

Abstract
We propose motif regressor for discovering sequence motifs upstream of genes that undergo expression changes in a given condition. The method combines the advantages of matrix-based motif finding and oligomer motif-expression regression analysis, resulting in high sensitivity and specificity. motif regressor is particularly effective in discovering expression-mediating motifs of medium to long width with multiple degenerate positions. When applied to Saccharomyces cerevisiae, motif regressor identified the ROX1 and YAP1 motifs from Rox1p and Yap1p overexpression experiments, respectively; predicted that Gcn4p may have increased activity in YAP1 deletion mutants; reported a group of motifs (including GCN4, PHO4, MET4, STRE, USR1, RAP1, M3A, and M3B) that may mediate the transcriptional response to amino acid starvation; and found all of the known cell-cycle regulation motifs from 18 expression microarrays over two cell cycles.
Keywords: sequence motif discovery‖microarray data‖correlation‖ transcription regulation
Direct experimental determination of transcription factor DNA-binding motifs (TFBM) is not practical or efficient in many biological systems. Therefore, computational algorithms such as the word-enumeration (1–4), the position-specific matrix update (5–7), and the dictionary (8) methods have been developed to identify putative motifs and guide experimentation. One of the most successful computational tactics for TFBM discovery is to cluster genes based on their expression profiles, and then search for motifs in the sequences upstream of tightly clustered genes (9). When noise is introduced into the cluster through spurious correlations, however, such an approach may result in false positives. A filtering method (10) based on the specificity of the motif occurrences has been shown to effectively eliminate false positives, but the sensitivity of the algorithm is still low in some cases. An iterative procedure for simultaneous clustering and motif finding has been suggested (11), but no effective algorithm has been implemented to demonstrate its advantage in biological data. Two novel methods for TFBM discovery via the association of gene expression values with oligomer motif abundances have been proposed (12, 13). They first conduct word enumeration and then use regression to check whether the genes whose upstream sequences contain a set of words have significant changes in their expression. These methods are effective for discovering conserved short motifs and sometimes interactions among them, but are not effective with longer motifs and may lose sensitivity in cases where TFBMs have multiple degenerate positions.‖
We present an alternative approach operating under the explicit assumption that, in response to a given biological condition, the effect of a TFBM is strongest among genes with the most dramatic increase or decrease in mRNA expression. We first use a fast and sensitive motif-finding method, mdscan (14), to generate a large set of motif candidates that are enriched in the DNA sequence upstream of genes with the highest fold change in mRNA level relative to a control condition. Then we verify each candidate motif by associating every gene's upstream sequence motif-matching score with its downstream expression measure (Fig. 1). In the motif-matching score, both the number of sites and the strength of matching are incorporated by using a third-order Markov background model and a position-specific motif matrix, thereby increasing both the sensitivity of the method and the specificity of the reported motifs.
Figure 1.

motif regressor strategy diagram.
Methods
Microarray and Sequence Data.
Saccharomyces cerevisiae microarray experiments in our study include Rox1p (15) and Yap1p (16) overexpression, YAP1 deletion (17), amino acid starvation response (16), α-factor synchronized cell-cycle progression (18), and chromatin immunoprecipitation (ChIP)-array experiments on Gcn4p (19), Rpd3p, Ume1p, and Ume6p (20). Yeast ORFs were ranked according to their relative change in mRNA level under each condition. We extracted up to 800 bp upstream of each ORF that was nonoverlapping with the adjacent ORF. Single repeats (e.g., AAAA…) of >10 bases and double repeats (e.g., ACACAC…) of >16 bases were removed.
Motif Discovery by mdscan.
