

Abstract
Aldolases have potential as tools for the synthesis of stereochemically complex carbohydrates. Here, we show that directed evolution can be used to alter the stereochemical course of the reaction catalyzed by tagatose-1,6-bisphosphate aldolase. After three rounds of DNA shuffling and screening, the evolved aldolase showed an 80-fold improvement in kcat/Km toward the non-natural substrate fructose 1,6-bisphosphate, resulting in a 100-fold change in stereospecificity. 31P NMR spectroscopy was used to show that, in the synthetic direction, the evolved aldolase catalyzes the formation of carbon—carbon bonds with unnatural diastereoselectivity, where the >99:<1 preference for the formation of tagatose 1,6-bisphosphate was switched to a 4:1 preference for the diastereoisomer, fructose 1,6-bisphosphate. This demonstration is of considerable significance to synthetic chemists requiring efficient syntheses of complex stereoisomeric products, such as carbohydrate mimetics.
There is a growing demand in the chemical and biotechnology industries for reliable and efficient methods for the production of enantiomerically pure compounds. Increasingly, synthetic chemists are exploiting enzymes in the asymmetric and stereoselective synthesis of chiral building blocks (1). However, suitable enzymes, with the required substrate specificity and stereospecificity, are often not available from natural sources. Directed evolution can provide a solution to this problem, and molecular biologists have successfully improved the properties of biocatalysts by altering activity (2), substrate specificity (3–5), and stability (6–9).
There are two general ways in which the stereoisomeric product of an enzyme-catalyzed reaction may be modified by directed evolution (Fig. 1). One approach is to evolve an enzyme, which accepts an unnatural stereoisomer as a substrate (Fig. 1A). By altering the substrate specificity in this way, the stereoisomeric product obtained is inevitably different from that produced by the wild-type enzyme. This approach has been used to produce mutant enzymes with an altered preference for the enantiomeric starting material favored in reactions catalyzed by d-2-keto-3-deoxy-6-phosphogluconate aldolase (10), a lipase (11), and a hydantoinase (12).
Figure 1.

Strategies for the directed evolution of enzymes that catalyze the formation of stereoisomeric products. (A) Modifying substrate specificity by direction evolution. (B) Modifying the stereochemical course of a reaction by directed evolution.
A fundamentally different approach would be to alter the stereochemical course of a bond-forming step (Fig. 1B) (13). This would enable the preparation of different possible stereoisomeric products from the same starting materials. To date, this approach has been used with enzymes that catalyze P—O bond formation (14), reduction of carbonyl groups (15), and hydroxylation of alkanes (16). Here, we apply directed evolution to the challenging problem of altering the stereochemical course of a C—C bond-forming step. The success of this approach would be of profound significance to synthetic chemists requiring efficient complementary syntheses of stereoisomeric complex molecules that cannot be accessed by using naturally occurring enzymes.
Aldolases catalyze the stereochemically controlled synthesis of carbon—carbon bonds and their use in the synthesis of complex molecules is well established (1, 17). One of the best studied aldolases is fructose 1,6-bisphosphate (FBP) aldolase (EC 4.1.2.13), a ubiquitous glycolytic enzyme, which catalyzes the reversible condensation of dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P) into fructose 1,6-bisphosphate. Aldolases may be classified according to their structure and reaction mechanism (18): the class I aldolases are usually multimers of (α/β)8-barrel subunits that utilize an active-site lysine residue in Schiff base formation during catalysis (19), whereas the class II aldolases have an absolute requirement for Zn2+ (20, 21). The crystal structure of the class II FBP aldolase from Escherichia coli showed that this enzyme also belongs to the (α/β)8 family of enzymes (22, 23).
Despite our growing understanding of the catalytic mechanism of the class II FBP aldolase (24–26), details regarding stereochemical control remain unclear. After abstraction of the 1-pro S proton from DHAP by Glu-182 (26, 27), attack of the DHAP ene-diolate intermediate on the Si-face of G3P in the FBP aldolases generates FBP (Fig. 2B). Attack of the same intermediate on the Re-face of G3P would generate the diastereoisomer, tagatose 1,6-bisphosphate (TBP) (Fig. 2A). Hence, TBP and FBP are both synthesized from DHAP and G3P, and they differ only in the stereochemistry at C4 (Fig. 2). The configuration of the product synthesized is therefore directly created in the carbon—carbon bond-forming step.
Figure 2.

