

Abstract
Context
Polycystic ovary syndrome (PCOS) is characterized by chronic hyperandrogenic anovulation and is associated with insulin resistance. Its pathogenesis is believed to be multifactorial, and abnormal gene regulation could be one contributing factor. Type 5 17 beta-hydroxysteroid dehydrogenase (17β-HSD5) appears to be the major testosterone-forming 17β-HSD isoenzyme in females.
Objective
To investigate the role of a potentially activating 17β-HSD5 gene (HSD17B5) variant in hyperandrogenism.
Design
Case report and case-control cohort study.
Setting
General Clinical Research Center.
Study subjects
A case of PCOS who had hyperthecosis associated with profound type B insulin resistance and an unusual, frankly male, testosterone response to a gonadotropin releasing hormone agonist test, 121 PCOS, and 128 population controls.
Interventions
Diagnostic.
Main outcome measures
Sequencing of HSD17B5 5′-flanking region and 9 exons, genotype/phenotype studies, and in vitro functional studies.
Results
Our case had a previously undescribed homozygous HSD17B5 variant (G-to-A substitution) -71 bp in the promoter region. Genotyping controls showed this to be a single nucleotide polymorphism (SNP-71G). Luciferase activity of a SNP-71G promoter construct was significantly higher than that of the wild-type, and electrophoretic mobility shift assays revealed that SNP-71G possessed significantly increased affinity to nuclear transcription factors. SNP-71G allele frequency (32.2% vs. 22.3%) and SNP-71G allele presence (53.7% vs. 38.3%) were significantly increased in PCOS (p=0.01). SNP-71G homozygosity tended to contribute about 20% to the plasma testosterone level.
Conclusions
SNP-71G is a functional polymorphism that may contribute to testosterone excess in a subset of PCOS patients.
Keywords: 17 beta-hydroxysteroid dehydrogenase (17β-HSD), Type 5 17β-HSD gene (HSD17B5), electrophoretic mobility shift assays (EMSA), polycystic ovary syndrome (PCOS), Sp1/Sp3, Single nucleotide polymorphism (SNP)
Introduction
Dysregulation of ovarian steroidogenesis seems to be responsible for polycystic ovary syndrome (PCOS). PCOS, a poorly understood chronic hyperandrogenism disorder, is the most common cause of anovulatory infertility, affecting about 5% of reproductive-age women (1). PCOS is also associated with insulin resistance and type 2 diabetes mellitus. The mechanism by which hyperinsulinemia is related to androgen excess and polycystic ovaries is unknown. In vitro studies have shown that insulin and insulin like growth factor-1 augment LH stimulation of steroidogenesis in ovary theca-interstitial (thecal) cells (2-5).
As with other hyperandrogenic disorders, the major circulating androgen is testosterone (6). Testosterone biosynthesis requires androgenic 17 beta-hydroxysteroid dehydrogenase (17β-HSD) activity, namely, types 3 and 5 17ß-HSD (17β-HSD3 & 5). The type 3 17β-HSD gene (HSD17B3) is mainly expressed in testis, where it is essential for sexual differentiation and development (7), but it is not expressed in the adrenal gland or ovary (8, 9). On the other hand, the type 5 17β-HSD gene (HSD17B5) is widely expressed and is found in the ovary and adrenal gland (8-13). Human HSD17B5 is composed of 9 exons spanning 16 kilobases and is located on chromosome 10p14,15 (11, 14). We recently reported that a binding site for the ubiquitous transcription factors Sp1/Sp3 in the HSD17B5 proximal promoter is necessary for gene activity (15).
In the present study, we suspected an activating HSD17B5 variant in a patient with the hyperthecosis form of PCOS due to profound type B insulin resistance because she had an unusual, frankly male testosterone response to a gonadotropin releasing hormone agonist (GnRHag) challenge test. We identified a variant adjacent to the Sp1/Sp3 binding site of the promoter. We determined genotype frequencies for this variant in a sample of 249 unrelated individuals and found that this is a single nucleotide polymorphism (SNP). Functional studies showed that this SNP moderately increases HSD17B5 gene promoter activity and increases its affinity for the transcriptional factors Sp1/Sp3. We also found an association of this SNP with PCOS.
Materials and methods
Identification of an HSD17B5 promoter variant in a patient with hyperthecosis
Case report
Our index case was a 44 year-old woman with systemic lupus erythematosis, type B insulin resistance (requiring 1200 U insulin daily together with troglitazone), insulin receptor antibodies, acanthosis nigricans, hirsutism, amenorrhea, and bilaterally enlarged ovaries that were ultrasonographically homogeneous. Her hormone profile showed basal testosterone levels ranging into the frankly male range (77-557 ng/dl) and free testosterone levels of 10-77 pg/ml (normal 3-10 pg/ml); steroid intermediates were otherwise low as expected for a glucocorticoid-treated patient (Table 1). In response to GnRHag test, she had a remarkably high testosterone response, and her ratios of plasma testosterone to androstenedione were 12-41 SD above normal (normal: 0.15 ± 0.085, SD, at baseline and 0.15 ± 0.069 post-GnRHag) (Table 1). Laparoscopy revealed enlarged ovaries; the histopathology was reported as marked hyperthecosis.
Table 1.
