and grown to approximately 70% confluence, before treatment with PPARγ ligands and PD153035, as specified below, and cultured for up to 6 days before analysis. A 0.1% (v/v) DMSO carrier control was included in all experiments. For some experiments, the medium was supplemented with calcium chloride to a final concentration of 2 mM calcium. Media with appropriate treatments were routinely replaced every 3 days. Experiments were conducted on up to three independent cell lines.
Reverse-transcribed polymerase chain reaction (RT-PCR)
RNA was extracted from freshly isolated urothelium using the RNeasy® Mini Kit (Qiagen Ltd., Crawley, UK) and from cell monolayers using Trizol™ (Invitrogen Ltd.). Isolation of RNA by chloroform extraction and isopropanol precipitation was performed as recommended by the manufacturers. The RNA was treated with DNase I to remove any DNA contamination (DNA-free™ kit, Ambion Europe Ltd., Huntingdon, UK). cDNA was synthesised using 1 μg of total RNA and the Superscript™ first-strand synthesis system (Invitrogen Ltd.), as outlined by the manufacturer. PCR was performed using Surestart Taq polymerase (Stratagene Europe, Amsterdam, The Netherlands), using primer pairs designed to amplify specific claudin products (Table 1). The primers for the internal control (GAPDH) were forward primer: 5′-GTCGGAGTCAACGGATTTGG-3′ and reverse primer: 5′-ATTGCTGATGATCTTGAGGC-3′. The optimal annealing temperature for each claudin was determined by running a PCR reaction with an annealing temperature gradient from 47.0 to 62.2°C in a PCR Express Thermal Cycler (Hybaid Ltd., Ashford, UK). PCR reactions were performed as follows: 95°C for 12 min, then 35 cycles of 95°C for 1 min, optimum annealing temp for 1 min and 72°C for 1 min, and a final extension at 72°C for 10 min. The PCR products were run on a 2% (w/v) agarose gel and visualised with ethidium bromide. Negative controls included a no-RT control for each RNA sample to check for DNA contamination and a no-template (water) control to check for PCR product contamination. Where primer sets did not span exon–intron boundaries, genomic DNA was included as a positive control.
TABLE 1.
Primers used for RT-PCR
| Claudin | Sequence of primers (5′-3′) | Product size (base pairs) |
|---|---|---|
| 1 | TTCTTCTTGCAGGTCTGGCT
CAGGAACAGCAAAGTAGGGC |
180 |
| 2 | CCTGGGATTCATTCCTGTTG
AGTTGGAGCGATTTCTCTGG |
190 |
| 3 | TACGTCTAAGGGACAGACGC
ACAGTCCATGCAGGTTGGTC |
239 |
| 4 | CTGGGAAGTGCAGAGTGGAT
AAGGAAGAGGAAAAACCCCA |
279 |
| 5 | CTGTTTCCATAGGCAGAGCG
AAGCAGATTCTTAGCCTTCC |
301 |
| 6 | CTCTCACTTAGGCTCTGCTG
AAATGTGAGTGTAAAGGGGG |
320 |
| 7 | TAAGTATGAGTTTGGCCCTG
TACAAAAAGCACACTCTCCC |
375 |
| 8 | TCTTCTCCCAGAGGCTTTTT
TTTGACTCAGGTACCCACAT |
390 |
| 10 | TCTGACAACAACAAAACACC
GAAAGCCACAACAGAAAATG |
333 |
Antibodies
All antibodies were titrated for use. Monoclonal antibodies against claudin 4 (clone 3E2C1), claudin 5 (clone 4C3C2), ZO1 (clone 1A12; used at 1.25 μg/ml for immunofluorescence and 5 μg/ml for immunohistochemistry) and rabbit polyclonal antibodies to occludin (PAD Z-T22; used at 2.5 μg/ml for either immunofluorescence or immunohistochemistry), claudin 1 (PAD JAY.8), claudin 3 (PAD Z23.JM) and claudin 7 (PAD ZMD.241) were from Zymed Laboratories, Inc. (supplied by Cambridge Bioscience, Cambridge, UK). Preliminary experiments were performed to characterise the suitability of the claudin antibodies for reactivity on human tissues by immunohistochemistry (claudins 3, 4, 5 and 7 used at concentrations of 1, 1, 10 and 2.5 μg/ml, respectively), immunofluorescence (claudins 1, 3, 4, 5 and 7 used at concentrations of 2.5, 2, 2, 4 and 2 μg/ml, respectively) and immunoblotting (claudins 1, 3, 4, 5 and 7 used at concentrations of 0.25, 0.2, 0.5, 0.5 and 0.25 μg/ml, respectively).
Mouse monoclonal antibodies against desmoplakin (clone DP2.15) (used at 1:40 as supplied) and cytokeratin 13 (CK13, clone IC7) (used at 1:80 as supplied) were purchased from ICN Biomedicals (Thane, UK). UPIIIa (clone AU1) (used at 1:80 as supplied) and E-cadherin (clone HECD1) (used at 0.2 μg/ml for immunofluorescence and 0.2 μg/ml for immunohistochemistry) were purchased from Progen Biotechnik (Heidelberg, Germany) and R&D systems (Abington, UK), respectively. Mouse monoclonal antibody against CK20 (clone IT-Ks 20.3) (used at 1:80 as supplied) was purchased by Chemicon International Ltd. (Harrow, UK) and against β-actin was purchased from Sigma-Aldrich Ltd.
Secondary antibodies for immunoblotting were anti-rabbit Ig conjugated to LI-COR IRDye™ 800 (Rockland, supplied by Tebu-Bio Ltd, Peterborough, UK) and anti-mouse Ig conjugated to Alexa Fluor® 680 (Molecular Probes, supplied by Cambridge Biosciences).
Immunofluorescence
Immunofluorescence was performed as described previously (Southgate and Trejdosiewicz, 1997). Briefly, cells were seeded at 2 × 105 cells/ml onto 12 well Teflon®-coated glass slides, treated as required and maintained for up to 6 days, with fresh medium (with appropriate treatment) on day 3. Slides were fixed in methanol and acetone (vol:vol), air-dried and incubated with titrated primary antibody for 16 h at 4°C. Excess primary antibody was removed by washing in PBS and slides were incubated with secondary antibodies conjugated to Alexa 488 (Molecular Probes, supplied by Cambridge Bioscience). To visualise nuclei, Hoechst 33258 (0.1 μg/ml; Sigma-Aldrich Ltd.) was included in the penultimate wash. Slides were observed on an Olympus BX60 microscope under epifluorescence illumination and appropriate filter sets.