Stereochemistry of the reaction catalyzed by aldolases. (A) The mechanism of TBP aldolase. The DHAP ene-diolate is formed after abstraction of the 1-proS proton from DHAP and polarization by the catalytic zinc cation. Attack of the activated DHAP C1 from its Si face onto the G3P C1 Re face generates the 3S, 4S product tagatose 1,6-bisphosphate, and proton donation by H-B (Asp-82) (32) converts the C4 carbonyl to a hydroxyl group, completing TBP synthesis. (B) The mechanism of FBP aldolase. The DHAP ene-diolate is formed after abstraction of the 1-proS proton from DHAP (27) by Glu-182 (26) and polarization by the catalytic zinc. Attack of the activated DHAP C1 from its Si face onto the G3P C1 Si face and proton donation by H-B (Asp-109) (24) convert the C4 carbonyl to a hydroxyl group, completing the synthesis of the 3S, 4R product fructose 1,6-bisphosphate. FBP and TBP are epimeric at C4, and this position is marked with an asterisk. R = CH(OH)CH2OPO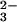 .
.
The stereochemical course of the reaction is controlled only by the relative free energy of the competing enzyme-bound transition states leading to FBP and TBP. We can therefore, for convenience, use the more readily assayed cleavage reactions during the evolution of an aldolase with reversed stereochemistry. The class II TBP aldolase from E. coli is highly specific for TBP in the cleavage direction and uses this substrate ≈300-fold better than its diastereoisomer FBP (Table 1). Sequence alignment and mutagenesis studies on the class II TBP aldolase, responsible for the reversible cleavage of TBP in E. coli, prompted an attempt to investigate and rationally alter the substrate specificity of FBP aldolase (28). Nine residues were highlighted as potentially impacting on substrate discrimination, and these residues were mutated into the corresponding residues in TBP aldolase. Surprisingly, despite abolishing FBP activity, none of the resulting mutant proteins produced an active TBP aldolase (28), suggesting that a more complex combination of substitutions is required to generate a change in the stereochemistry of carbon—carbon bond formation, highlighting the subtleties of enzyme redesign needed. Here, we report the successful use of directed evolution to generate an aldolase capable of catalyzing a carbon—carbon bond formation with reversed stereochemistry by the addition of a reactive intermediate to the opposite diastereotopic face of its aldehyde substrate.
Table 1.
Steady-state kinetic parameters for wild-type and mutant aldolases
| Enzyme | FBP cleavage
|
TBP cleavage
|
kcat/Km (FBP)/ kcat/Km (TBP) | ||||
|---|---|---|---|---|---|---|---|
| kcat, min−1 | Km, mM | kcat/Km, min−1⋅mM | kcat, min−1 | Km, mM | kcat/Km, min−1⋅mM | ||
| FBPA | 630 ± 4 | 0.17 ± 0.003 | 3,700 | 0.9 ± 0.08 | 0.35 ± 0.06 | 2.6 | 1,423 |
| TBPA (AgaY) | 4.1 ± 0.35 | 1.3 ± 0.32 | 3.2 | 280 ± 8 | 0.26 ± 0.03 | 1,080 | 0.003 |
| 1-20F8 (D104G) | 3 ± 0.21 | 0.26 ± 0.05 | 12 | 29 ± 2.4 | 0.15 ± 0.03 | 193 | 0.06 |
| 1-20H6 (S106G) | 9.6 ± 0.45 | 0.37 ± 0.05 | 26 | 380 ± 8 | 0.05 ± 0.004 | 7,600 | 0.0034 |
| 1-24H12 (H26Y) | 24 ± 0.96 | 0.27 ± 0.03 | 89 | 38 ± 3.4 | 0.018 ± 0.005 | 2,533 | 0.04 |
| 2-5C1 (H26Y/D104G/V121A) | 39 ± 0.98 | 0.16 ± 0.01 | 244 | 16 ± 0.36 | 0.013 ± 0.001 | 1,231 | 0.20 |
| 3-23B5 (H26Y/D104G/ V121A/P256L) | 36 ± 0.81 | 0.16 ± 0.01 | 230 | 13 ± 0.14 | 0.016 ± 0.001 | 800 | 0.3 |
The steady-state kinetic parameters of the wild-type and mutant enzymes for FBP and TBP cleavage were measured by using a coupled enzyme assay (29). Kinetic parameters (±standard error of the fit) were determined by fitting the data to the Michaelis–Menten equation by using the program kaleidagraph (Abelbeck Software, Reading, PA). FBPA, FBP aldolase; TBPA, TBP aldolase.