Hormonal profile of our index case compared to control and classic PCOS females.
| GnRHag test (leuprolide 10 μg/kg) | ||||||
|---|---|---|---|---|---|---|
| Basal* | Peak* | |||||
| Patient | Control females** (Mean ± SD.) | Classic PCOS** (Mean ± SD.) | Patient | Control females** (Mean ± SD.) | Classic PCOS** (Mean ± SD.) | |
| Testosterone (ng/dl) | 109 | 12.1 ± 8.9 | 71.7 ± 43 | 690 | 19.8 ± 12 | 105 ± 51 |
| Androstenedione (ng/dl) | 83 | 77.4 ± 30 | 257 ± 98 | 231 | 135 ± 55 | 375 ± 162 |
| Estradiol (pg/ml) | 75 | 64.1 ± 30 | 73 ± 19 | 268 | 375 ± 128 | 515 ± 253 |
| 17-hydroxyprogesterone (ng/dl) | 43 | 24 ± 10.3 | 90 ± 38 | 774 | 160 ± 53 | 518 ± 240 |
| 17-hydroxypregnenolone (ng/dl) | 14 | 11.3 ± 4.7 | 26 ± 11 | 98 | 36.2 ± 13 | 78 ± 41 |
| Dehydroepiandrosterone (DHEA, ng/dl) | 53 | 68.5 ± 21 | 114 ± 38 | 143 | 89 ± 30 | 153 ± 54 |
| DHEA-SO4 (μg/dl) | 11 | 35.2 ± 20 | - | 14 | 35 ± 18 | - |
| LH (IU/L) | 3 | 11.2 ± 3.1 | 24.1 ± 6.5 | 54.5 | 148 ± 55 | 245 ± 85 |
| FSH (IU/L) | 2.3 | 6.8 ± 1.5 | 5.6 ± 1.3 | 17.5 | 67 ± 24 | 58 ± 27 |
Multipliers to convert from mass to SI units are androstenedione 0.0348, 17-hydroxypregnenolone 0.0301, 17-hydroxyprogesterone 0.0303, DHEA 0.0347, DHEA-SO4 27.2, and testosterone 0.0347 in nmol/L, respectively; free testosterone 3.47 and estradiol 3.67 in pmol/L, respectively.
Subjects
The PCOS study population consisted of 121 Caucasian, African-American, Hispanic, and Asian subjects found to have PCOS associated with functional ovarian hyperandrogenism in our Clinical Research Center over the past 15 years (16). PCOS was defined consistent with Rotterdam criteria: otherwise unexplained evidence of hyperandrogenism (specifically, plasma free testosterone above the upper limit of normal for reproductive age women, 10 pg/ml) and menstrual irregularity and/or a polycystic ovary (17). Additionally, all had an elevated plasma free testosterone (above 10 pg/ml or 34.7 pmol/L) and functional ovarian hyperandrogenism according to either dexamethasone suppression test or GnRH agonist criteria (18). Women with any other cause of oligomenorrhea and hyperandrogenism were excluded. As a control group, 128 random population subjects were also studied. The study was approved by the University of Chicago Institutional Review Board. Written consent was obtained from all participants and clinical and laboratory information was stored in a database.
Genomic DNA sequence
Genomic DNA was extracted from blood, and a 1386-bp 5′-flanking region was amplified by PCR using the primers 5′-AAAGCTTTCAATGATTTTATAT-3′ and 5′-AACACGAACCTTACAACCCAAT -3′. Five μl of PCR products were pretreated according to the manufacturer’s protocol (USB Corporation, Cleveland, OH). Briefly, five μl of PCR amplification mixtures were treated with 1 μl each of shrimp alkaline phosphatase (2 u/μl) and exonuclease (10 u/μl) at 37°C for 20 min and the enzymes then inactivated by heating to 80°C for 15 min. The treated PCR products were directly sequenced using the ABI dye terminator cycle sequencing technique with specific HSD17B5 gene primers and GeneAmp PCR system 2700 thermal cycler (Perkin-Elmer). The sequencing reaction was performed using the following thermoprofile: 3 min at 95 °C, then 30 seconds at 95 °C, 30 seconds at 50 °C, and 4 min at 72 °C for a total 35 cycles. The samples were separated on an Applied Biosystems PRISM 377 DNA Sequencer and analyzed using the ABI PRISM Sequence Navigator according to the manufacture’s instructions.
Analysis of the 17ß-HSD5 -71G variant
Restriction fragment length polymorphism assay (RFLP)
A two-step PCR-based RFLP assay was used to determine the genotype of at the promoter region of interest. The first PCR reaction was done using a set of primers HSD17B5 (F1): 5′-GAATAATTTAATATAGAGATT-3′, HSD17B5 (R1): 5′- AACACGAACCTTACAACCCAAT -3′ to generate a 468-bp fragment. The first PCR reaction was carried out using 50 ng of genomic DNA, 1x Taq buffer, 1 μM each primer, 250 μM 2′-deoxynucleoside 3′-triphosphate, and 0.25 μl (5 units/μl) of Taq polymerase (Promega) in a final 25-μl volume. PCR amplification was carried out for 35 cycles by heat denaturing at a temperature of 95°C for 30 s, annealing at a temperature of 45°C for 30 s, and primer extension at 72°C for 60 s, with a final step at 72°C for 7 min. PCR products were verified by agarose gel electrophoresis. One μl of PCR product was used for a second PCR reaction using nested primers to amplify a 170-bp fragment. These were: HSD17B5 (F2): 5′-CAATTTTCTCCACAGACCATATAAGACCAGCT -3′ and HSD17B5 (R2): 5′-TCCCTGTCACTTGTCTGACTAGC -3′. Mismatches (underlined) were introduced in the forward primer to generate a restriction site (Puv II) in the variant allele, but not in the wild type. Twenty μl of the nested PCR product was digested with 0.5 units of Pvu II (Promega) according to the manufactory at 37°C for 2 h. Then, the digests were electrophoresed on a 2.5% agarose gel and stained with ethidium bromide to generate allele-specific fragments: A/A = 170 bp, G/G = 142 and 28 bp, and A/G = 170, 142, and 28 bp.
Effect of the variant -71G (vs wild-type -71A) on function of the HSD17B5 gene
Rat thecal cell culture
Thecal cells were obtained from hypothysectomized rats following the procedure described by Magoffin and Erickson (19). Briefly, ovaries from 20 day-old hypophysectomized female Sprague-Dawley rats were removed on the fourth postoperative day, minced and thecal cells dispersed with collagenase and deoxyribonuclease. The dispersed cells were cultured on 6 well plates at a concentration of 300,000 cells/well in McCoy’s 5a medium (without serum), supplemented with L-glutamine (2 mM), insulin (6.25 μg/ml), transferrin (6.25 μg/ml), selenium (6.25 ng/ml), and antibiotics (penicillin G, streptomycin sulfate, and amphotericin B from GIBCO) and cultured for 48 h at 37°C under a water-saturated atmosphere of 95% air.