Immunohistochemistry
Immunohistochemistry was performed on paraffin wax-embedded tissue sections using the StreptABComplex/Horseradish Peroxidase system from Dako Cytomation (Ely, UK), as previously described (Varley et al., 2004b). Blocking steps were included to neutralise endogeneous peroxidase and avidin-binding activities and sections were boiled for 10 min in 10 mM citric acid buffer, pH 6.0 to retrieve antigens lost during tissue processing. After overnight incubation in primary antibody at 4°C, slides were washed, incubated in biotinylated secondary antibodies and a streptavidin biotin horseradish peroxidase complex (Dako Cytomation) and visualised using a diaminobenzidine substrate reaction (Sigma-Aldrich Ltd.). The sections were counterstained with haemotoxylin, dehydrated and mounted in DPX (Sigma-Aldrich Ltd). Negative and positive antibody specificity controls were included in all experiments.
Western blot analysis
Cell cultures were treated in situ with lysis buffer (25 mM HEPES pH 7.4, 125 mM NaCl, 10 mM NaF, 10 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 0.2% (w/v) SDS, 0.5% (w/v) sodium deoxycholate acid, 1% (w/v) Triton X-100, 1 μg/ml aprotinin, 10 μg/ml leupeptin and 100 μg/ml phenylmethylsulfonyl fluoride) and the lysates were sheared by passing three times through a 21-gauge needle. After 30 min on ice, the lysates were microcentrifuged at 10,000g for 30 min at 4°C and the protein concentrations of supernatants were measured by Coomassie assay (Pierce, supplied by Perbio Science UK Ltd., Cheshire, UK). Cell extracts were resolved on 12.5% SDS–polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were incubated with titrated primary antibodies for 16 h at 4°C. Bound antibody was detected with Alexa Fluor® 680 and LI-COR IRDye™ 800 conjugated secondary antibodies and visualised using the Odyssey™ Imaging System (LI-COR Biosciences UK Ltd., Cambridge).
Immunoprecipitations
Cells were treated with lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% (w/v) Triton X-100, 1% (w/v) sodium deoxycholate acid, 50 mM NaF, 1 mM sodium orthovanadate, 1 μg/ml aprotinin, 10 μg/ml leupeptin and 100 μg/ml phenylmethylsulfonyl fluoride) and the protein concentration of the lysates was determined as outlined above. For immunoprecipitations, 500 μg of protein lysate was rotated with the relevant antibody on spin-columns overnight at 4°C, as recommended by the manufacturer (Catch and Release kit, Upstate Ltd., Dundee, UK). The immunoprecipitated protein was eluted from the column in a denaturing elution buffer (Catch and Release kit, Upstate) and resolved on SDS–PAGE by western blot analysis.
siRNA
siRNA templates were designed for three regions that were homologous for PPARγ1 (NM_138712), PPARγ2 (NM_015869), PPARγ3 (NM_138711) and PPARγ4 (NM_005037) using the Ambion siRNA template design tool (www.ambion.com). siRNA templates were for region 1: 5′-AAGACATTCCATTCA-CAAGAACCTGTCTC-3′ and 5′-AATTCTTGTGAATGGAAT-GTCCCTGTCTC-3′; region 2: 5′-AAATCACCATTCGTTA-TCTATCCTGTCTC-3′ and 5′-AAATAGATAACGAATGGT-GATCCTGTCTC-3′; region 3: 5′-AATCTGACACCTAA-GAAATTTCCTGTCTC-3′ and 5′-AAAAATTTCTTAGGTG-TCAGACCTGTCTC-3′. The templates were used to make PPARγ siRNA complexes using the Silencer™ siRNA construction kit (Ambion Ltd., Huntingdon, Cambridgeshire, UK).
For transfections, NHU cells were seeded at 4 × 105 cells/ml onto six-well Primaria® plates (Becton-Dickinson, supplied by VWR International Ltd., Poole, UK) and incubated for 16 h until approximately 50% confluent. A cocktail mixture of the three target PPARγ siRNAs was incubated with Oligofectamine™ reagent (Invitrogen Ltd.) in KSFM (without supplements) at ambient temperature for 20 min. The cells were washed with KSFM and the siRNA mixture was added to the cells. After 7 h incubation at 37°C with 5% CO2 in air, KSFMc was added to the cells with PPARγ agonists or inhibitors as outlined in the figure legend. Protein was extracted after 6 days. As controls, cells were incubated with transfection agent, Oligofectamine™, alone in the presence or absence of TZ and PD153035.
Promoter and protein sequence analysis
The transcriptional start site for each claudin gene (NCBI accession numbers: claudin 1, BC012471; claudin 3, NM_001306; claudin 4, NM_001305; claudin 5, NM_003277 and claudin 7, NM_001307) was retrieved from NCBI (http://www.ncbi.nlm.nih.gov) or by linking to UCSC Genome Browser (http://genome.ucsc.edu). A 2,000 bp region upstream of the transcriptional start site for each claudin was used to predict any peroxisome proliferator response elements (PPRE), using a high affinity defined matrix as previously described (Varley et al., 2004a).
The protein sequences for Homo sapiens claudin 4 (NP_001296) and claudin 5 (AAH02404) were retrieved from NCBI.
RESULTS
Expression of tight junction proteins in human urothelium
The immunolocalisation of TJ component proteins was related to differentiated position within the urothelium, with respect to basal, intermediate and superficial zones. Expression of claudin 3 was confined to tightly-focused ‘kissing points’ between superficial cells (3/3 cases; Fig. 1A,B; arrowed), except in a fourth case, where weak expression was also observed at the intercellular borders of upper intermediate cells (Fig. 1). Claudins 4 and 5 were strongly expressed at basolateral junctions of superficial cells in all four cases studied; claudin 4 was also expressed by intermediate and basal cell layers, becoming less intercellular and more cytoplasmic towards the basal cell layer. Claudin 7 was intensely expressed in all but the superficial layer of the urothelium, localising to intercellular borders, particularly in the upper intermediate cells (4/4 cases; Fig. 1A,B).
Fig. 1.