Methods
Bacterial Strains and Expression Vectors.
E. coli strain KM3 was as reported (29). Electrocompetent E. coli XL1 blue were from Stratagene. The expression vector pKagaY was as reported (28) and pKK223-3 was from Pharmacia.
Library Construction.
DNA shuffling was carried out according to the method of Stemmer (30) with slight modifications. Two oligonucleotides, 5′-GAG GAT GAA TTC ATG AGC ATT ATC TCC ACT-3′ and 5′-AAA AAC AAG CTT TTA TGC TGA AAT TCG ATT-3′, were used to amplify the agaY gene by using pKagaY (28) as template. The PCR reaction was carried out in a 50-μl reaction mixture consisting of 100 pmol each primer, 0.5 μg of plasmid, 0.2 mM each dNTP, and 2.5 units of Pfu DNA polymerase (Promega) in the reaction buffer provided by the manufacturer. Amplification involved an initial denaturation step at 95°C for 5 min followed by cycling at 95°C for 1 min, 55°C for 1 min, and 72°C for 2 min for 30 cycles, then a final extension for 2.5 min at 72°C. PCR product was purified by using the Wizard PCR Purification system (Promega). Purified agaY gene (≈1 μg) was incubated at 20°C for 20 min in the digestion buffer provided. Digestion was started by the addition of 0.1–1.0 units of DNase I and the reaction incubated at 20°C for 30 min. Reactions were terminated by heat treatment at 80°C for 15 min. Fragments of ≈50–150 bp in length were gel purified by using the QIAex II gel extraction kit (Qiagen, Chatsworth, CA). Purified fragments were submitted to PCR by using 2.5 units of TaqDNA polymerase, containing 1.5 mM MgCl2 and 0.2 mM each dNTP. The PCR cycle consisted of an initial denaturation step at 95°C for 5 min followed by cycling at 95°C for 1 min, 50°C for 1 min, and 72°C for 1 min for 30 cycles, then a final extension for 2.5 min at 72°C. Aliquots of reassembled agaY genes were used as the template for a final PCR amplification reaction, under conditions identical to the first PCR reaction used to amplify the wild-type agaY gene. The PCR product was gel purified and ligated into pKK223-3 by using the EcoRI and HindIII restriction sites. Ligation mixtures were transformed into electrocompetent E. coli XL1 blue.
Library Screening for FBP Cleavage.
Individual colonies were used to inoculate wells of a 96-well plate containing 150 μl of 2TY medium supplemented with 50 μg/ml ampicillin and 0.3 mM ZnCl2. After cell growth at 37°C for 18 h with shaking at 250 rpm, 100 μl of each culture was transferred to a 96-V-well plate, centrifuged at 1,000 × g and 4°C for 20 min, and the supernatants removed. The original plates were stored. Each cell pellet was resuspended in 100 μl of 50 mM Tris⋅HCl (pH 8.0) containing 1 mg/ml lysozyme, 0.1% (vol/vol) Triton X-100, and 0.1 M potassium acetate. The plates were frozen and thawed at room temperature. Cell debris was collected by centrifugation at 1,000 × g and 4°C for 20 min and 50 μl of each cleared supernatant transferred to a new 96-well plate. An equal volume of 50 mM Tris⋅HCl (pH 8.0) containing 0.1 M potassium acetate, 0.5 mg/ml NAD+, 0.3 mg/ml nitroblue tetrazolium, 0.02 mg/ml phenazine methosulphate, 1.5 mg/ml sodium arsenate, 20 μg/ml glyceraldehyde 3-phosphate dehydrogenase (Boehringer Mannheim), and varying concentrations of fructose 1,6-bisphosphate (typically 0.25–5 mM final concentration) was added. Reactions were monitored visually over 30–60 min. Those clones showing positive reactions were grown in 3 ml of 2TY medium supplemented with 50 μg/ml ampicillin and 0.3 mM ZnCl2, by using the original cultures as inoculum.
Quantitative Screening of Cell Lysates.
Lysates from 3 ml of overnight cultures were prepared by using a scaled-up version of the above protocol. Aliquots (typically 50 μl) were assayed for cleavage of fructose 1,6-bisphosphate and tagatose 1,6-bisphosphate by using a standard coupled assay (28) and the data normalized to the optical density at 600 nm of each culture.
