

Abstract
The identification of antigens associated with tumor destruction is a major goal of cancer immunology. Vaccination with irradiated tumor cells engineered to secrete granulocyte–macrophage colony stimulating factor generates potent, specific, and long-lasting antitumor immunity through improved tumor antigen presentation by dendritic cells and macrophages. A phase I clinical trial of this immunization strategy in patients with disseminated melanoma revealed the consistent induction in distant metastases of dense T and B cell infiltrates that effectuated substantial tumor necrosis and fibrosis. To delineate the target antigens of this vaccine-stimulated tumor destruction, we screened a melanoma cDNA expression library with postimmunization sera from a long-term responding patient (K030). High-titer IgG antibodies recognized melanoma inhibitor of apoptosis protein (ML-IAP), a caspase antagonist containing a single baculoviral IAP repeat and a COOH-terminal RING domain. Although K030 harbored antibodies to ML-IAP at the time of study entry, multiple courses of vaccination over 4 years increased antibody titers and elicited isotype switching. Moreover, lymphocyte infiltrates in necrotic metastases included CD4+ and CD8+ T cells specific for ML-IAP, as revealed by proliferation, tetramer, enzyme-linked immunospot, and cytotoxicity analysis. Whereas melanoma cells in densely infiltrated lesions showed strong ML-IAP expression by immunohistochemistry, lethal disease progression was associated with the loss of ML-IAP staining and the absence of lymphocyte infiltrates. These findings demonstrate that ML-IAP can serve as a target for immune-mediated tumor destruction, but that antigen-loss variants can accomplish immune escape.
There is convincing evidence that some patients with malignant melanoma generate cellular and humoral antitumor responses. T lymphocytes and sera from patients recognize a large number of melanoma antigens (1, 2). The presence of brisk T cell infiltrates in primary lesions or lymph node metastases is correlated with prolonged survival (3–5). Nonetheless, a role for host immunity in inhibiting melanoma formation or attenuating disease progression remains to be established and the potential usefulness of the characterized antigens for melanoma diagnosis, prognosis, and therapy needs to be delineated.
One strategy to identify antigens linked with immune-mediated tumor destruction involves the detailed analysis of patients achieving durable responses to immunotherapy (6–10). We demonstrated that vaccination with irradiated tumor cells engineered to secrete granulocyte–macrophage colony-stimulating factor (GM-CSF) stimulates potent, specific, and long-lasting antitumor immunity in multiple murine models (11). Vaccination enhances tumor antigen presentation by mature, CD11b+, CD8α− dendritic cells and macrophages; CD4+ and CD8+ T lymphocytes, CD1d-restricted NK1.1+ T cells, and antibodies contribute to tumor rejection (11–14). A recent phase I clinical trial of this immunization scheme in patients with metastatic melanoma revealed the consistent augmentation of antitumor immunity without significant toxicity (15). Whereas metastases resected before therapy disclosed minimal host reactions in all cases, metastases resected after vaccination revealed dense T and B cell infiltrates that provoked extensive tumor necrosis (at least 80%) in 11 of 16 patients examined. T lymphocytes harvested from the necrotic lesions produced a broad range of cytokines and manifested cytotoxicity to autologous tumor cells. High-titer IgG antibodies reactive with melanoma determinants were demonstrable by flow cytometry and Western analysis.
By screening a melanoma cDNA expression library with postimmunization sera, we previously identified ATP6S1, a putative accessory unit of the vacuolar H+-ATPase complex, as a target for high-titer IgG antibodies in several vaccinated patients (16). Potent humoral responses to ATP6S1 were correlated with tumor destruction although the cellular reactions were not documented. Using the same technical approach, we now delineate the caspase antagonist melanoma inhibitor of apoptosis protein (ML-IAP) (17–20) as a target for high-titer IgG antibodies in subject K030, a patient who attained a partial clinical response and a survival of 4.5 years, despite visceral metastases on protocol entry. Consistent with the pathologic evidence of a coordinated humoral and cellular host reaction, we further show that necrotic metastases contained CD4+ and CD8+ T cells specific for ML-IAP. Finally, the association of lethal disease progression with the outgrowth of antigen-loss tumor variants, along with the absence of lymphocyte infiltrates, suggests that immunity to ML-IAP contributed to tumor destruction.