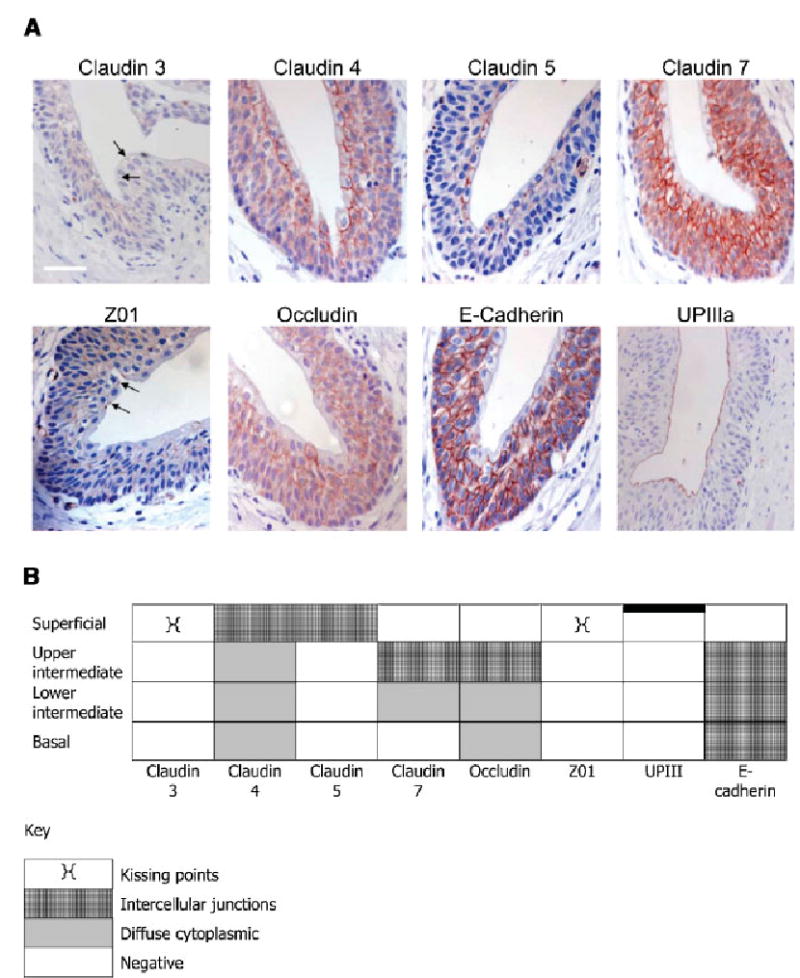
A: Immunohistochemical localisation of tight junction proteins in ureteric urothelium. Expression of claudin 3, 4, 5, 7, ZO1, occludin, E-cadherin and UPIIIa was determined by immuno-peroxidase in paraffin wax-embedded sections of normal human ureter. Sections were counterstained with haematoxylin. Scale bar = 100 μm for all except UPIIIa where scale bar = 50 μm. B: Schematic diagram of the localisation of the tight junction proteins in human urothelium.
ZO1 expression was sparse and limited to the ‘kissing points’ between superficial cells (4/4 cases; Fig. 1A,B; arrowed), where it most closely mimicked claudin 3. Occludin showed a very similar pattern of expression to claudin 7, being expressed throughout basal and intermediate cell layers, but primarily restricted to intercellular sites in the upper intermediate cell layer (2/2 cases).
The intercellular adhesion molecule, E-cadherin, and the differentiation-specific protein UPIIIa, were used as control antigens for immunohistochemical studies. E-cadherin was confined to intercellular junctions throughout the urothelium, but was absent from the most basal and most apical surfaces, whereas UPIIIa was confined to the apical surface of the urothelium, demonstrating an intact superficial cell layer (Fig. 1A).
By RT-PCR, freshly-isolated normal human ureteric urothelium taken from two independent samples revealed amplification products for claudins 1, 2, 4, 5 and 7, with one sample also expressing claudins 3, 8 and 10. Expression of claudin 6 was not detected (Table 2).
TABLE 2.
Summary of claudin expression patterns
| Claudin
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 10 | ||
| In situ | RT-PCR | + | + | −/+ | + | + | − | + | −/+ | −/+ | |
| IHC | NA | NA | + | + | + | NA | + | NA | NA | ||
| In vitro:-baseline | RT-PCR | + | + | + | + | + | − | + | −/+ | − | |
| WB | + | NA | −/+ | + | − | NA | + | NA | NA | ||
| In vitro: differentiating (+TZ +PD153035) | RT-PCR | = | ↘ | ↑ | = | ↘ | − | = | −/= | − | |
| WB | ↘ | NA | ↑ | ↑ | ↑ | NA | ↗ | NA | NA | ||
NA, no antibody available; ↑, major increase after treatment; ↘, small decrease after treatment; ↗, small increase after treatment; =, expression stays constant after treatment; +, present; −, absent; −/+, −/=, variable expression.
Claudin transcript expression by cultured NHU cells and effects of TZ and EGF signalling
We have shown previously that treatment of NHU cells with the PPARγ agonist TZ, in the presence of the EGF receptor inhibitor PD153035, induces expression of the terminal differentiation-associated uroplakin genes (Varley et al., 2004a). We used this protocol to assess semi-quantitatively (relative to GAPDH amplification) whether TZ and/or PD153035 had any effect on claudin gene expression. The results were confirmed in a total of two independent NHU cell lines.
Control NHU cell cultures maintained on KSFMc remained as non-stratified monolayers even when maintained for up to 6 days post-confluence. Analysis by RT-PCR revealed expression of claudins 1, 2, 3, 4, 5 and 7, with one line also showing expression of claudin 8. Claudins 6 and 10 were not expressed (Fig. 2; Table 2). Expression of claudins 5 and 8 appeared to diminish with time in culture.
Fig. 2.
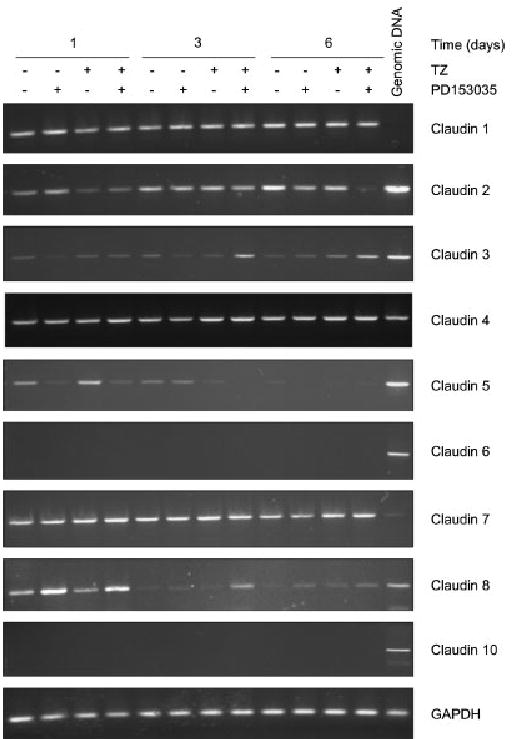
RT-PCR of claudin mRNA expression in NHU cells and the effect of TZ and PD153035 on claudin expression. NHU cells were pretreated for 24 h in the absence or presence of TZ (1 μM) and incubated in media with or without PD153035 (1 μM). Media were changed every 3 days with fresh PD153035 added. RNA was exacted at 1, 3 and 6 days, cDNA was generated and RT-PCR was performed as outlined in the methods, using claudin primers and GAPDH as the internal control. The PCR products were electrophoresed on a 2% agarose gel and visualised using ethidium bromide. Similar results were obtained with a second independent NHU cell line.
Treatment with TZ and PD153035 was associated with increased expression of claudin 3 mRNA at days 3 and 6 post-treatment (Fig. 2). There was inhibition of claudin 2 mRNA after 6 days of treatment with TZ and PD153035, whereas expression of claudins 1, 4 and 7 was unchanged, irrespective of treatment or time in culture (Fig. 2 and Table 2). Treatment of cells with TZ alone was without apparent effect, whereas PD15035 alone induced an increase in claudin 5 and a decrease in claudin 8 transcripts only (Fig. 2).