Purification of the Class II TBP Aldolase.
The wild-type and mutant enzymes were expressed and purified to homogeneity as described (28). Protein concentration was determined by the bicinchoninic acid method by using BSA as standard.
Determination of Product Specificity by 31P and 1H NMR Spectroscopy.
Triose phosphate isomerase (50 units) was used to generate G3P in situ from DHAP (initial concentration 15 mM) in 50 mM Tris⋅HCl (pH 8.0). The time course of the reaction was followed in an experiment containing 100 μg of purified mutant aldolase 3-23B5 with 31P-NMR spectra recorded as a function of time on a Bruker (Billerica, MA) DRX500 instrument. Control experiments with the wild-type FBP and TBP aldolases contained 10 μg of enzyme in otherwise identical experiments. Under these conditions, the switch in the stereochemical course of the catalyzed reaction could be distinguished from thermodynamic factors, because the production of FBP and TBP was observed at low conversions before equilibration of the products was significant.
All chemical shifts were measured in parts per million relative to external 85% phosphoric acid.
Results and Discussion
Evolution of FBP Aldolase Activity on the TBP Aldolase Framework.
Crystallographic studies of the class II FBP and TBP aldolases have revealed details of interactions between the ene-diolate transition state analogue, phosphoglycolohydroxamate, and the enzymes (31, 32). To date, however, details of the interactions between glyceraldehyde 3-phosphate and the enzymes are lacking, and the mechanism by which they control the stereochemistry of the bond-forming steps remains unclear. Thus, initial efforts to alter this enzyme chemistry involved random mutation of the entire enzyme. Random mutations were introduced throughout the wild-type TBP aldolase (agaY) gene (28) by DNA shuffling essentially according to the method of Stemmer (30) (see Methods) to create a library of agaY variants with, on average, a single amino acid change per gene product. After cloning of the mutant agaY library, transformants were screened for the ability to cleave FBP. The protein library was prepared by lysis of resuspended cell pellets collected from cultures of individual colonies grown in 96-well microtiter plates. The cleavage of FBP into DHAP and G3P was coupled via a NAD+-dependent dehydrogenase to further redox reactions involving phenazine methosulphate and nitroblue tetrazolium. On reduction, by the generated NADH, nitroblue tetrazolium forms a purple formazan dye that can be detected visually. Potential positives identified by using this semiquantitative screen were then regrown from single colonies and assayed with FBP and TBP by using standard enzyme-coupled assays (28, 29) to determine specificity. Plasmids were prepared from confirmed positives and used to transform the fda-deficient strain E. coli KM3 (29) and lysates assayed again.
After screening of ≈3,000 first-generation variants with 1 mM FBP, three mutants, designated 1-20F8, 1-20H6, and 1-24H12, were confirmed to exhibit higher activity toward FBP than the wild-type TBP aldolase (Fig. 3). Two of the three variants also showed a marked decrease in activity toward TBP. The genes encoding these three variants were pooled and again subjected to DNA shuffling to produce the second-generation library. To enhance the stringency of the screen, we assayed this library at 4-fold lower substrate concentration (Fig. 3). Approximately 800 generation two variants were screened in this manner. Despite the lower substrate concentration used, one variant, designated 2-5C1, was found to have further dramatically improved specificity toward FBP as compared with the first-generation variants. This single variant was subjected to DNA shuffling to generate the third-generation library. A total of 3,600 variants of the third-generation library were screened for FBP cleavage with 0.06 mM FBP as substrate, and one variant (3-23B5) was identified with a further increase in activity toward FBP.
Figure 3.

Aldolase activities of clones selected during screening. Mutant TBP aldolases were expressed in E. coli KM3 and cleared cell lysates were prepared. Lysate samples were assayed for FBP (black bars) and TBP aldolase activity (gray bars) by using 1 mM substrate for the first-generation and site-directed variants (D104G/S106G and H26Y/D104G/S106G), 0.25 mM substrate for 2-5C1, and 0.06 mM substrate for 3-23B5. Activities were normalized to the OD600 of each culture. Data shown are the average of three independent cultures.
DNA Sequences of Evolved Aldolases.