Materials and Methods
Clinical Protocols.
Sera, lymphocytes, and tumor samples were obtained from K030 on Institutional Review Board/Food and Drug Administration/Recombinant DNA Advisory Committee-approved Dana–Farber Partners Cancer Care clinical protocols. The trial of vaccination with irradiated autologous melanoma cells engineered to secrete GM-CSF by retrovirus-mediated gene transfer was previously reported (15). The trial of vaccination with irradiated autologous melanoma cells engineered to secrete GM-CSF by adenovirus-mediated gene transfer will be described elsewhere. Sera and peripheral blood mononuclear cells were also obtained from healthy blood bank donors at the Dana–Farber Cancer Institute.
Library Construction and Screening.
A melanoma cell line was established from a heavily infiltrated metastasis of subject K008 (16). Total RNA was isolated from the line by using guanidine isothiocyanate, and mRNA was selected with two rounds of oligo(dT)-cellulose. A cDNA expression library was constructed in the Lambda Zap vector by using a commercial cDNA library kit (Stratagene) according to the manufacturer's procedures. Plaques (1 × 106) were screened with precleared (against Escherichia coli and lambda phage lysates) postvaccination (3 months) sera from patient K030 at a 1:1,000 dilution in TBST (50 mM Tris/138 mM NaCl/2.7 mM KCl/0.05% Tween 20, pH 8.0). Positive plaques were detected with an alkaline phosphatase-conjugated polyclonal goat anti-human pan-IgG antibody (Jackson ImmunoResearch) diluted 1:1,000 in TBST and 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium (BCIP/NBT) as a color-development substrate (Promega). Reactive clones were plaque-purified, and the excised phagemids were sequenced.
Serology.
GST-ML-IAP and GST full-length recombinant proteins were produced with the pGEX 5X-3 vector (Amersham Pharmacia) according to the manufacturer's procedures. ELISA plates (Corning, Acton, PA) were coated overnight at 4°C with 500 ng of GST-ML-IAP or GST protein in a carbonate buffer, pH 9.6. The wells were blocked overnight at 4°C with 2% nonfat dried milk (NFDM)/PBS, washed, and incubated in duplicate with 50 μl of patient serum diluted 1:100–1:10,000 in 2% NFDM/PBS for 2 h at room temperature. A polyclonal goat anti-human pan IgG (Jackson ImmunoResearch, 1:1,000 dilution), monoclonal mouse anti-IgG1 (Zymed, 1:500 dilution), or monoclonal mouse anti-IgG4 (Southern Biotechnology Associates, 1:500 dilution) conjugated to alkaline phosphatase in 2% NFDM/PBS was added at room temperature for 1 h, and the plate was developed with p-nitrophenyl phosphate (Sigma). The values reported were the mean absorbance at 405 nm of GST-ML-IAP. All time points showed no reactivity against GST.
T Cell Assays.
Necrotic metastases were processed to single-cell suspension by mechanical digestion and placed into short-term culture (RPMI medium 1640 + 10% FCS). Nonadherent cells were harvested after several hours and were cryopreserved for immune analysis, and ≈25% of these suspensions were CD3+ T lymphocytes, with equal proportions of CD4+ and CD8+ cells. The melanoma cell line (M34) was established from the adherent cells of the heavily infiltrated calf metastasis.
Proliferative responses to ML-IAP were evaluated by first sorting CD8− cells from the processed calf metastasis and expanding these cells in media supplemented with 10 units/ml IL-2 for 4 weeks to obtain a 95% pure population of CD4+ T cells. Five replicates of 1 × 105 cells were then incubated with GST-ML-IAP or GST (1 μg/ml) for 4 days and thymidine uptake was measured. CD4+ T cells from a healthy donor served as a control.