Constructs of human HSD17B5 promoter/luciferase
To construct point mutants of HSD17B5, PCR reactions were performed using a forward primer containing the desire mutation (Table 2) and reversed primer [(5′-GGGCCCAGATCTCCCTGTCACTTGTCTGACTAGC-3′), a Bag II site, underlined, was introduced for facilitative cloning] and HSD17B5 promoter construct (-1060 bp) as the template (15). All of the PCR-amplified fragments containing point mutants were ligated into pGEM-T easy vector and sequenced to ensure fidelity of the amplified sequences. Inserts were subcloned into the Sal I and Bgl II sites of a promoterless luciferase expression vector (pGL3-Basic vector). All the constructs used in the present study were of identical length, −82 bp to +68, which is 5′ contiguous to the translation initiation Met codon; base numbers are counted from the transcriptional start site (11). Plasmid DNA isolation was carried out on a QIAGEN-tip 500 column according to the manufacturer’s protocol.
Table 2.
Sense oligonucleotides used for site-directed mutagenesis constructs.
| Oligonucleotides | sequence (5′ to 3′) |
|---|---|
| -82 | -82 GGATCCGTCGACTAAGACTGCCTATGTACCTCCT -61 |
| -82mut1 | -82 GGATCCGTCGACTAAGACTGCCTGTGTACCTCCTCCTAC –56 |
| -82mut2 | -82 GGATCCGTCGACTAAGACTGCCTCTGTACCTCCTCCTAC -56 |
| -82mut3 | -82 GGATCCGTCGACTAAGACTGCCTAAGTACCTCCTCCTAC -56 |
| -82mut4 | -82 GGATCCGTCGACTAAGACTGCCTATGTCCCTCCTCCTAC -56 |
| -82mut5 | -82 GGATCCGTCGACTAAGACTGCCTATGTAACTCCTCCTAC -56 |
| -82mut6 | -82 GGATCCGTCGACTAAGACTGCCTATGTACTTCCTCCTAC -56 |
| -82mut7 | -82 GGATCCGTCGACTAAGACTGCCTATGTACCCCCTCCTAC -56 |
| -82mut8 | -82 GGATCCGTCGACTAAGACTGCCTATGTACCTTCTCCTAC -56 |
| -82mut9 | -82 GGATCCGTCGACTAAGACTGCCTATGTACCTCTTCCTAC -56 |
| -82mut10 | -82 GGATCCGTCGACTAAGACTGCCTATGTACCTCCTCTCAC -56 |
All the constructs were containing an extension from +68 to the base number indicated in the construction name; base numbers are counted from the transcriptional start site (14). Mutated bases are shown in boldface and underlined. Restriction site for Sal I (italicized) was attached to oligonucleotides for cloning.
Transient transfection and luciferase activity assays
For transfection experiments, cells were subcultured onto 6 well culture dishes (300,000 cells/well) and used 48 hr later. Cells were transfected with 1.5 μg of constructs containing the wild-type and mutants and 0.2 μg of the pCI β-galactosidase expression plasmid as a control for transfection efficiency. Transfections were performed using 3 μl Fugene (Roche Molecular Biochemicals, Indianapolis, IN) reagent for each well of cells in DMEM/F12 medium (2.1 ml). After 24 hours of incubation at 37 °C in a 5% CO2 incubator, the transfected cells were washed twice in PBS and were lysed in 300 μl of lysis buffer (Promega, Madison, WI) (15).
The luciferase activity assays were performed on 20 μl of cell lysate using a Promega kit. ß-Galactosidase assays were performed on 100 μl of cell lysate by adding a diluted sample to an equal volume of Assay 2X buffer. Samples were incubated for 16 hours, the reactions were terminated by addition of sodium carbonate, and absorbance was read at 420 nm with a spectrophotometer (15).
Electrophoretic mobility shift assays (EMSA)
To generate double-stranded DNA for -71G or -71A fragment probes, PCR reactions were performed using biotin-labeled forward oligonucleotides 5′-/5Bio/ACAGACCATATAAGACTGCC-3′ and reverse primer (5′-GCTTCTCCTCAGAGATTACAAA-3′ (Integrated DNA Technologies, Coralville, IA), and HSD17B5 promoter constructs containing -71G and -71A as the templates (15). The PCR fragments containing the -71G or -71A were purified after digestion by Hpa1, which generated 48 bp biotin-labeled probe containing Sp1/Sp3 binding element. The reaction mix (10 μl) contained a range of concentrations of rat theca cell nuclear proteins and 2 μl gel shift binding 5 X buffer, and was preincubated at 22 °C for 10 min prior to adding 1 pmol of probes. After a further 20-min incubation, samples were separated on a native 4% polyacrylamide gel and then transferred to a nylon membrane. The positions of the biotin end-labeled oligonucleotides were detected by a chemiluminescent reaction with streptavidin-horseradish peroxidase according to the manufacturer’s instruction (Bright-Star system, Ambion), visualized and analyzed using ChemiDoc XRS system and Quantity One software (Bio-Rad) (15).
Statistical analysis
Data are expressed as mean ± S.D. Chi square test was used to test for deviation of genotype distribution from Hardy–Weinberg equilibrium and to determine whether there were any significant differences in allele or genotype frequencies between cases and controls (http://ihg.gsf.de). Statistical significance in the luciferase activity experiments among constructs and in the clinical data among genotypes, as well as differences between allelic variants of EMSA results, were assessed by ANOVA followed by Scheffe’s post-hoc test. A P value <0.05 was considered statistically significant.
Results
Identification of a new variant in the human HSD17B5 gene promoter
To search for mutations or polymorphisms in the 5′-flanking region of the human HSD17B5 gene, a 1386-bp fragment corresponding to the HSD17B5 5′-flanking region was amplified and sequenced from genomic DNA from our index patient and a control subject. Comparison of the patient with control revealed the existence of an A-to-G substitution -71 bp from the transcription initiation site of the HSD17B5 gene (Fig. 1). Gene bank search revealed this to be a previously undescribed substitution. Our patient was homozygous for the -71G variant in the HSD17B5 promoter. The results were confirmed by RFLP.