Using every occurring w-mer (5 ≤ w ≤ 15 in our studies) in the top k (default as 100) sequences as a seed, mdscan (14) finds all w-mers in the top sequences that are similar to the seed and constructs from them a motif matrix. A pair of w-mers is said to be “similar” if they share at least m matched positions, where the probability for two random w-mers to share ≥m matched positions is ɛ (default as 0.0015). All of the motif matrices are evaluated by a semi-Bayesian scoring function, and the 50 highest-scoring motifs are saved. Each retained motif matrix is refined by adding or removing w-mers in the top K (default as 500, this includes the original k) sequences to increase the motif score. Motifs with average frequency of the consensus bases <0.7 are eliminated. If several motifs of the same width share similar consensus, the one with the highest score is retained. Here, two consensuses are said to be “similar” if they share ≥m matched positions, allowing frame shifts and considering both forward and reverse-complements. mdscan reports up to 30 distinct motifs.
Sequence Motif-Matching Scoring.
We determine how well the upstream sequence of a gene g matches a motif m, in terms of both degree of matching and number of sites, by the following function:
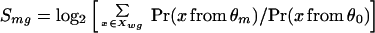 |
1 |
where θm is the probability matrix of motif m of width w, θ0 is the third-order Markov model estimated from all of the yeast intergenic sequences, and Xwg is the set of all w-mers in the upstream sequence of gene g. Using a motif matrix and a background with Markov dependency greatly improves the sensitivity and specificity of the scoring function (14).
Linear Regression.
For each motif reported by mdscan, we first fit the simple linear regression:
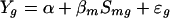 |
2 |
where Yg is the log2-expression value of gene g, Smg is defined in (1), and ɛg is the gene-specific error term. The baseline expression α and the regression coefficient βm will be estimated from the data. A significantly positive or negative βm indicates that upstream sequences containing motif m are correlated with enhanced or inhibited downstream gene expression, respectively.
The candidate motifs with a significant P value (P ≤ 0.01) for the simple linear regression coefficient βm are retained and used by the stepwise regression procedure to fit a multiple regression model:
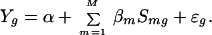 |
3 |
Stepwise regression begins with Model (3) with only the intercept term, and adds at each step the motif that gives the largest reduction in residual error. After adding each new motif mi, the model is checked to remove the ones whose effects have been sufficiently explained by mi. The final model is reached when no motif can be added with a statistically significant coefficient.
Results
The Discovery of Single Motifs That Influence Gene Expression.
To test the validity of motif regressor, we sought to identify the TFBMs of Rox1p and Yap1p by examining the upstream regions of genes with highest fold change in expression upon the overexpression of Rox1p (15) and Yap1p (16) (Table 1). Rox1p is a heme-induced TF that recognizes YYNATTGTTY (21) and represses genes normally expressed in hypoxic conditions. We used mdscan to search the upstream sequences of the 10, 25, 50, and 100 most repressed genes ranked by fold change, respectively, to generate up to 30 candidate motifs for each width from 5 to 15 bases. We used five times the number of input sequences for refinement. The correct motif, as defined by previous studies (21), was the top-ranked motif discovered for input sequence sizes 10, 25, and 50, but was not top-ranked when using 100 sequences. In contrast, the top-ranked motifs found by motif regressor for all input sequence sizes (10, 25, 50, 100) had consensuses that matched the known ROX1-binding consensus. For all input sequence sizes, at least 8 of the top 10 motifs had a motif consensus that matched perfectly with the known ROX1 consensus, all with very low (<10−9) regression P values (unadjusted for multiple testing). These results (Table 1) were then compared with those obtained by other motif-finding algorithms. We applied alignace (9) to search the upstream sequences of 10, 25, 50, and 100 most repressed genes ranked by fold change, respectively, specifying the correct motif width and using all other default parameters. We failed to find any motifs resembling the known ROX1-binding consensus for any input sequence size. Similarly, we ran meme (6) for input of 10, 25, 50, and 100 sequences, respectively, allowing motif width to vary between 5 and 15 and each sequence to contain 0-n repetitions of a motif. Again, meme did not report any motifs with consensus similar to the known ROX1-binding consensus.
Table 1.