Effect of TZ and PD153035 on claudin protein expression in cultured NHU cells
Immunoblotting with antibodies against claudins 1, 3, 4, 5 and 7 revealed that baseline claudin expression in proliferating, sub-confluent NHU cell cultures comprised principally of claudin 1. At confluency, small amounts of claudin 4 were also detected (Fig. 3).
Fig. 3.
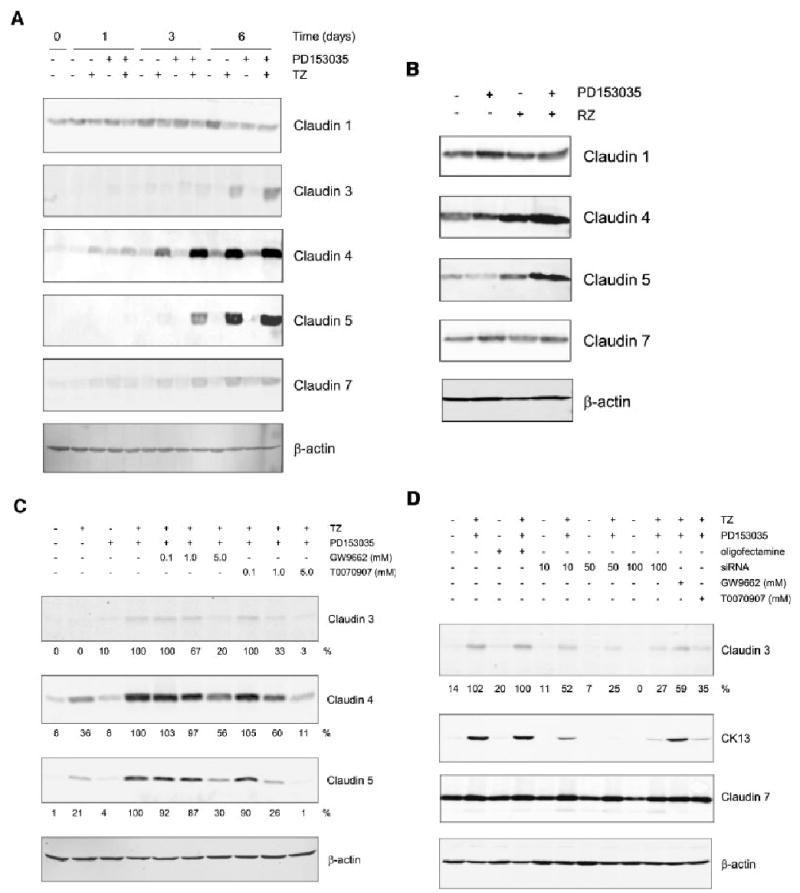
Effect of PPARγ ligands (TZ and RZ) and PD153035 on claudin protein expression in NHU cells. NHU cell were treated for 24 h with 1 μM TZ (A) or RZ (B), in the presence or absence of PD153035 (1 μM) and protein was extracted at 0, 1, 3 and 6 days for cells treated with TZ (A) and at 6 days for cell treated with RZ (B). Media were replaced every 3 days with fresh PD153035 added. Cell extracts (20 μg) were resolved on 12.5% SDS–polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were incubated with pre-titrated primary antibodies for 16 h at 4°C, as indicated. Bound antibody was detected with fluorescent-conjugated secondary antibodies and quantified using an infrared imaging system (LI-COR Odyssey). β-actin was used as an internal loading control. Similar results were obtained in three (TZ) and two (RZ) independent NHU cell lines. C: NHU cells were pretreated for 3 h with or without the PPARγ antagonists GW9662 or T0070907 (0.1, 1.0, 5.0 μM) and PD153035 (1 μM) as indicated. Next, cells were treated in the presence or absence of TZ (1 μM) for 24 h, followed by treatment with or without PD153035 (1 μM) for 6 days, all in the presence of PPARγ antagonists (as indicated). After 6 days, protein was extracted and 20 μg was separated per lane on a 12.5% SDS–polyacrylamide gel, transferred onto a nitrocellulose membrane and incubated 16 h at 4°C in the presence of the primary antibody, as indicated, and visualised as above. D: NHU cells were treated with or without TZ (1 μM) for 24 h in the presence of PPARγ siRNA (10, 50 or 100 nM) or GW9662 (5 μM) or T0070907 (5 μM) as indicated. Next, media were replaced with or without PD153035 and GW9662 or T0070907 as indicated and again on day 3. After, 6 days protein was extracted and separated on SDS–PAGE as outlined in (C).
Following treatment of cultured NHU cells with TZ, a maximal induction of claudin 3 (13.3-fold above basal), claudin 4 (4.3-fold above basal) and claudin 5 (de novo induction) was seen after 6 days (Fig. 3A). Expression of claudin 3 (21.9-fold), claudin 4 (6.7-fold) and claudin 5 (de novo induction) was enhanced when cells were treated with both TZ and PD153035, with upregulation of claudin 4 preceding the appearance of claudins 3 and 5. Claudins 1 and 7 showed only minor changes in expression related to treatment (Fig. 3A and Table 2). By contrast, treatment with PD153035 alone resulted in no major change in claudin expression. These results were verified in a total of three independent cell lines. The changes in claudin expression were confirmed using a second PPARγ agonist, rosiglitazone (RZ) (Fig. 3B).
To verify that the changes in expression of claudins 3, 4 and 5 were mediated via a PPARγ-specific response, NHU cells were treated with PPARγ-specific antagonists, GW9662 and T0070907, prior to treatment with TZ and PD153035. Both T0070907 and GW9662 inhibited the induction of claudins 3, 4 and 5 expression by TZ and PD153035 in a dose-responsive manner, with maximal inhibitions of approximately 97%, 89% and 99%, respectively, being induced by 5 μM T0070907 (Fig. 3C). The involvement of PPARγ was corroborated using PPARγ siRNA, which demonstrated that PPARγ siRNA specifically attenuated the induction of claudin 3 and a control CK13 gene by TZ and PD153035 treatment, but did not knock down expression of claudin 7, which was included as a specificity control. The effect was maximal (75% inhibition compared to cells treated with oligofectamine alone) when cells were incubated with 50 nM PPARγ siRNA (Fig. 3D).
Promoter analysis revealed that there were potential PPRE binding sites located upstream of the predicted transcriptional start site of claudin 3 (+729 to +747) and claudin 7 (+22 to +40 and +102 to +120), but not claudin 1 or 5. The only predicted PPRE binding site for claudin 4 was on the negative strand (−563 to −581).
Effects of TZ and EGF signalling on the localisation of tight junction proteins
As combined TZ and PD153035 treatment induced changes in claudin mRNA and protein expression, indirect immunofluorescence was used to determine any changes in the localisation patterns of TJ proteins. As a positive control, cytokeratin markers of transitional and terminal urothelial differentiation (CK13 and CK20, respectively) were included and confirmed our previous report (Varley et al., 2004b) that co-treatment with TZ and PD153035 resulted in upregulation of CK13 (data not shown) and de novo expression of CK20 (Fig. 4). Expression of the adherens junction protein, E-cadherin, was also examined.