Fig. 1.
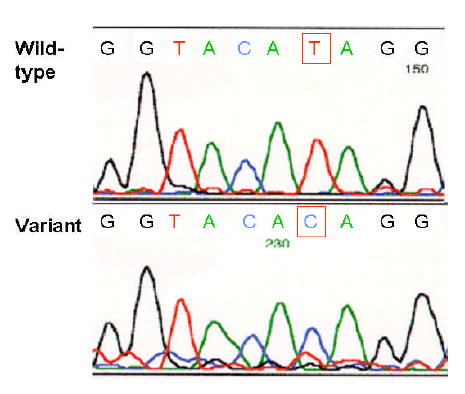
The automatic DNA sequence of the hyperthecosis patient and normal control HSD17B5 genes; the sequence primer was antisense. A homozygous single-base change at bp -71 was found, that encodes a variant, -71G.
Demonstration that the -71G HSD17B5 variant is a SNP-71G associated with PCOS
A total of 121 subjects with PCOS and 128 population subjects were genotyped for the -71G variant by RFLP (Fig. 2). The ethnic distribution of the PCOS patients was 48.8% Caucasian (Ca), 39.7% Afro-American (Af), 3.3% Hispanic (Hi), 7.4% Asian (As); and other 0.8%; that of controls was 55.4% Ca, 38.8% Af, 3.3% Hi, 5.8.4% As, and other 2.5%. The distribution of alleles among each group is shown in Table 3 and is consistent with Hardy-Weinberg equilibrium among ethnic groups. These studies show that -71G variant is a SNP.
Fig. 2.

Restriction fragment length polymorphism assay. 170 bp PCR fragments were digested with Pvu II and separated by agarose gel electrophoresis. Representative agarose gel electrophoresis of Pvu II RFLP of the HSD17B5 gene variants. 28 bp fragments generated from G-variants were not seen because too low on gel.
Statistical analysis was performed to test whether this association with PCOS is significant. The SNP-71G allele frequencies were significantly more frequent than in the control group, with an odds ratio (OR) of 1.66 (95% confidence interval (CI) 1.114-2.475; P= 0.0124). The odds ratio for bearing the G allele was based on the data in Tables 3 for the two largest ethnic groups within our study populations. The odds ratio for Caucasians with PCOS bearing the G allele was 2.251 (95% CI: 1.287-3.937; P=.0041). That for Afro-Americans bearing the G allele was 1.195 (95% CI: 0.635-2.247); this odds ratio is neither significant nor significantly different from that of Caucasians. Hispanic and Asian groups are too small for statistical analysis.
Table 3.
Genotype and allele frequencies for HSD17B5 SNP-71G in PCOS and controls
| Total | N | A/A | G/A | G/G | P value | OR (95% CI) |
|---|---|---|---|---|---|---|
| PCOS | 121 | 46.3% | 43.0% | 10.7% | 0.0124 | 1.66 (1.114-2.475) |
| Control | 128 | 61.7% | 32% | 6.25% | ||
| Caucasian | ||||||
| PCOS | 59 | 39% | 47.5% | 13.6% | 0.0045 | 2.251(1.287-3.937) |
| Control | 67 | 65.7% | 26.9% | 7.5% | ||
| African-American | ||||||
| PCOS | 48 | 47.9% | 43.8% | 8.3% | 0.581 | 1.195 (0.635-2.247) |
| Control | 47 | 51.1% | 44.7% | 4.2% | ||
Analysis of variance (ANOVA) was performed on the baseline plasma testosterone level of the PCOS patients to begin to explore the relationship of genotype to endocrine phenotype (Fig. 3). The data suggest that SNP-71G homozygosity contributes about 20% to this parameter. However, the difference is not statistically significant.
Fig. 3.
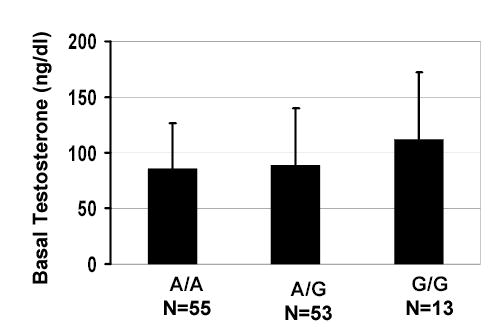
Relationship between plasma testosterone and genotype in PCOS patients. A tendency to elevation in the G/G group is seen.
Functional effect of the SNP-71G of the HSD17B5 gene
To investigate whether the G-to-A substitution has an effect on gene expression, transfection experiments in rat theca cells were carried out with each allelic promoter-reporter gene construct. To characterize more precisely the contributions of the SNP and proximate nucleotides to promoter activity, we cloned a series of point mutation of -82/+68 constructs of the HSD17B5 promoter. The reporter gene expression driven by the G allelic HSD17B5 promoter was 70% higher (P<0.05) than reporter gene expression directed by the A allelic promoter. However, other constructs containing mutations between -70 to -59 had significantly decreased promoter activity (P<0.05), except for -82-70A and -82-64C. We conclude that the SNP-71G of HSD17B5 gene has significantly but modestly increased promoter activity (Fig. 4).
Fig. 4.