Motifs discovered from Rox1p and Yap1p overexpression experiments
| Method | Rox1p overexpression | Yap1p overexpression |
|---|---|---|
| alignace | AAAAAAAAAAAAAAAAAAAAAAAA | AAGAAGAAAA |
| Best 10 motifs (10 input sequences) | AAAAAAAAAAAG | GAAGAGAAGAA |
| AAGGAAAAAAAGAAAAAAAAA | AAAGAAGAAA | |
| AAAAAAAAGAAAAGAAAAAAA | CATTTCTAATCT | |
| AAGGAAAAAAGAAA | GAAAAGCG | |
| AAGAAAAAAA | AAGAGGAG | |
| GCGCCCCGGA | AAAAAAAGAAG | |
| GAGCGCTCATGCCGCTGTTTT | GAAAAAAAAG | |
| AAAATAAAAAAAAAAAAA | AAAAAAAGAAA | |
| CTGCGGAAAA | AAAATGAAAAATG | |
| meme | TTTTTTTCTTTT | CATTACTAATCA |
| All motifs (10 input sequences) | TTCCGCGGA | AAAGAGGTG |
| mdscan | TTGTT (w = 5) | TTACT (w = 5) |
| Best motif at each width (10 input sequences) | TATTGT (w = 6) | GAAGAA (w = 6) |
| CTATTGT (w = 7) | TTACTAA (w = 7) | |
| CTATTGTT (w = 8) | TTACTAAT (w = 8) | |
| ATCTATTGT (w = 9) | GATTACTAA (w = 9) | |
| TATGTACGTA (w = 10) | GATTACTAAT (w = 10) | |
| GTACGTATGTA (w = 11) | GATTACTAATC (w = 11) | |
| TCTCTTGCCTTT (w = 12) | TCATTACTAAGC (w = 12) | |
| AGGACAAAAGGAA (w = 13) | GATTACTAATCAC (w = 13) | |
| TATATACATATATA (w = 14) | ATTATTAATCAAAT (w = 14) | |
| ATATATACGTATATA (w = 15) | GATTACTAATCACAT (w = 15) | |
| mdscan | GTTTG (w = 5) | AAGGG (w = 5) |
| Best motif at each width (100 input sequences) | CTATTG (w = 6) | CCGCGG (w = 6) |
| TATTGTT (w = 7) | AAGGGGA (w = 7) | |
| CTATTGTT (w = 8) | GTTACCCG (w = 8) | |
| GCGATGAGC (w = 9) | CACACACCA (w = 9) | |
| GCGATGAGCT (w = 10) | CTTACTAATCCA (w = 10) | |
| TGCGATGAGCT (w = 11) | ACACATATATATA (w = 11) | |
| ATGCGATGAGCT (w = 12) | CACACACACACACA (w = 12) | |
| GCGATGAGCTGAG (w = 13) | ATATATATATATATA (w = 13) | |
| ACACACACACACAC (w = 14) | CACACACACACACACAC (w = 14) | |
| CACACACACACACAC (w = 15) | TATATATATATATATATA (w = 15) | |
| motif regressor | TCTATTGTT (0) | ATGATTACTAATCA (5.58e-11) |
| Top 10 motifs (regression P value) (100 input sequences) | TTTCTATTGT (0) | TGCTTACTAATC (1.39e-10) |
| CTATTGTTTTC (0) | GATTACTAATC (3.30e-10) | |
| ACTTCTATTGT (0) | GTGATTACTAATC (3.26e-9) | |
| TTTCTATTGTTTT (0) | CTTACTAATC (1.48e-7) | |
| TTTCTATTGTTTTT (0) | TTACTAATC (4.23e-6) | |
| CTATTGTT (1.11e-16) | TCCTGCCCCTT (5.55e-6) | |
| ATTGT (1.20e-14) | TCCCATTAC (4.80e-5) | |
| GGTGGC (1.38e-11) | GCGCCCCTTAC (1.68e-4) | |
| TATTGTT (1.04e-10) | TCCCTCTCCCTT (1.92e-4) |
Motifs reported by alignace, meme, mdscan, and motif regressor for two microarray experiments. Both alignace and meme used only the top 10 genes, and incorporating more genes led to worse results. mdscan ranked the correct motifs as the best in several widths when the top 10 sequences were used, but failed when 100 sequences were used. motif regressor ranked the correct motifs as number 1 for all settings. The most difficult case, using the top 100 genes to find motifs and the top 500 genes to refine them, is reported. Boldface indicates agreement between a discovered motif and the known motif consensus.