The complete DNA sequences of each of the first-generation variants were determined. Each variant possessed a single amino acid mutation: D104G, S106G, and H26Y in 1-20F8, 1-20H6, and 1-24H12, respectively. The second-generation variant 2–5C1 possessed two of the first-generation mutations in combination (H26Y + D104G), along with an additional mutation of V121A. A double H26Y/D104G mutant constructed by site-directed mutagenesis showed identical activity to the 2-5C1 variant, demonstrating that the V121A mutation was neither beneficial for the switch in stereochemistry nor detrimental to the enzyme activity (data not shown). The third-generation variant, 3-23B5, possesses an additional nucleotide change resulting in a fourth amino acid mutation, P256L. Nowhere in our screening procedures was the double D104G/S106G mutant or the triple H26Y/D104G/S106G mutant found. Asp-104 and Ser-106 may be too close in the TBP aldolase sequence for efficient recombination to occur, although recombination between such closely separated mutations has been previously observed during DNA shuffling (33). Thus, the double and triple mutants may not have been selected by our screening system, because these combinations of mutations were not fairly represented in the second-generation library. To test this, the TBP aldolase mutants D104G/S106G and H26Y/D104G/S106G were constructed by site-directed mutagenesis and were expressed in E. coli KM3. Assays of lysates prepared from these variants indicated that the mutants did not show improved specificity toward FBP in comparison to any of the variants selected so far (Fig. 3), indicating that these mutants were not selected because of their poor fitness. In addition, saturation mutagenesis of the wild-type enzyme was conducted at positions 26, 104, 106, and 256 (data not shown). However, subsequent screening failed to identify any further improved variants.
Characterization of Evolved Aldolases.
To understand the mechanism underlying the switch in activity observed during the course of evolution, the variants 1-20F8, 1-20H6, 1-24H12, 2-5C1, and 3-23B5 were purified from the FBP aldolase-deficient strain of E. coli, KM3, and characterized by steady-state kinetics with either FBP or TBP as substrate as described (28) (Table 1). All first-generation mutants selected have significantly improved catalytic efficiency toward the non-native substrate, FBP, when compared with the wild-type TBP aldolase. The H26Y (1-24H12) mutant shows almost a 30-fold improvement in catalytic efficiency (as judged by the ratio of kcat/Km) with FBP as compared with the wild-type TBP aldolase. At the same time, this mutant showed a 7-fold decrease in kcat with the natural substrate, TBP, but an unexpected 14-fold decrease in Km, resulting in an enzyme that was 2-fold more efficient with TBP as substrate compared with the wild-type enzyme. This single mutation therefore appears to result in general decreases in the Km measured for either substrate; nevertheless, the increase in kcat with FBP, combined with the decrease in kcat for TBP, results in an enzyme with a switch in the stereochemistry of the reaction toward that of FBP aldolase of 12-fold. Similar results were obtained for the other two first-round variants, both of which showed significant but less dramatic switches in reaction stereochemistry. The kinetic parameters for these mutants are shown in Table 1.
The second-generation mutant H26Y/D104G/V121A (2-5C1) clearly contains an important combination of mutations. This mutant shows an 80-fold improvement in catalytic efficiency with FBP, resulting from a 10-fold increase in kcat and an 8-fold decrease in Km with FBP, compared with the wild-type TBP aldolase. This variant also shows a further decrease in kcat with TBP (18-fold lower compared with wild-type), but again the Km measured for TBP was 20-fold lower that the wild-type TBP aldolase.
The third-generation mutant H26Y/D104G/V121A/P256L (3–23B5) shows little change in activity with respect to kcat and Km with FBP, compared with its second-generation parent 2-5C1. However, the introduction of the P256L mutation leads to a large decrease in kcat for TBP (22-fold compared with the wild-type TBP aldolase) and an apparent Km 16-fold lower than that of the wild-type enzyme, resulting in an enzyme that is less efficient with TBP than the wild-type TBP aldolase.
The wild-type TBP aldolase has a high specificity for TBP over FBP {[(kcat/Km)TBP/(kcat/Km)FBP] = 340}. In contrast, the third-generation evolved aldolase (3-23B5) is less specific toward TBP {[(kcat/Km) TBP/(kcat/Km)FBP] = 3.5}, resulting in a 100-fold change in stereospecificity. It is interesting to note that the Km for TBP decreased in parallel with that of FBP and, for this reason, the specificity constant of the evolved aldolase was not completely inverted in favor of FBP. For the mutants analyzed here, therefore, it appears that the stepwise generation of a switch in reaction stereochemistry is occurring via an aldolase with generally broader stereospecificity. Nonetheless, this process results in an evolved enzyme that is highly optimized toward the turnover of the new substrate, FBP, with a 220-fold switch in kcat toward FBP cleavage. Given that this results from a change in the stereochemical course of the reaction, this represents a dramatic alteration of the enzyme function.