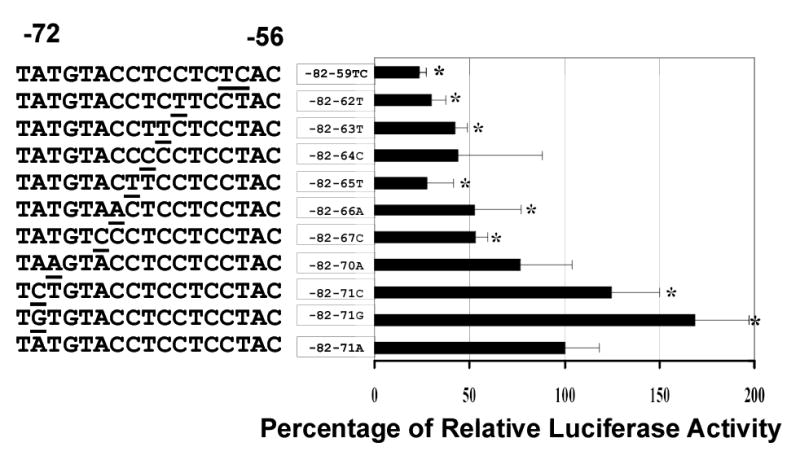
Analysis of single nucleotide mutants of the human HSD17B5 promoter–luciferase constructs. Rat thecal cells were transfected with the indicated plasmids, and luciferase activity was measured. Values represent means ± SD. of three separate determinations, performed in triple. *P<0.05 compared with wild-type control (-82-71A). The Sp1/Sp3 binding site is underlined in the control sequences.
We have proven that Sp1/Sp3 binds to a core sequence of CCT repeats that is necessary for HSD17B5 promoter activity (15). The SNP-71G is close to the Sp1/Sp3 binding site. To investigate if the presence of SNP-71G also increased the affinity of nuclear proteins (Sp1/Sp3) to this region of the HSD17B5 promoter, EMSAs were performed. EMSAs were carried out with rat thecal cell nuclear protein with labeled double-stranded oligonucleotides containing either the wild-type sequence (A/A) or the SNP-71 sequence (G/G). Interestingly, as predicted by the promoter studies, the EMSAs suggested that binding affinity of oligonucleotides containing SNP-71 to a range of concentrations of nuclear proteins was much higher than that of the wild-type sequence (Fig. 5, A). In order to precisely quantitate the binding affinity because the probes bind to multiple isoforms of Sp1/Sp3 (15), we quantified residual free probes over a range of increasing concentrations of nuclear proteins (Fig. 5, B & C). The results confirmed that SNP-71G possesses significantly higher affinity than the wild type (p=0.04) (Fig. 5, B & C). The results were verified using a fragment of pGl3 basic sequences as a control (Fig. 5, B). These data suggest that SNP-71 can greatly enhance the binding affinity of this site for the nuclear transcription factors.
Fig. 5.
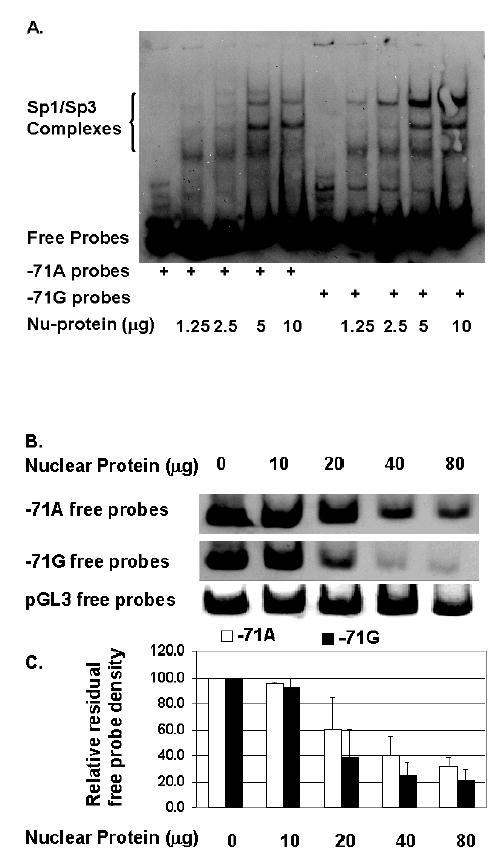
The HSD17B5 promoter containing SNP-71G has increased affinity for transcription factors Sp1/Sp3.
A. EMSA autoradiograph. Varying concentrations of nuclear proteins were incubated with biotin-labeled probes containing either SNP-71G or -71A wild-type sequence.
B. Residual free probe of an EMSA.
C. Relative densitometry of bands of residual free probe. Each value is the average of three independent experiments means ± SD. The lesser residual free -71G than -71A probes with higher nucleoprotein concentrations indicates higher binding affinity of nucleoprotein for the –71G (P=0.04).
Discussion
PCOS appears to be a complex trait with contributions from both polygenic heritable and non-heritable factors (20). Dominant inheritance of polycystic ovaries and testosterone excess has been suggested to occur (21, 22), and familial clustering has been reported (23, 24). Although there is vigorous debate about whether PCOS is fundamentally a neuroendocrine or ovarian disorder (1), evidence is accumulating in favor of the latter (9, 25, 26). Thus, PCOS seems to usually arise as a complex genetic disorder in which an intrinsic ovarian genetic trait interacts with other congenital or cellular environmental factors to cause abnormal regulation (dysregulation) of steroidogenesis. Linkage and association studies have suggested gene linkages for the steroidogenic enzymes 17-hydroxylase/C17,20-lyase (CYP17) (27) and cholesterol side-chain cleavage enzyme (P450scc or CYP11α) (28), the insulin variable number of tandem repeats (VNTR) (28), and follistatin (29), as well as chromosome 19p13.3 in the insulin receptor gene region (30, 31). CYP17, follistatin, and VNTR have subsequently been excluded (32-35). However, CYP11α 5′-UTR (tttta)n variants were confirmed to be associated with both PCOS and testosterone levels in women with PCOS, but the latter association was not observed in healthy women (36), which led to the postulate that additional genetic and/or environmental factors have an epistatic effect on the CYP11α genotype, allowing its hyperandrogenic action to be expressed only in patients with PCOS and not in healthy subjects.