Yap1p is a transcriptional activator required for oxidative stress tolerance, and is known to recognize the DNA sequence TTACTAA (22). Genes induced by Yap1p overexpression presented another interesting case for comparing motif-finding methods. alignace reported a motif ranked fourth with a consensus that differs from the reported YAP1 consensus (22) by one base for input sequence sizes 10 and 25, but did not find any motifs resembling the YAP1 motif for input sequence sizes 50 and 100. meme ranked the correct YAP1 motif first for input sequence size 10, and third for input sequence sizes 25 and 50, but failed to find the YAP1 motif for input sequence size 100 (Table 1). mdscan outperformed both alignace and meme, finding and ranking the correct motif first over a range of motif widths when using 10, 25, and 50 input sequences, respectively. With 100 input sequences, mdscan found and ranked the correct motif first at motif width w = 10. motif regressor also performed strongly, with at least the top 6 of the top 10 motifs containing motif consensuses that matched the known YAP1 consensus for all input sequence sizes.
We also analyzed the expression data from YAP1 deletion mutants (17), expecting to find the YAP1 motif among the sequences upstream of down-regulated genes. To our surprise, motif regressor found only one significant motif (P = 0.0075) partially matching the YAP1 motif, with a consensus CGTTACCCTCC. Using the sequences upstream of induced genes, however, motif regressor found many significant motifs matching the GCN4 consensus TGACTCA (21). It has been previously shown that Gcn4p can bind efficiently to YAP1 sites TTACTAA (22), so it is possible that the activity of Gcn4p is increased in YAP1 mutants to partially compensate for the lack of Yap1p. Increased Gcn4p activity would then lead to the induced expression of many Gcn4p targets, and our discovery of the GCN4 motif. Among the 135 Gcn4p intergenic targets identified by Gcn4p ChIP-array experiments (19), 95 showed increased downstream expression in YAP1 mutants (P < 10−6). Furthermore, among the 118 up-regulated genes with >1.5-fold change in YAP1 mutants, 20 were identified as Gcn4p target downstream genes by genome-wide ChIP assays (19) (P < 10−12).
The Discovery of Multiple Motifs That Influence Gene Expression.
motif regressor was applied to the sequences upstream of yeast genes whose expression changed after 30 min of amino acid starvation (16). mdscan found 414 motifs of width 5–15 from the most induced genes and most repressed genes, respectively. The simple linear regression step screened out 179 insignificant motifs (P > 0.01). The stepwise regression on the remaining 235 motifs yielded 25 that were significant. The resulting model had an R-square of 19.8%, implying that together the 25 motifs might account for 19.8% of the variation in genomic expression. Stepwise regression overestimates the true R-square due to its selective use of explanatory variables (www.stata.com/support/faqs/stat/stepwise.html). However, we performed a simulation study by using 6,000 genes and 400 independent motifs, which found the overestimation of R-square under these conditions to be small, ≈1%.