Synthetic Utility of the Evolved Aldolase.
Because the evolved third-generation variant 3-23B5 showed a 4-fold higher kcat for the cleavage of FBP relative to TBP, we anticipated that FBP would be the major product of the condensation reaction catalyzed by this mutant. To assess the synthetic potential of the evolved aldolase, the enzymic condensation of DHAP and G3P was monitored in real-time by 31P NMR spectroscopy (Fig. 4). Because the two phosphate groups in FBP and TBP resonate at distinct chemical shifts, the stereochemistry of the bond-forming event can be directly monitored. Under the conditions used, the reaction is under kinetic control, so the experiment directly probes the ability of the evolved aldolase to catalyze the formation of carbon—carbon bonds with different stereochemistry. The formation of TBP by the wild-type TBP aldolase is shown in Fig. 4A. The results demonstrate the high specificity of the enzyme in generating the single-product TBP, where the new carbon—carbon bond has 3S, 4S stereochemistry. By contrast, when incubated with the same substrates under identical conditions (Fig. 4B), the evolved aldolase (3-23B5) produces substantial quantities of FBP and only small amounts of TBP (in ≈4:1 ratio), confirming that the stereochemistry of the enzyme reaction has been altered such that the new bond generated has the 3S, 4R stereochemistry. The evolved enzyme must therefore preferentially catalyze the attack of the DHAP enediolate intermediate on the Si-face of G3P, generating a new carbon—carbon bond with 3S, 4R stereochemistry. The demonstration that directed evolution may be used to overturn a >99:<1 preference for the formation of one diastereoisomer to a 4:1 preference for another diastereoisomer is remarkable. Indeed, the level of stereocontrol exhibited by the mutant enzyme is synthetically useful and demonstrates that enzymes may be evolved to enable the synthesis of different stereoisomeric products from the same starting materials. This example demonstrates that directed evolution is a viable and exciting addition to the armory of the synthetic chemist.
Figure 4.

31P-NMR spectroscopy time course of the evolved aldolase catalyzed condensation of DHAP and G3P. (A) Wild-type AgaY TBP aldolase. (B) The evolved variant, 3-23B5. 31P resonances in the spectra are labeled by color: DHAP, blue; inorganic phosphate, red; FBP, yellow; TBP, magenta.
Implications for Stereochemical Control.
We have shown that the introduction and recombination of single amino acid changes by directed evolution can give rise to a class II aldolase that catalyzes an aldol reaction with an altered stereochemistry of reaction. The recently solved crystal structure of the TBP aldolase (32) allows us to locate the positions of the amino acid mutations (His-26, Asp-104, Ser-106, and Pro-256) in the evolved aldolase (Fig. 5). His-26 lies in a short loop that connects β1 and α2 and is close to Arg-257, which likely forms part of the C6-phosphate-binding site (32). It is likely, therefore, that the H26Y mutation subtly affects the position of bound G3P in the evolved aldolase. Both Asp-104 and Ser-106 are completely conserved in the FBP and TBP aldolases and lie close to the catalytic zinc-binding site. Mutations in this area may slightly reorientate the enediolate plane with respect to the incoming carbonyl of G3P. The mutation P256L is located at the N terminus of helix α10 and, as such, is located in the active site of the adjacent subunit. Mutation of this proline may alter the conformation of Arg-257 and the α9–α10 loop, which is implicated in substrate binding. Thus, these mutations may alter the substrate-binding pocket sufficiently to allow rotation of the G3P plane relative to the DHAP enediolate to permit attack by the opposite face of the incoming G3P molecule.
Figure 5.