Our index patient had the hyperthecosis form of PCOS associated with profound type B insulin resistance, which is a rare disorder (37). Enlarged ovaries and hyperandrogenism (range 83-1033 ng/dl) are common in patients with type B insulin resistance (37), this association is likely related to insulin resistance, as often seen in PCOS; however, the molecular mechanism is unknown. Because of our patient’s frankly male testosterone response to a GnRH agonist challenge test, we searched for sequences in the promoter of the HSD17B5 gene that might be responsive to both gonadotropins and insulin. This led us to identify a previously undescribed homozygous variant (G-to-A substitution) in this patient. The variant is located -71 bp from the transcription initiation site and a few bp upstream from Sp1/Sp3 binding core sequence (15). There is no study to date that has screened for mutations or polymorphisms in the human HSD17B promoter region in PCOS patients, and the mechanism by which HSD17B5 is regulated in PCOS patients is unknown. Most PCOS patients, in addition to being hyperandrogenic, are also insulin resistant and/or obese, and insulin resistance plays a major role in PCOS (1, 38 ).
In order to define whether this variant is a mutation or a polymorphism, we have genotyped 121 PCOS patients and 128 population controls. Our data revealed this to be a single nucleotide polymorphism (SNP-71G). Genotype/phenotype co-relationship studies revealed that there was a significant increased frequency of SNP-71G in PCOS. However, this increased SNP-71G frequency was seen in Caucasian, but not in African-American PCOS patients. Other ethnic groups were too small for statistical analysis. Plasma testosterone data suggest that SNP-71G homozygosity contributes about 20% to the plasma testosterone levels. However, this difference is not statistically significant, possibly since the S.D. is wide and the fraction of patients homozygous for SNP-71G is relatively small. In addition, PCOS may well be a phenotypic expression affected by the interaction of many genes; it was not surprising that the levels of testosterone in PCOS patients showed a wide range. Nevertheless, our studies suggest that the SNP-71G is clinical relevant and may be involved in the control of the expression of the HSD17B5 under certain conditions, particularly in cases with severe hyperinsulinemia. Clearly, our genotyping results are preliminary and are exploratory in nature; therefore, our results should be interpreted with caution while awaiting replication.
Because this polymorphism is near the Sp1/Sp3 responsive element, which is essential for the regulation of HSD17B5 expression (15), it is possible that SNP-71G may affect the binding affinity of this responsive element and modulate the stimulation of testosterone production. In this regard, we found a higher reporter activity (70%) and higher binding affinity of nucleoproteins by the SNP-71G HSD17B5 promoter than that of the wild-type (A/A) promoter in rat thecal cells. This would be expected to result in higher levels of HSD17B5 mRNA and protein and s subsequent increase of testosterone biosynthesis. Notably, a similar SNP (SNP309, a G to T substitution) in the promoter of the MDM2 gene, an oncogene product that inhibits the ability of p53 to activate transcription, has been demonstrated to increase the binding affinity of the transcription factor Sp1, which results in a similar increase in promoter activity (60%), high levels of MDM2 RNA and protein, and attenuation of the p53 signaling; this SNP is associated with accelerated tumor formation in both hereditary and sporadic cancers (39). In addition, decreased repression of the CYP17 promoter by a nuclear transcription factor was reported to be a potential mechanism contributing to increased steroidogenesis in PCOS theca cells (40). Our experiments provide the further biological evidence that transcriptional dysregulation of steroidogenic genes may be implicated in PCOS, which could result in higher levels of HSD17B5 mRNA and protein and a subsequent increase of testosterone biosynthesis.
Sp1/Sp3 are ubiquitously expressed transcription factors that play a key role in maintaining basal transcription of many genes. The observation that insulin stimulates thecal cell production of androgens (3-5) supports that hypothesis that insulin stimulation plays a significant role in the etiology of hyperandrogenism in insulin-resistant women with PCOS. Furthermore, insulin and IGF-1 have been shown to regulate Sp1 and Sp3 expression levels as well as increase transcriptional binding affinity (41, 42 ). Several mechanisms exist by which Sp1 alters gene activity in response to insulin (41). These include the possibility that Sp1 acts alone in mediating the effects of insulin; Sp1 co-operatively interacts with other insulin-responsive transcription factors; and dissociation of Sp1 from an insulin-responsive promoter site, where it is necessary for basal activity, permits the actions of another factor or factors to modulate gene activity in response to insulin. Although the mechanisms underlining the association between hyperandrogenism and hyperinsulinemia are not entirely understand, our present studies provide a possible linkage between testosterone biosynthesis and insulin or IGF-1 effects by altering HSD17B5 expression through Sp1/Sp3. However, further studies are needed to demonstrate the up-regulation of insulin or IGF-1 on HSD17B5 expression through Sp1/Sp3.
In conclusion, we have identified a polymorphism in the HSD17B5 promoter that appears to be a novel genetic marker associated with a small subset (about 10%) of PCOS. Since we initially identified SNP-71G in a rare form of PCOS due to lupus erythematosis-related, autoantibody-mediated severe insulin resistance, it is possible that the SNP-71G effect in ordinary PCOS is enhanced by the presence of insulin resistance, which plays an important role in the pathogenesis of PCOS. To our knowledge, this is the first candidate gene polymorphism that may directly contribute to a phenotypic aspect of PCOS, namely, the plasma testosterone level. The present study supports the hypothesis that SNPs in the testosterone biosynthetic pathway can contribute to the genetic variation which underlies the phenotypic variation seen in an individual’s susceptibility to PCOS.
Acknowledgments
We thank Dr. Carole Ober for the control samples, Drs. Lawrence Layman and Nick Sauter for referring the patient, Dr. Xiefei Du for skillful technical assistance, and the Clinical Research Center staff at the University of Chicago RR-00055 CCRC).
Footnotes
This work was supported by NIH grants RO1-HD39267 (R.L.R. and K.Q.), U54-04185 (R.L.R.), K08-HD043279-01 (K.Q.), RO1-DK15070 (S.R.), DK205995 (R.L.R., D.A.E. and S.R.), RR00055 (R.L.R., D.A.E. and S.R.), and the Children’s Research Foundation (K.Q.).