The 25 motifs can be classified into 15 different DNA patterns (Fig. 2). Eight of these patterns (STRE, GCN4, M3A, M3B, MET4, PHO4, RAP1, and URS1) were previously known, and together they have an R-square of 17.6%. The stress response element STRE and the GCN4 motif, which regulates amino acid biosynthesis, are known to positively regulate transcription during amino acid starvation. Two patterns involved in RNA processing (M3A and M3B; ref. 24) have been previously found in genes repressed under environmental stress (web supplement to ref. 16; http://genome-www.stanford.edu/yeast_stress/images/sfigure0.html). Met4p, with its auxiliary factors Cbf1p, Met28p, Met31p, and Met32p, regulates the transcriptional activation of genes involved in sulfur metabolism, especially the sulfur amino acid pathway (25). Pho4p, together with Pho2p, are the master transcription regulators of the PHO genes responsible for the scavenging and uptake of inorganic phosphate under phosphate-limiting conditions (26). Rap1p is the primary regulator of the yeast ribosomal protein genes (RPGs), and the RAP1 motif occurs in most of the RPG promoters (27). Rap1p is required for the heavy transcription of RPGs under favorable growth conditions, and for the repression of these genes under unfavorable conditions (28). Finally, the URS1 site is known to be present in the promoters of many yeast genes induced under stress conditions (29). From the eight discovered known motifs, we gain insight to the cell's amino acid starvation response: slowing cell growth (M3A, M3B, and RAP1), responding to general environmental stress (STRE and URS1), initiating nutrient scavenging and production as evidenced by the phosphate and sulfur regulatory pathways (PHO4 and MET4), and promoting amino acid biogenesis (GCN4). Although the remaining seven unknown patterns (9 discovered motifs) accounted for only 2.2% additional variation, the two most significant motifs from simple linear regression (motif 19 and 10) still have simple regression R-square of 5% and 1.33%, respectively, suggesting their biological relevance to the amino acid starvation response.
Figure 2.

Motifs discovered from the amino acid starvation microarray experiment. The motif regressor multiple regression model reported a total 25 significant motifs active in amino acid starvation response. The 25 motifs can be organized into 15 groups, 8 of which represent previously known TF motifs. The identity of the known motifs reflects how the cell responds to amino acid starvation: slowing cell growth (M3A, M3B, and RAP1), responding to general environmental stress (STRE and URS1), and initiating phosphate (PHO4), sulfur (MET4), and amino acid (GCN4) biogenesis and metabolism. Motif matrices are represented as sequence logos (23).
The M3B motif has been discovered by many computational algorithms (4, 10, 12), and it was recently suggested to be bound by the histone deacetylase and repressor Rpd3p and its associated proteins Ume1p and Ume6p (20). We applied motif regressor to targets separately selected by ChIP-array experiments on Rpd3p, Ume1p, and Ume6p, respectively (20). motif regressor found URS1 to be the most significant motif for Ume6p, RAP1 for Ume1p, and M3B for Rpd3p. Because targets of Rpd3p and Ume1p overlap by >50%, M3B and RAP1 motifs are found significant for both Ume1p and Rpd3p. This finding suggests that Rpd3p and Ume1p might have cooperative binding interactions, and that Rpd3p, Ume1p, and Ume6p might be involved in regulating transcription during amino acid starvation.
To determine the false-positive rate of motif regressor, we assigned randomly the observed expression fold changes to yeast genes, ran mdscan to find motifs of width 5–15, and screened the results by simple linear regressions. This process was repeated 100 times, giving a total of 40,324 motifs, among which 1,398 (3.5%) had regression P values <0.01. This number is slightly higher than the expected 1% because the genes used to find candidate motifs are also used to fit the regression. By comparison, 235 of the 414 candidate motifs (56.8%) from the amino acid starvation had regression P values <0.01, among which we expect <15 (3.5% of 414) to occur by chance.
The Discovery of Multiple Motifs That Influence Gene Expression over Many Time Points.
We ran motif regressor on expression data from 18 time points over two complete yeast cell cycles, starting from release from alpha-factor arrest in M/G1 (18), and obtained a total of 273 significant motifs (including overlapping motifs). Many motifs are found at the time points at which they are known to have the strongest effects. For example, MBF motifs are found during G1 with a strong inducing effect, and during G2 with a strong repressing effect, but are not found in M or S phases. To examine these motifs' effects over all time points, we regressed for each motif at each time point the gene expression values against the upstream sequence motif-matching scores. With each motif represented by a vector of 18 simple regression coefficients, we hierarchically clustered the 273 motifs into 20 groups based on their Euclidean distances (Fig. 3).