Fig. 4.
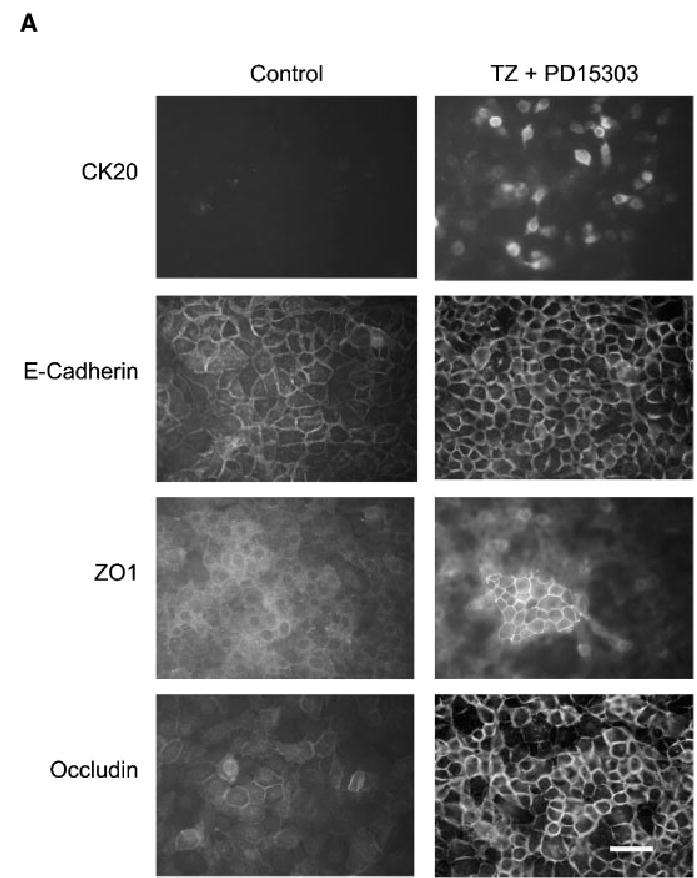
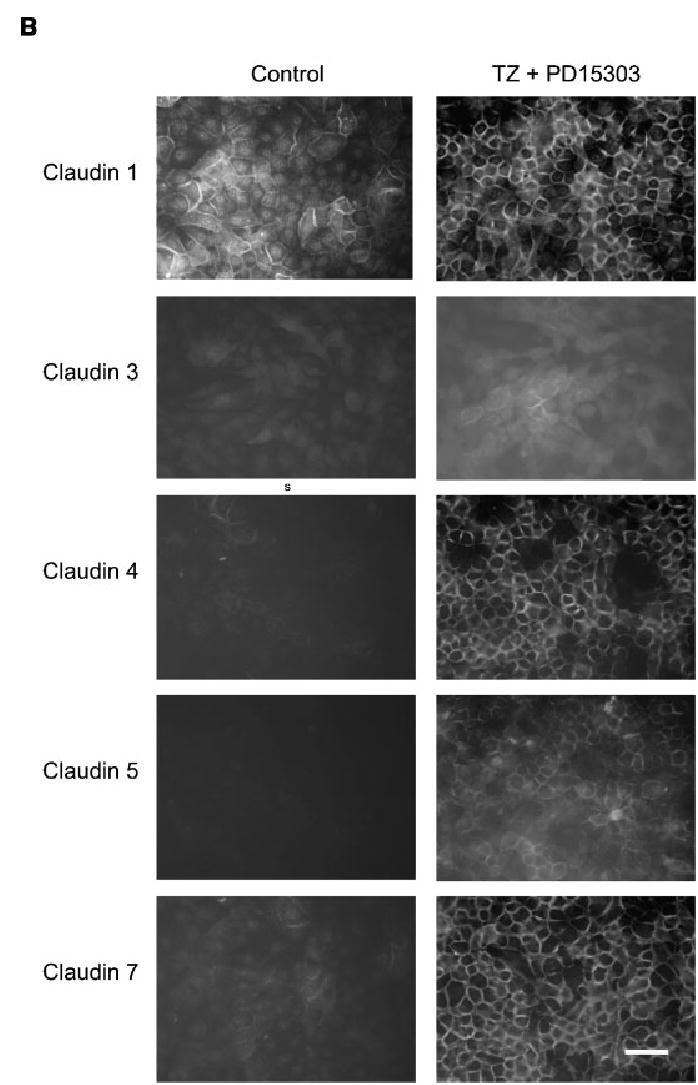
Influence of TZ and PD153035 on the localisation of the tight junction proteins. NHU cells were seeded at 2 × 105 cells/ml onto glass slides, allowed to adhere and treated with or without TZ (1 μM) for 24 h. Subsequently, cells were treated in the absence or presence of PD153035 (1 μM) and slides were fixed after 1, 3 and 6 days. Media were replaced every 3 days with fresh PD153035 added. Immunofluorescence was performed using the antibodies indicated. The images depicted in the figure are from slides fixed at 6 days. Scale bar, 30 μm. Similar results were obtained in a further two independent NHU cell lines.
In confluent NHU cell cultures, E-cadherin was predominantly localised to intercellular borders with some cytoplasmic fluorescence, but became exclusively localised to intercellular borders after treatment with TZ and PD153035 (Fig. 4). These results were reproduced in a total of three independent NHU cell lines.
In control cultures maintained in KSFMc alone, there was very little evidence of localisation of TJ component proteins to intercellular junctions (Fig. 4). In NHU cells, the majority of TJ proteins were weak and cytoplasmic, with the exception of claudin 1 whose expression was predominantly nuclear, with a small proportion localised to cell borders (Fig. 4).
Combined treatment of NHU cells with both TZ and PD153035 resulted in localisation of occludin and claudins 1 and 7 to intercellular borders, ZO1 to large patches between apposing cell borders and the de novo expression of claudins 3, 4 and 5 at the cell junctions (Fig. 4). The effect was maximal at day 6, although all proteins studied showed some relocalisation by 24 h, with the exception of claudins 3, 5 and ZO1, where relocalisation first became apparent after 3 days post-treatment (Table 3 and Fig. 4; data shown for day 6 of treatment). Immunofluorescence data was in agreement with the amounts of protein detected by Western blot analysis.
TABLE 3.
Effects of TZ and PD153035 on the localisation of tight junction proteins in NHU cells after 6 days treatment (n =3)
| DMSO control | PD153035 (1 μM) | TZ (1 μM) | TZ +PD153035 (1 μM each) | Ca2+ (2 mM) | |
|---|---|---|---|---|---|
| ZO1 | −/+ | + | + | ++ | ++ |
| Occludin | ++ | +++ | ++/+++ | +++ | +++ |
| E-cadherin | +++ | +++ | +++ | +++ | +++ |
| Claudin 1 | + | +++ | + | +++ | + |
| Claudin 4 | −/+ | ++ | + | +++ | −/+ |
| Claudin 5 | − | −/+ | −/+ | +++ | − |
| Claudin 7 | + | ++ | ++ | +++ | ++ |
The number of cells that expressed the specific tight junction proteins at the borders were expressed as: −, negative; −/+, few positive cells per field of view/well; +, 5%–30%; ++, 30%–60%; +++, 60%–100%.