The active site of the wild-type class II TBP aldolase (AgaY). The locations of a number of active site residues and the positions of the residues mutated during the evolution of the new enzyme are shown. Asp-104 and Ser-106 form a hydrogen bond network with His-83 (one of the ligands to the catalytic zinc cation) and through this to the substrate analogue, phosphoglycolohydroxamate (cyan), and Asp-82, the residue responsible for protonation at C4 (32). Changes in this network are likely to produce subtle changes in the orientation of the two substrates, leading to altered stereochemistry of bond formation. His-26 lies between the active site and the α-10 to α-11 loop of the adjacent subunit (residues 255–258 of this loop are shown in green). This loop contains Pro-256 and contributes Arg-257, responsible for binding the C6 phosphate of the substrate. The changes found during evolution of altered stereochemistry (P256L and H26Y) are likely to cause subtle repositioning of Arg-257 and hence the G3P end of the substrate to result in the stereochemical changes observed. The coordinates are taken from Protein Data Bank (PDB ID code 1GVF) (32) and the figure was made by using MOLSCRIPT (34) and RASTER3D (35).
None of the residues identified from this directed evolution experiment is identical to those targeted in our previous alignment and structure guided study (28). Moreover, these positions are either completely conserved (in the case of D104, S106, and P256) or semiconserved (in the case of H26) across all members of the FBP and TBP aldolases. The work has thus shown that directed evolution can be used to invert the stereochemistry of a new stereogenic center formed in an enzyme-catalyzed reaction. A consequence of this demonstration is that complementary biocatalysts can be generated that catalyze the conversion of the same starting materials into stereoisomeric (diastereo- or enantiomeric) products. This finding is of profound significance to synthetic chemists requiring efficient and complementary syntheses of complex targets, which are stereoisomers of the products of the reactions catalyzed by wild-type enzymes. Moreover, it shows that directed evolution is able to produce solutions to complex molecular problems distinct from those found in nature.
Acknowledgments
We thank Prof. W. Fessner (Technische Universität, Darmstadt, Germany) for the gift of tagatose bisphosphate. We are grateful to Dr. Bill Hunter (Dundee) for access to the TBP aldolase PDB file before publication; Simon Barrett for assistance with the NMR experiments; Stuart Warriner for help with the preparation of the figures; and David Harrison, Mike McPherson, and Sheena Radford for useful discussions. This work was supported by the Biotechnology and Biological Sciences Research Council, the Engineering and Physical Sciences Research Council, and the Wellcome Trust and is a contribution from the Astbury Centre for Structural Molecular Biology.
Abbreviations
- FBP
fructose 1,6-bisphosphate
- TBP
tagatose 1,6-bisphosphate
- DHAP
dihydroxyacetone phosphate
- G3P
glyceraldehyde 3-phosphate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wong C-H, Whitesides G M. Enzymes in Synthetic Organic Chemistry. Oxford: Pergamon; 1994. [Google Scholar]
- 2.Buchholz F, Angrand P O, Stewart A F. Nat Biotechnol. 1998;16:657–662. doi: 10.1038/nbt0798-657. [DOI] [PubMed] [Google Scholar]
- 3.Kumamaru T, Suenaga H, Mitsuoka M, Watanabe T, Furukawa K. Nat Biotechnol. 1998;16:663–666. doi: 10.1038/nbt0798-663. [DOI] [PubMed] [Google Scholar]
- 4.Crameri A, Raillard S A, Bermudez E, Stemmer W P C. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- 5.Matsumura I, Ellington A D. J Mol Biol. 2001;305:331–339. doi: 10.1006/jmbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Arnold F H. Protein Eng. 1999;12:47–53. doi: 10.1093/protein/12.1.47. [DOI] [PubMed] [Google Scholar]