Figure 3.

Motif clusters from cell cycle expression time series experiments. The 273 significant motifs reported by motif regressor over two cell cycles are clustered by motif coefficients over the 18 time points. Motif coefficients can be interpreted as the putative influence a particular motif has on the expression of downstream genes. The 20 resulting clusters include the known cell cycle-related TF motifs MCB, SCB, SFF, MCM1, and SWI5. Other motif clusters also have coefficients that fluctuate with the cell cycle, such as STE12, STRE, groups of motifs that resemble MCB and SCB, and some novel G1 motifs. Five motif clusters have coefficients that do not fluctuate with the cell cycle, including M3B and some motifs of unknown function. The clusters were ordered by first appearance of their cell-cycle influence. This figure was produced using treeview software (30).
Fifteen of the 20 motif clusters have putative influences that fluctuate with the cell cycle. Among them are the binding motifs of well-known cell-cycle regulators MCM1, SWI5, MCB, SCB, and SFF (Fig. 4a). Our findings that SWI5 promotes expression at M/G1, SCB promotes expression at G1, and MCB promotes expression at G1 agree well with established data (29). MCM1 was known to promote expression in both G2/M and M/G1, and our findings are consistent with the latter. SFF is thought to function throughout cell cycle, but has been demonstrated to have an inductive influence in S/G2, which our observations support. A number of clusters contain motifs such as ACGCGC and GACGC that resemble both MCB (consensus ACGCGT) and SCB (consensus CGCGAAA). One possibility is that these variant sites match the real MCB or SBF targets well enough to score highly, causing these motifs counted as significant in regression by the MCB/SCB targets. Another explanation is that Mbp1p and Swi4p, which recognize MCB and SCB respectively, are partially redundant regulators and the two proteins recognize each other's binding sites (31). Mbp1p and Swi4p could indeed bind sequences resembling MCB and SCB (although perhaps not optimally) and induce transcription enough to make regression significant.
Figure 4.

Motif effects (coefficients) during the cell cycle. (a) Known cell- cycle-related motifs MCM1, SWI5, MCB, SCB, and SFF have coefficients that fluctuate with the cell cycle. (b) Other cell cycle motifs (STE12, STRE, and motifs in Cluster4 and Cluster6) influence expression through the cell cycle, but to a lesser extent than the known cell cycle regulators. (c) Non-cell-cycle motifs, M3B, and motifs in Cluster18, Cluster19, and Cluster20 showed sharp, low- amplitude fluctuations that correlate to a known experimental artifact that resulted from differential processing of odd- and even-numbered time points (G. Sherlock, personal communication).
We found the STE12 motif, which was correlated with an inductive effect, in the earliest time points. This result is expected because release from mating pheromone was used to synchronize the cells in this experiment and Ste12p is the key transcriptional activator for pheromone-induced transcription. We also found that STE12 retained its putative inductive effect in the second G1 cycle, which is not likely to be explained by the synchronization method, but is consistent with its reported joint role with MCM1 to activate a subset of G1-induced genes (32). The putative strong inductive effect of STRE observed immediately after cell-cycle arrest can be explained as a stress response to the centrifugation and handling required for the release from cell-cycle arrest. A milder inductive effect of STRE is also seen in the following cell cycle throughout G1 and S phases (Fig. 4b). Although ≈50% of yeast genes contain the STRE motif in their promoters (16) and many STRE genes are also regulated by a host of other TFs, this result suggests further experiments to determine whether STRE indeed induces gene expression during G1/S.
Influences of five motif clusters do not vary with the cell cycle. They include two clusters for the M3B motif, which was also found in the amino acid starvation experiment and is present in upstream of genes involved in RNA processing. The variation in the coefficient of the other three motif clusters, especially cluster 19, seemed to correlate with a known experimental artifact (G. Sherlock, personal communication; Fig. 4c). The biological significance of these motifs is thus difficult to interpret.