Effects of Changes in Exogenous Calcium
We have previously shown that increased exogenous calcium (0.9–4 mM) induces stratification and the formation of adherens junctions and desmosomes, but does not induce terminal differentiation in cultured NHU cells (Southgate et al., 1994). It has recently been reported that TZ may trigger the release of calcium from internal stores by a PPARγ-independent non-genomic mechanism (Palakurthi et al., 2001). To assess whether the changes in localisation and/or upregulation of claudin expression induced by TZ and PD153035 showed any similarity to changes induced by near-physiological exogenous calcium concentrations, we compared NHU cells grown in medium containing 2 mM Ca2+ to cells treated with TZ and PD153035.
NHU cells grown in medium containing 2 mM Ca2+ developed a very different morphology compared to controls or cells treated with TZ and PD153035. Control NHU cell cultures formed a typical epithelioid ‘cobblestone’ monolayer, with some stratification only apparent following treatment with PD153035. By contrast, in the presence of 2 mM Ca2+, cells were compact, striated and stratified. Significantly, cells grown in 2 mM Ca2+ showed desmosome formation, as seen by the relocalisation of the desmoplakin II protein, whereas no desmosomes were formed in NHU cells treated with TZ and PD153035. Unlike cultures treated with TZ and PD153035, NHU cells maintained in 2 mM Ca2+ showed no increase in expression of claudins 4 or 5, CK13 or CK20 differentiation antigens, and no relocalisation of claudin 1. However, both 2 mM Ca2+ and combined TZ/PD153035 treatments showed a comparable increase in expression of ZO1 and occludin proteins, with relocalisation to intercellular borders (Table 3).
These differences suggest that the induced expression of claudins 4 and 5 by TZ and PD153035 was not directly linked to local changes in calcium concentration affecting the development of intercellular junctional complexes.
Involvement of ubiquitination in the stabilisation of claudin 4 protein
The data presented in Figures 2 and 3A suggest that combined treatment with TZ and PD153035 regulates the abundance of claudins 4 and 5 at the post-transcriptional level. This could occur via a protein stabilisation mechanism. Protein sequence analysis for claudin 4 (NP_001296) and claudin 5 (AAH02404) revealed that both claudins have a number of lysine residues in the amino termini of the protein sequence. As the final stages of ubiquitination involves the transfer of ubiquitin to the reactive lysine residue on the protein destined for degradation (Ciechanover and Ben-Saadon, 2004), this suggested that both proteins could be degraded by ubiquitination.
To determine if claudin 4 and 5 proteins were affected by the ubiquitination-proteasome pathway, NHU cells were treated with a proteasome inhibitor, MG132 (Fig. 5). In the presence of MG132, the amounts of claudin 4 protein were increased some eight-fold at 24 h, to levels comparable to cells treated with TZ and PD153035. By contrast, claudin 5 protein levels were not significantly affected when cells were treated with MG132 or TZ and PD153035 at 24 h. After 48 h, claudin 5 protein was induced by TZ and PD153035, but still there was no increase in claudin 5 in the presence of MG132.
Fig. 5.
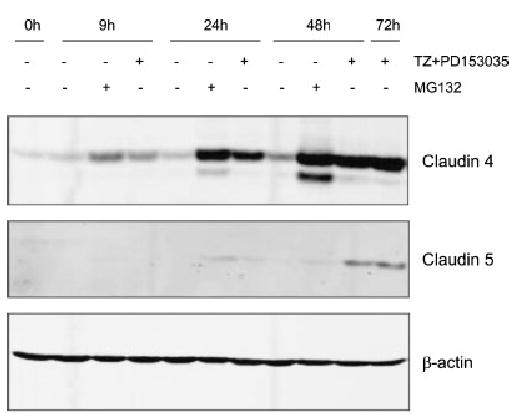
Involvement of proteasomes in stabilisation of claudin 4. NHU cells were treated with or without TZ (1 μM) for 24 h followed by treatment in the absence or presence of PD153035 (1 μM) or MG132 (12.5 μM) for 0, 9, 24 and 48 h. Cell extracts (20 μg) were resolved on 10% SDS–polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were incubated with titrated primary antibodies, fluorescent-conjugated secondary antibodies and the signal quantified by infrared imaging, as described in Figure 3. β-actin was used as an internal loading control. NHU cells treated with TZ, followed by PD153035 for 72 h, were used as a positive control.
Heterodimer partners of claudin 4
To determine whether the observed upregulation of claudin 5 was an indirect effect mediated by heterodimerisation with stabilised claudin 4, TZ- and PD153035-treated NHU cells were immunoprecipitated with anti-claudin 4 antibody, followed by Western blot analysis of the immunoprecipitates for the presence of claudins. The results demonstrated that claudin 5, but not claudins 3 and 7, was indeed associated with claudin 4 (Fig. 6), supporting the immunohistochemical observations of claudin 4 and 5 co-localization in situ.
Fig. 6.
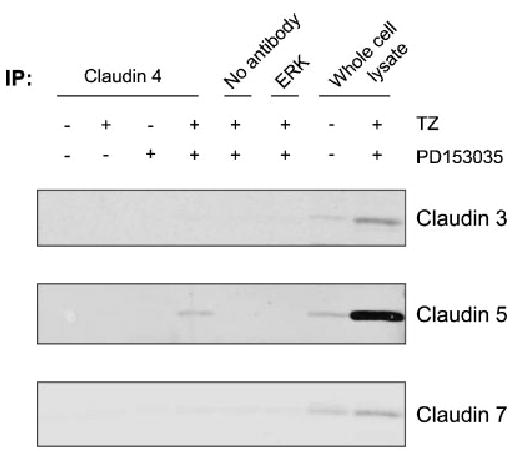
Immunoprecipitation of claudin 4. NHU cells were treated in the presence or absence of TZ (1 μM) for 24 h followed by treatment with or without PD153035 (1 μM) for 6 days. Cell extracts (500 μg) were rotated with an antibody for claudin 4, an irrelevant antibody (ERK), or no antibody on spin-columns, as indicated. As a positive control, 20 μg of whole cell lysate was used. The immunoprecipitated protein was eluted from the column and resolved on SDS–PAGE by Western blot analysis. Membranes were incubated with antibodies against claudin 3, 5 or 7, fluorescent-conjugated secondary antibodies and the signal quantified by infrared imaging, as described in Figure 3.