- 7.Giver L, Gershenson A, Freskgard P O, Arnold F H. Proc Natl Acad Sci USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akanuma S, Yamagishi A, Tanaka N, Oshima T. Protein Sci. 1998;7:698–705. doi: 10.1002/pro.5560070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherry J R, Lamsa M H, Schneider P, Vind J, Svendsen A, Jones A, Pedersen A H. Nat Biotechnol. 1999;17:379–384. doi: 10.1038/7939. [DOI] [PubMed] [Google Scholar]
- 10.Fong S, Machajewski T D, Mak C C, Wong C. Chem Biol. 2000;7:873–883. doi: 10.1016/s1074-5521(00)00035-1. [DOI] [PubMed] [Google Scholar]
- 11.Liebeton K, Zonta A, Schimossek K, Nardini M, Lang D, Dijkstra B W, Reetz M T, Jaeger K E. Chem Biol. 2000;7:709–718. doi: 10.1016/s1074-5521(00)00015-6. [DOI] [PubMed] [Google Scholar]
- 12.May O, Nguyen P T, Arnold F H. Nat Biotechnol. 2000;18:317–320. doi: 10.1038/73773. [DOI] [PubMed] [Google Scholar]
- 13.Jaeger K E, Reetz M T. Curr Opin Chem Biol. 2000;4:68–73. doi: 10.1016/s1367-5931(99)00054-x. [DOI] [PubMed] [Google Scholar]
- 14.Jiang R T, Dahnke T, Tsai M D. J Am Chem Soc. 1991;113:5485–5486. [Google Scholar]
- 15.Nakajima K, Kato H, Oda J, Yamada Y, Hashimoto T. J Biol Chem. 1999;274:16563–16568. doi: 10.1074/jbc.274.23.16563. [DOI] [PubMed] [Google Scholar]
- 16.van Den Heuvel R H, Fraaije M W, Ferrer M, Mattevi A, van Berkel W J. Proc Natl Acad Sci USA. 2000;97:9455–9460. doi: 10.1073/pnas.160175897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fessner W. Curr Opin Chem Biol. 1998;2:85–97. doi: 10.1016/s1367-5931(98)80040-9. [DOI] [PubMed] [Google Scholar]
- 18.Rutter W J. Fed Proc. 1964;23:1248–1257. [PubMed] [Google Scholar]
- 19.Horecker B L, Tsolas O, Lai C Y. In: The Enzymes. 3rd Ed. Boyer P D, editor. Vol. 7. New York: Academic; 1972. pp. 213–258. [Google Scholar]
- 20.Harris C E, Kobes R D, Teller D C, Rutter W J. Biochemistry. 1969;8:2442–2454. doi: 10.1021/bi00834a029. [DOI] [PubMed] [Google Scholar]
- 21.Kobes R D, Simpson R T, Vallee B L, Rutter W J. Biochemistry. 1969;8:585–588. doi: 10.1021/bi00830a018. [DOI] [PubMed] [Google Scholar]
- 22.Blom N, Tetreault S, Coulombe R, Sygusch J. Nat Struct Biol. 1996;3:856–862. doi: 10.1038/nsb1096-856. [DOI] [PubMed] [Google Scholar]
- 23.Cooper S J, Leonard G A, McSweeney S M, Thompson A W, Naismith J H, Qamar S, Plater A, Berry A, Hunter W N. Structure (Cambridge, UK) 1996;4:1303–1315. doi: 10.1016/s0969-2126(96)00138-4. [DOI] [PubMed] [Google Scholar]
- 24.Plater A R, Zgiby S M, Thomson G J, Qamar S, Wharton C W, Berry A. J Mol Biol. 1999;285:843–855. doi: 10.1006/jmbi.1998.2376. [DOI] [PubMed] [Google Scholar]
- 25.Qamar S, Marsh K, Berry A. Protein Sci. 1996;5:154–161. doi: 10.1002/pro.5560050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zgiby S, Plater A R, Bates M A, Thomson G J, Berry A. J Mol Biol. 2002;315:147–156. doi: 10.1006/jmbi.2001.5237. [DOI] [PubMed] [Google Scholar]
- 27.Rose I A. J Am Chem Soc. 1958;80:5835–5836. [Google Scholar]
- 28.Zgiby S M, Thomson G J, Qamar S, Berry A. Eur J Biochem. 2000;267:1858–1868. doi: 10.1046/j.1432-1327.2000.01191.x. [DOI] [PubMed] [Google Scholar]
- 29.Berry A, Marshall K E. FEBS Lett. 1993;318:11–16. doi: 10.1016/0014-5793(93)81317-s. [DOI] [PubMed] [Google Scholar]
- 30.Stemmer W P C. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 31.Hall D R, Leonard G A, Reed C D, Watt C I, Berry A, Hunter W N. J Mol Biol. 1999;287:383–394. doi: 10.1006/jmbi.1999.2609. [DOI] [PubMed] [Google Scholar]
- 32.Hall D R, Bond C S, Leonard G A, Watt C I, Berry A, Hunter W N. J Biol Chem. 2002;277:22018–22024. doi: 10.1074/jbc.M202464200. [DOI] [PubMed] [Google Scholar]
- 33.Raillard S, Krebber A, Chen Y, Ness J E, Bermudez E, Trinidad R, Fullem R, Davis C, Welch M, Seffernick J, et al. Chem Biol. 2001;8:891–898. doi: 10.1016/s1074-5521(01)00061-8. [DOI] [PubMed] [Google Scholar]
- 34.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 35.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]