Discussion
Previously described approaches for regulatory motif discovery either extract the motif features from the upstream sequences with little help from microarray-derived expression values (9, 10), or conduct feature selection based solely on the correlation between short oligomer motif occurrences and expression values (12, 13). motif regressor uses mdscan as a feature extraction tool to find candidate motif matrices and then uses correlation analysis to select motifs relevant to changes in gene expression. Although our feature selection step resembles reducer (12) and the filtering method (10), our adoption of matrix-based motif finding and linear regression enhances both the sensitivity and the specificity. Microarray data from a single experiment can be rapidly analyzed to determine what motifs might be influencing the changes observed. For experiments with multiple time point measurements, the clustering of motif regression coefficients over time course provides a method to quickly recognize the effects of different TFs on the process being studied. With minor modifications, motif regressor can also be applied to ChIP-array experiments.
For experimental biologists, motif regressor is a useful catalyst for planning insightful and directed biological experiments. For example, an experiment is suggested by the analysis of the YAP1 deletion experiment, in which the GCN4 motif was found upstream of up-regulated genes. One possible explanation for this result is that Gcn4p activity is increased in the absence of Yap1p, and the natural targets of Gcn4p are expressed at relatively higher levels. Alternatively, a subset of Gcn4p targets could be induced indirectly through a secondary pathway initiated by YAP1 deletion not dependent on Gcn4p. A ChIP array experiment may determine whether in YAP1 mutants Gcn4 protein actually binds to targets normally bound by Yap1p. Indeed, results obtained through computational approaches must always be tested for biological and mechanistic relevance in vivo.
The method described here assumes that the most interesting motifs are those that cause the most dramatic changes in gene expression under a given condition. Therefore, it may not be ideal for discovering motifs that specify consistent, but subtle, changes in expression. Another assumption is that a given motif can specify only one type of regulation at a given time point, either induction or repression. To detect motifs that may facilitate the binding of both activators and repressors, we may need to use the absolute value of the log-expression values as the dependent variable in the regression, or conduct regression for induced or repressed genes separately.
Acknowledgments
We thank Wei Wang for advice during algorithm development, and Douglas Brutlag, Audrey Gasch, Arkady Khodursky, Nobuo Ogawa, Gavin Sherlock, and the two anonymous referees for insight and help during the preparation of this manuscript. This research was supported by National Institutes of Health Grant 1F37LM07626-01 (to E.M.C.), the Helen Hay Whitney Foundation (to J.D.L.), National Institutes of Health Grant R01 HG02518-01 (to J.S.L.), and National Science Foundation Grant DMS-0204674 (to J.S.L.).
Abbreviations
- TFBM
transcription factor DNA-binding motif
- ChIP
chromatin immunoprecipitation
Footnotes
Throughout the article, we use italicized, all capital designations (YAP1) for genes, first-letter capitalized designations ending with “p” (Yap1p) to represent proteins, and all capital, nonitalic designations (YAP1) to represent DNA binding motifs and sites bound by the respective protein.