References
- 1.Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocrin Rev. 1995;16:322–353. doi: 10.1210/edrv-16-3-322. [DOI] [PubMed] [Google Scholar]
- 2.Cara JF, Rosenfield RL. Insulin-like growth factor-I and insulin potentiate luteinizing hormone-induced androgen synthesis by rat ovarian theca-interstitial cells. Endocrinol. 1988;123:733–739. doi: 10.1210/endo-123-2-733. [DOI] [PubMed] [Google Scholar]
- 3.Cara JF, Fan J, Azzarello J, Rosenfield RL. Insulin-like growth factor-I enhances luteinizing hormone binding to rat ovarian theca-interstitial cells. J Clin Invest. 1990;86:560–565. doi: 10.1172/JCI114745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbieri RL, Smith S, Ryan KJ. The role of hyperinsulinemia in the pathogenesis of ovarian hyperandrogenism. Fertil Steril. 1988;50:197–212. doi: 10.1016/s0015-0282(16)60060-2. [DOI] [PubMed] [Google Scholar]
- 5.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83:2001–5. doi: 10.1210/jcem.83.6.4886. [DOI] [PubMed] [Google Scholar]
- 6.Rosenfield RL. Plasma testosterone binding globulin and indexes of the concentration of unbound androgens in normal and hirsute subjects. J Clin Endocrinol Metab. 1971;32:717–28. doi: 10.1210/jcem-32-6-717. [DOI] [PubMed] [Google Scholar]
- 7.Mendonca B, Arnhold I, Bloise W, Andersson S, Russell D, Wilson J. 17b-Hydroxysteroid dehydroghenase 3 deficiency in women. J Clin Endocrinol Metab. 1999;84:802–804. doi: 10.1210/jcem.84.2.5477. [DOI] [PubMed] [Google Scholar]
- 8.Qin K, Rosenfield RL. Expression of 17ß-hydroxysteroid dehydrogenase type 5 in human ovary. A pilot study. J Soc Gyn Invest. 2000;7:61–64. doi: 10.1016/s1071-5576(99)00067-2. [DOI] [PubMed] [Google Scholar]
- 9.Nelson VL, Qin K, Rosenfield RL, Wood JR, Penning TM, Legro RS, Strauss JF, 3rd, McAllister JM. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:5925–5933. doi: 10.1210/jcem.86.12.8088. [DOI] [PubMed] [Google Scholar]
- 10.Qin K, New MI, Cheng KC. Molecular cloning of multiple cDNAs encoding human enzymes structurally related to 3 alpha-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol. 1993;46:673–9. doi: 10.1016/0960-0760(93)90308-j. [DOI] [PubMed] [Google Scholar]
- 11.Khanna M, Qin K, Wang RW, Cheng KC. Substrate specificity, gene structure, and tissue-specific distribution of multiple human 3 alpha-hydroxysteroid dehydrogenases. J Biol Chem. 1995;270:20162–8. doi: 10.1074/jbc.270.34.20162. [DOI] [PubMed] [Google Scholar]
- 12.Dufort I, Rheault P, Huang XF, Soucy P, Luu-The V. Characteristics of a highly labile human type 5 17 beta-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:568–74. doi: 10.1210/endo.140.2.6531. [DOI] [PubMed] [Google Scholar]
- 13.Luu-The V. Analysis and characteristics of multiple types of human 17beta-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol. 2001;76:143–51. doi: 10.1016/s0960-0760(00)00155-2. [DOI] [PubMed] [Google Scholar]
- 14.Khanna M, Qin K, Klisak I, Belkin S, Sparkes RS, Cheng KC. Localization of multiple human dihydrodiol dehydrogenase (DDH1 and DDH2) and chlordecone reductase (CHDR) genes in chromosome 10 by the polymerase chain reaction and fluorescence in situ hybridization. Genomics. 1995;25:588–90. doi: 10.1016/0888-7543(95)80066-u. [DOI] [PubMed] [Google Scholar]
- 15.Qin K, Rosenfield RL. Characterization of the basal promoter element of the human type 5 17beta-hydroxysteroid dehydrogenase gene. Biochim Biophys Acta. 2005;1728:115–25. doi: 10.1016/j.bbaexp.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Rosenfield RL, Barnes RB, Ehrmann DA. Studies of the nature of 17-hydroxyprogesterone hyperresponsiveness to gonadotropin releasing hormone agonist challenge in functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1994;79:1686–1692. doi: 10.1210/jcem.79.6.7989476. [DOI] [PubMed] [Google Scholar]
- 17.Workshop REA-SPC. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Ehrmann DA, Rosenfield RL, Barnes RB, Brigell DF, Sheikh Z. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med. 1992;327:157–162. doi: 10.1056/NEJM199207163270304. [DOI] [PubMed] [Google Scholar]
- 19.Magoffin DA, Erickson GF. Primary culture of differentiating ovarian androgen-producing cells in defined medium. J Biol Chem. 1982;257:4507–13. [PubMed] [Google Scholar]
- 20.Legro RS, Strauss JF. Molecular progress in infertility: polycystic ovary syndrome. Fertil Steril. 2002;78:569–76. doi: 10.1016/s0015-0282(02)03275-2. [DOI] [PubMed] [Google Scholar]
- 21.Carey A, Waterworth D, Patel K, White D, Little J, Novelli P, Franks S, Williamson R. Polycystic ovaries and premature male pattern baldness are associated with one allele of the steroid metabolism gene CYP 17. Hum Molec Genet. 1994;3:1873–1876. doi: 10.1093/hmg/3.10.1873. [DOI] [PubMed] [Google Scholar]
- 22.Legro RS, Driscoll D, Strauss JF, 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A. 1998;95:14956–60. doi: 10.1073/pnas.95.25.14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferriman D, Purdie A. The inheritance of polycystic ovarian disease and a possible relationship to premature balding. Clin Endocrinol. 1979;11:291–300. doi: 10.1111/j.1365-2265.1979.tb03077.x. [DOI] [PubMed] [Google Scholar]