DISCUSSION
This is the first detailed report of expression of TJ proteins in human urothelium and describes specific localisation patterns for claudins 3, 4, 5, 7, ZO1 and occludin in situ. In addition, our study presents evidence that urothelium in situ expresses claudins 1, 2, 8 and 10, at least at the mRNA level. Our results extend previous studies that have demonstrated expression of ZO1 by human urothelia and ZO1, occludin and claudins 4, 8, 12 by rodent urothelia (Acharya et al., 2004; Slobodov et al., 2004).
A majority of TJ proteins were not restricted to the ‘kissing points’ of the terminal junctions between the superficial cells. Indeed, several component proteins were expressed in basal and intermediate cell zones, although localisation at intercellular sites occurred primarily in the upper intermediate and superficial cell zones. This phenomenon has been observed previously in other epithelial tissues and implies that the claudins may have extended roles in regulating paracellular permeability within stratified tissues, although an alternative role in cell adhesion has also been suggested (Gregory et al., 2001; Langbein et al., 2002; Mysorekar et al., 2002; Acharya et al., 2004).
Our immunohistochemical results indicate a coordinated pattern of TJ-associated protein expression relating to differentiation stage in situ (Fig. 1B): claudins 4 and 5 are expressed at a later stage of differentiation than claudin 7 and occludin, but prior to claudin 3, which, together with ZO1, is implicated in the terminal junction of the superficial cells. Thus, the claudins can be considered alongside the cytokeratins and uroplakins as urothelial differentiation markers. This raises the question as to what extent the differences in phenotype between basal, intermediate and superficially-positioned urothelial cells in situ reflect a coordinated and hierarchical differentiation programme.
NHU monolayer cell cultures offer one approach to study this question. In low (0.09 mM) calcium medium, NHU cells express a proliferative, wound-response phenotype driven via an EGFR-activated autocrine loop (Varley et al., 2005). Here, we demonstrate that this proliferative phenotype is associated with expression of transcripts for claudins 1, 2, 3, 4, 5, 7 and 8 variably. However, expression of claudin 3, 4 and 7 proteins was low and claudin 5 protein was not detected, even when NHU cells were very confluent. Furthermore, with the exception of claudin 1, there was no evidence of claudin localisation to intercellular borders in NHU cells propagated in low calcium medium. Increasing exogenous [Ca2+] to 2 mM results in desmosome formation, but the cells do not undergo cytodifferentiation (Southgate et al., 1994; Varley et al., 2004a) and, as demonstrated above, there was no induction of claudin protein expression or claudin relocalisation to TJs. By contrast, when EGFR-mediated proliferation is blocked, activation of PPARγ initiates a sequence of events culminating in expression of the uroplakins (Varley et al., 2004a) and cytokeratin markers of terminal urothelial cytodifferentiation (Varley et al., 2004b). Crucially, such differentiating NHU cells show a pattern of TJ-associated changes that mirrors the normal tissue counterpart in situ.
Our study indicates that the changes in claudin expression are regulated through both transcriptional and post-translational mechanisms. It appears that claudin 3 gene expression may be transcriptionally regulated directly by PPARγ, as analysis of the claudin 3 gene promoter revealed consensus PPARγ-binding response elements (PPRE) and claudin 3 mRNA was detected within 3 days of PPARγ activation and was specifically knocked-down by PPARγ siRNA. This is in marked contrast to the differentiation-associated induction of uroplakin gene expression, which occurs with delayed kinetics and probably involves induction of PPARγ-responsive intermediary transcription factors; a view supported by our findings that no PPRE were detected in the uroplakin gene promoters (Varley et al., 2004a). This is akin to the PPARγ-dependent differentiation programme identified for adipocytes, where receptor activation initiates a programme of transcriptional changes leading directly and indirectly (via other transcription factors) to the expression of the mature adipocyte phenotype (Forman et al., 1995; Kliewer et al., 1995).
The expression of claudins 4 and 5 was induced by the PPARγagonists and enhanced when PD153035 was also present, in agreement with previous observations that inhibition of downstream EGFR signalling is permissive for PPARγ-mediated differentiation (Varley et al., 2004a,b). However, the upregulation of claudins 4 and 5 was seen at protein, but not mRNA levels, suggestive of post-translational stabilisation. Both proteins have multiple lysine residues in their amino termini that are potential sites for modification by ubiquitination or acetylation. It has also been reported that the cytoplasmic tails of claudin proteins could be important in protein stability (Van Itallie et al., 2004). We have demonstrated that ubquitination could be responsible for the degradation of claudin 4, as the proteosome inhibitor, MG132, induced rapid accumulation of claudin 4. By contrast, claudin 5 did not appear to accumulate when proteosome processing was inhibited, suggesting an alternative mechanism for stabilisation. The tendency for claudins 4 and 5 to co-localise in situ suggest a non-covalent association between the proteins, thereby resulting in an indirect stablisation of the claudin 5 protein. Furthermore, we found strong evidence for the formation of claudin 4/5 heterodimers by immunoprecipitation analysis. Thus, it appears that protein stabilisation represents a third control mechanism for protein expression during PPARγ-mediated urothelial cell differentiation.
In addition to changes in transcript and protein expression, changes in the localisation of the TJ proteins (claudins, occludin and ZO1) were seen after treatment of NHU cells with TZ and PD153035. However, there were differences in the kinetics for relocalisation, with claudins 1, 4, 7 and occludin being localised to intercellular borders by day 1, claudin 5 by day 3 and claudin 3 and ZO1 by day 6. This again appears to reflect the differentiation progression stages in situ, with occludin, claudin 4 and 7 showing localisation to intercellular borders in upper intermediate cells and basolateral junctions of superficial cells, claudin 5 localising to the basolateral junctions of superficial cells, and ZO1 and claudin 3 restricted to the ‘kissing points’ between superficial cells.
The claudins are implicated in regulating the paracellular properties of different epithelia and hence the differentiation-associated changes in claudin expression are likely to have major consequences for urothelial barrier function. In MDCK cells, forced expression of claudin 2 is associated with decreased transepithelial resistance (Furuse et al., 2001), whereas overexpression of claudins 1, 4 and 8 resulted in an increased TER (McCarthy et al., 2000; Van Itallie et al., 2001; Yu et al., 2003). Furthermore, transfection of claudin 8 into MDCK cells was associated with a selective down-regulation of claudin 2 expression (Yu et al., 2003). It has been shown that claudin 8 acts as a cation barrier, whereas claudin 2 forms a cation-selective channel which is capable of transforming a barrier from ‘tight’ to ‘leaky’ (Furuse et al., 2001; Amasheh et al., 2002). Thus, our observed downregulation of claudin 2 during terminal urothelial differentiation may be an important factor in the development of the very high TER values associated with effective urothelial barrier function.
In summary, we have presented results that begin to define the composition of TJs in urothelium and in cultured NHU cells. Furthermore, we have identified that differentiation is accompanied by changes in transcription, protein expression and localisation of TJ proteins and suggest that this is part of a co-ordinated, integrated differentiation programme that ultimately is important for urinary barrier function. In a number of diseases of the bladder, such as cancer and interstitial cystitis, the urothelium fails to form a functional barrier. Understanding how TJ proteins are regulated and how this is integrated into the development of a fully differentiated, functional urothelium could have implications for treatment of diseases of the bladder and other epithelial tissues.