References
- 1.van Helden J, Andre B, Collado-Vides J. J Mol Biol. 1998;281:827–842. doi: 10.1006/jmbi.1998.1947. [DOI] [PubMed] [Google Scholar]
- 2.Vilo J, Brazma A, Jonassen I, Robinson A, Ukkonen E. Proc Int Conf Intell Syst Mol Biol. 2000;8:384–394. [PubMed] [Google Scholar]
- 3.Sinha S, Tompa M. Proc Int Conf Intell Syst Mol Biol. 2000;8:344–354. [PubMed] [Google Scholar]
- 4.Hampson S, Kibler D, Baldi P. Bioinformatics. 2002;18:513–528. doi: 10.1093/bioinformatics/18.4.513. [DOI] [PubMed] [Google Scholar]
- 5.Lawrence C E, Altschul S F, Boguski M S, Liu J S, Neuwald A F, Wootton J C. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- 6.Grundy W N, Bailey T L, Elkan C P. Comput Appl Biosci. 1996;12:303–310. doi: 10.1093/bioinformatics/12.4.303. [DOI] [PubMed] [Google Scholar]
- 7.Stormo G D, Hartzell G W., III Proc Natl Acad Sci USA. 1989;86:1183–1187. doi: 10.1073/pnas.86.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bussemaker H J, Li H, Siggia E D. Proc Natl Acad Sci USA. 2000;97:10096–10100. doi: 10.1073/pnas.180265397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth F P, Hughes J D, Estep P W, Church G M. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 10.Hughes J D, Estep P W, Tavazoie S, Church G M. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- 11.Holmes I, Bruno W J. Proc Int Conf Intell Syst Mol Biol. 2000;8:202–210. [PubMed] [Google Scholar]
- 12.Bussemaker H J, Li H, Siggia E D. Nat Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 13.Keles S, Van Der Laan M, Eisen M B. Bioinformatics. 2002;18:1167–1175. doi: 10.1093/bioinformatics/18.9.1167. [DOI] [PubMed] [Google Scholar]
- 14.Liu X S, Brutlag D L, Liu J S. Nat Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 15.Gasch A P, Huang M, Metzner S, Botstein D, Elledge S J, Brown P O. Mol Biol Cell. 2001;12:2987–3003. doi: 10.1091/mbc.12.10.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasch A P, Spellman P T, Kao C M, Carmel-Harel O, Eisen M B, Storz G, Botstein D, Brown P O. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes T R, Roberts C J, Dai H, Jones A R, Meyer M R, Slade D, Burchard J, Dow S, Ward T R, Kidd M J, et al. Nat Genet. 2000;25:333–337. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- 18.Spellman P T, Sherlock G, Zhang M Q, Iyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee T I, Rinaldi N J, Robert F, Odom D T, Bar-Joseph Z, Gerber G K, Hannett N M, Harbison C R, Thompson C M, Simon I, et al. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 20.Kurdistani S K, Robyr D, Tavazoie S, Grunstein M. Nat Genet. 2002;31:248–254. doi: 10.1038/ng907. [DOI] [PubMed] [Google Scholar]
- 21.Zhu J, Zhang M Q. Bioinformatics. 1999;15:607–611. doi: 10.1093/bioinformatics/15.7.607. [DOI] [PubMed] [Google Scholar]
- 22.Fernandes L, Rodrigues-Pousada C, Struhl K. Mol Cell Biol. 1997;17:6982–6993. doi: 10.1128/mcb.17.12.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider T D, Stephens R M. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tavazoie S, Hughes J D, Campbell M J, Cho R J, Church G M. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 25.Blaiseau P L, Thomas D. EMBO J. 1998;17:6327–6336. doi: 10.1093/emboj/17.21.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogawa N, DeRisi J, Brown P O. Mol Biol Cell. 2000;11:4309–4321. doi: 10.1091/mbc.11.12.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieb J D, Liu X, Botstein D, Brown P O. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- 28.Warner J R. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 29.Gailus-Durner V, Chintamaneni C, Wilson R, Brill S J, Vershon A K. Mol Cell Biol. 1997;17:3536–3546. doi: 10.1128/mcb.17.7.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;25:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon I, Barnett J, Hannett N, Harbison C T, Rinaldi N J, Volkert T L, Wyrick J J, Zeitlinger J, Gifford D K, Jaakkola T S, Young R A. Cell. 2001;106:697–708. doi: 10.1016/s0092-8674(01)00494-9. [DOI] [PubMed] [Google Scholar]
- 32.Oehlen L J, McKinney J D, Cross F R. Mol Cell Biol. 1996;16:2830–2837. doi: 10.1128/mcb.16.6.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]