- 24.Hague W, Adams J, Reeders S, Peto T, Jacobs H. Familial polycystic ovaries: a genetic disease? Clin Endocrinol. 1988;29:593–605. doi: 10.1111/j.1365-2265.1988.tb03707.x. [DOI] [PubMed] [Google Scholar]
- 25.Gilling-Smith C, Willis DS, Beard RW, Franks S. Hypersecretion of androstenedione by isolated theca cells from polycystic ovaries. J Clin Endocrinol Metab. 1994;79:1158–1165. doi: 10.1210/jcem.79.4.7962289. [DOI] [PubMed] [Google Scholar]
- 26.Nelson VL, Legro RS, Strauss JF, 3rd, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946–57. doi: 10.1210/mend.13.6.0311. [DOI] [PubMed] [Google Scholar]
- 27.Casey ML, MacDonald PC, Andersson S. 17 beta-Hydroxysteroid dehydrogenase type 2: chromosomal assignment and progestin regulation of gene expression in human endometrium. J Clin Invest. 1994;94:2135–41. doi: 10.1172/JCI117569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waterworth DM, Bennett ST, Gharani N, McCarthy MI, Hague S, Batty S, Conway GS, White D, Todd JA, Franks S, Williamson R. Linkage and association of insulin gene VNTR regulatory polymorphism with polycystic ovary syndrome. [see comments] Lancet. 1997;349:986–90. doi: 10.1016/S0140-6736(96)08368-7. [DOI] [PubMed] [Google Scholar]
- 29.Urbanek M, Legro RS, Driscoll DA, Azziz R, Ehrmann DA, Norman RJ, Strauss JF, 3rd, Spielman RS, Dunaif A. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc Natl Acad Sci U S A. 1999;96:8573–8. doi: 10.1073/pnas.96.15.8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucci S, Futterweit W, Concepcion ES, Greenberg DA, Villanueva R, Davies TF, Tomer Y. Evidence for association of polycystic ovary syndrome in caucasian women with a marker at the insulin receptor gene locus. Journal of Clinical Endocrinology & Metabolism. 2001;86:446–9. doi: 10.1210/jcem.86.1.7274. [DOI] [PubMed] [Google Scholar]
- 31.Urbanek M, Du Y, Silander K, Collins FS, Steppan CM, Strauss JF, 3rd, Dunaif A, Spielman RS, Legro RS. Variation in resistin gene promoter not associated with polycystic ovary syndrome. Diabetes. 2003;52:214–7. doi: 10.2337/diabetes.52.1.214. [DOI] [PubMed] [Google Scholar]
- 32.Franks S, Gharani N, Waterworth D, Batty S, White D, Williamson R, McCarthy M. Genetics of polycystic ovary syndrome. Mol Cell Endocrinol. 1998;145:123–8. doi: 10.1016/s0303-7207(98)00178-6. [DOI] [PubMed] [Google Scholar]
- 33.Urbanek M, Wu X, Vickery KR, Kao LC, Christenson LK, Schneyer A, Legro RS, Driscoll DA, Strauss JF, 3rd, Dunaif A, Spielman RS. Allelic variants of the follistatin gene in polycystic ovary syndrome. J Clin Endocrinol Metab. 2000;85:4455–61. doi: 10.1210/jcem.85.12.7026. [DOI] [PubMed] [Google Scholar]
- 34.Liao WX, Roy AC, Ng SC. Preliminary investigation of follistatin gene mutations in women with polycystic ovary syndrome. Mol Hum Reprod. 2000;6:587–90. doi: 10.1093/molehr/6.7.587. [DOI] [PubMed] [Google Scholar]
- 35.Powell BL, Haddad L, Bennett A, Gharani N, Sovio U, Groves CJ, Rush K, Goh MJ, Conway GS, Ruokonen A, Martikainen H, Pouta A, Taponen S, Hartikainen AL, Halford S, Zeggini E, Jarvelin MR, Franks S, McCarthy MI. Analysis of multiple data sets reveals no association between the insulin gene variable number tandem repeat element and polycystic ovary syndrome or related traits. J Clin Endocrinol Metab. 2005;90:2988–93. doi: 10.1210/jc.2004-2485. [DOI] [PubMed] [Google Scholar]
- 36.Diamanti-Kandarakis E, Bartzis MI, Bergiele AT, Tsianateli TC, Kouli CR. Microsatellite polymorphism (tttta)(n) at -528 base pairs of gene CYP11alpha influences hyperandrogenemia in patients with polycystic ovary syndrome. Fertil Steril. 2000;73:735–41. doi: 10.1016/s0015-0282(99)00628-7. [DOI] [PubMed] [Google Scholar]
- 37.Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore) 2002;81:87–100. doi: 10.1097/00005792-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Ehrmann DA, Jeppe S, Byrne M, Karrison T, Rosenfield RL, Polonsky K. Insulin secretory defects in polycystic ovary syndrome. Relationship to insulin sensitivity and family history of non-insulin-dependent diabetes mellitus. J Clin Invest. 1995;96:520–527. doi: 10.1172/JCI118064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, Strong LC, Lozano G, Levine AJ. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 40.Wickenheisser JK, Nelson-DeGrave VL, Quinn PG, McAllister JM. Increased cytochrome P450 17alpha-hydroxylase promoter function in theca cells isolated from patients with polycystic ovary syndrome involves nuclear factor-1. Mol Endocrinol. 2004;18:588–605. doi: 10.1210/me.2003-0090. [DOI] [PubMed] [Google Scholar]
- 41.Samson SL, Wong NC. Role of Sp1 in insulin regulation of gene expression. J Mol Endocrinol. 2002;29:265–79. doi: 10.1677/jme.0.0290265. [DOI] [PubMed] [Google Scholar]
- 42.Li T, Chen YH, Liu TJ, Jia J, Hampson S, Shan YX, Kibler D, Wang PH. Using DNA microarray to identify Sp1 as a transcriptional regulatory element of insulin-like growth factor 1 in cardiac muscle cells. Circ Res. 2003;93:1202–9. doi: 10.1161/01.RES.0000104085.76261.02. [DOI] [PubMed] [Google Scholar]
- 43.Rosenfield RL, Perovic N, Ehrmann DA, Barnes RB. Acute hormonal responses to the gonadotropin releasing hormone agonist leuprolide: dose-response studies and comparison to nafarelin. J Clin Endocrinol Metab. 1996;81:3408–3411. doi: 10.1210/jcem.81.9.8784105. [DOI] [PubMed] [Google Scholar]