Acknowledgments
J.S. is supported by York Against Cancer.
Footnotes
Contract grant sponsor: NIH; Contract grant number: IR21 DK066075.
References
- Acharya P, Beckel J, Ruiz WG, Wang E, Rojas R, Birder L, Apodaca G. Distribution of the tight junction proteins ZO-1, occludin, and claudin-4, -8, and -12 in bladder epithelium. Am J Physiol Renal Physiol. 2004;287(2):F305–F318. doi: 10.1152/ajprenal.00341.2003. [DOI] [PubMed] [Google Scholar]
- Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, Fromm M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115(Pt 24):4969–4976. doi: 10.1242/jcs.00165. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004;14(3):103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Furuse M, Furuse K, Sasaki H, Tsukita S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153(2):263–272. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Betanzos A, Nava P, Jaramillo BE. Tight junction proteins. Prog Biophys Mol Biol. 2003;81(1):1–44. doi: 10.1016/s0079-6107(02)00037-8. [DOI] [PubMed] [Google Scholar]
- Gregory M, Dufresne J, Hermo L, Cyr D. Claudin-1 is not restricted to tight junctions in the rat epididymis. Endocrinology. 2001;142(2):854–863. doi: 10.1210/endo.142.2.7975. [DOI] [PubMed] [Google Scholar]
- Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD. Expression of peroxisome proliferator-activated receptor gamma (PPARgamma) in human transitional bladder cancer and its role in inducing cell death. Neoplasia. 1999;1(4):330–339. doi: 10.1038/sj.neo.7900050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Meyers S, Liang FX, Deng FM, Kachar B, Zeidel ML, Sun TT. Role of membrane proteins in permeability barrier function: Uroplakin ablation elevates urothelial permeability. Am J Physiol Renal Physiol. 2002;283(6):F1200–F1207. doi: 10.1152/ajprenal.00043.2002. [DOI] [PubMed] [Google Scholar]
- Issemann I, Prince RA, Tugwood JD, Green S. The retinoid X receptor enhances the function of the peroxisome proliferator activated receptor. Biochimie. 1993;75(3–4):251–256. doi: 10.1016/0300-9084(93)90084-6. [DOI] [PubMed] [Google Scholar]
- Jain S, Pulikuri S, Zhu Y, Qi C, Kanwar YS, Yeldandi AV, Rao MS, Reddy JK. Differential expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) and its coactivators steroid receptor coactivator-1 and PPAR-binding protein PBP in the brown fat, urinary bladder, colon, and breast of the mouse. Am J Pathol. 1998;153:349–354. doi: 10.1016/s0002-9440(10)65577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami S, Arai G, Hayashi T, Fujii Y, Xia G, Kageyama Y, Kihara K. PPARgamma ligands suppress proliferation of human urothelial basal cells in vitro. J Cell Physiol. 2002;191:310–319. doi: 10.1002/jcp.10099. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358(6389):771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Langbein L, Grund C, Kuhn C, Praetzel S, Kartenbeck J, Brandner JM, Moll I, Franke WW. Tight junctions and compositionally related junctional structures in mammalian stratified epithelia and cell cultures derived therefrom. Eur J Cell Biol. 2002;81(8):419–435. doi: 10.1078/0171-9335-00270. [DOI] [PubMed] [Google Scholar]
- McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Skare IB, Lynch RD, Schneeberger EE. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J Cell Sci. 2000;113(Pt 19):3387–3398. doi: 10.1242/jcs.113.19.3387. [DOI] [PubMed] [Google Scholar]
- Mysorekar IU, Mulvey MA, Hultgren SJ, Gordon JI. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J Biol Chem. 2002;277(9):7412–7419. doi: 10.1074/jbc.M110560200. [DOI] [PubMed] [Google Scholar]
- Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61(16):6213–6218. [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. The tight junction: A multifunctional complex. Am J Physiol Cell Physiol. 2004;286(6):C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Slobodov G, Feloney M, Gran C, Kyker KD, Hurst RE, Culkin DJ. Abnormal expression of molecular markers for bladder impermeability and differentiation in the urothelium of patients with interstitial cystitis. J Urol. 2004;171(4):1554–1558. doi: 10.1097/01.ju.0000118938.09119.a5. [DOI] [PubMed] [Google Scholar]
- Southgate J, Trejdosiewicz LK. Immunolabelling of cells and tissues: Approaches and pitfalls. Human Reproduction. 1997;12(Suppl 5):65–75. [Google Scholar]
- Southgate J, Hutton KA, Thomas DF, Trejdosiewicz LK. Normal human urothelial cells in vitro: Proliferation and induction of stratification. Lab Invest. 1994;71(4):583–594. [PubMed] [Google Scholar]
- Southgate J, Masters JR, Trejdosiewicz LK. 2002. Culture of Human Urothelium. In: Freshney RI, Freshney MG, editors. Culture of epithelial cells. 2nd edn. New York: J Wiley and Sons, Inc. pp 381–400.
- Tsukita S, Furuse M. Claudin-based barrier in simple and stratified cellular sheets. Curr Opin Cell Biol. 2002;14(5):531–536. doi: 10.1016/s0955-0674(02)00362-9. [DOI] [PubMed] [Google Scholar]
- Van Itallie C, Rahner C, Anderson JM. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest. 2001;107(10):1319–1327. doi: 10.1172/JCI12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Colegio OR, Anderson JM. The cytoplasmic tails of claudins can influence tight junction barrier properties through effects on protein stability. J Membr Biol. 2004;199(1):29–38. doi: 10.1007/s00232-004-0673-z. [DOI] [PubMed] [Google Scholar]
- Varley CL, Stahlschmidt J, Lee WC, Holder J, Diggle C, Selby PJ, Trejdosiewicz LK, Southgate J. Role of PPARgamma and EGFR signalling in the urothelial terminal differentiation programme. J Cell Sci. 2004a;117(Pt 10):2029–2036. doi: 10.1242/jcs.01042. [DOI] [PubMed] [Google Scholar]
- Varley CL, Stahlschmidt J, Smith B, Stower M, Southgate J. Activation of peroxisome proliferator-activated receptor-gamma reverses squamous metaplasia and induces transitional differentiation in normal human urothelial cells. Am J Pathol. 2004b;164(5):1789–1798. doi: 10.1016/s0002-9440(10)63737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley C, Hill G, Pellegrin S, Shaw NJ, Selby PJ, Trejdosiewicz LK, Southgate J. Autocrine regulation of human urothelial cell proliferation and migration during regenerative responses in vitro. Exp Cell Res. 2005;306(1):216–229. doi: 10.1016/j.yexcr.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem. 2003;278(19):17350–17359. doi: 10.1074/jbc.M213286200. [DOI] [PubMed] [Google Scholar]