MHC class I-restricted responses were evaluated by first identifying candidate HLA-A*0201 (one of the alleles expressed by K030) binding peptides by using standard algorithms (21). Two peptides (Sigma-Genosys, The Woodlands, TX), JS34 (SLGSPVLGL) and JS90 (RLASFYDWPL), showed high-affinity binding in T2 cell-based assays (22). Soluble HLA-A2 tetramers were prepared as described (23) with β2-microglobulin and JS34, JS90, or L11 (LLFGYPVYV), a peptide derived from human T-lymphotrophic virus-1 Tax, and conjugated to phycoerythrin. Cells harvested from metastases were stained with tetramers and FITC-conjugated anti-CD8 mAb (DAKO) for 30 min at room temperature and analyzed on an EPICS XL (Coulter).
For enzyme-linked immunospot (ELISPOT) analysis, ImmunoSpotplates (Cellular Technology) were coated overnight at 37°C with 10 μg/ml anti-IFN-γ mAb (Mabtech). Cells harvested from metastases were mixed 1:1 with peripheral blood mononuclear cells (as a source of antigen-presenting cells) and plated at 2 × 105 cells per well with 1 μg/ml JS34, JS90, L11 Tax, or HIV-1 reverse transcriptase-polymerase I476 (ILKEPVHGV) peptide. After 24 h at 37°C, the wells were washed and then incubated with 1 μg/ml biotin-conjugated anti-IFN-γ mAb (Mabtech) followed by streptavidin-alkaline phosphatase (Mabtech). Spots were developed by using BCIP/NBT as a color development substrate and counted with an ImmunoSpot microplate reader (Cellular Technology, Cleveland).
For cytotoxicity analysis, the ML-IAP coding sequence was inserted into the pMFG retroviral vector, and high-titer stocks of vesicular stomatitis virus (VSV)-G pseudotyped viral particles were generated as described (11, 24). M34 cells were transduced with the virus to obtain autologous target cells that expressed high levels of ML-IAP. Nonadherent cells harvested from metastases were cultured with irradiated M34-ML-IAP cells in media plus 10 units/ml IL-2 for 3 weeks and were then evaluated in a 4-h 51Cr release assay using M34 and M34-ML-IAP cells as targets. Percent specific lysis was calculated using the following formula: (experimental release − spontaneous release)/(maximal release − spontaneous release).
Anti-ML-IAP mAb.
Mice were immunized with formalin-treated GST-ML-IAP, and hybridomas were generated by using standard techniques. A clone specific for ML-IAP (3F9, IgG1) and suitable for use in tissue sections and Western analysis was identified. For immunohistochemistry, 5-μm-thick formalin-fixed, paraffin-embedded tissue sections were deparaffinized, pretreated with 10 mM sodium citrate, pH 6.0 (Zymed), in a steam pressure cooker (Decloaking Chamber, BioCare Medical, Walnut Creek, CA) per the manufacturer, and washed in distilled water. Next, slides were pretreated with Peroxidase Block (DAKO) for 5 min at room temperature in a hydrated chamber to quench endogenous peroxidase activity, and were blocked with a 1:5 dilution of goat serum in 50 mM Tris, pH 7.4, for 20 min. The slides were then incubated with a 1:50 dilution of 3F9 in 50 mM Tris, pH 7.4, and 3% goat serum for 1 h, washed, and incubated with a goat anti-mouse Ig horseradish peroxidase-conjugated antibody (Envision Detection Kit, DAKO) for 30 min. The slides were developed with a diaminobenzidine chromogen kit (DAKO) per the manufacturer, placed in an enhancing solution (Zymed) for 5 min, and counterstained with hematoxylin. In some experiments, mAbs to CD4, CD8, or CD20 were used.
Results
Clinical Course.
Patient K030 was a 58-year-old female initially diagnosed with adenocarcinoma of the breast in 1990. She underwent high-dose chemotherapy and peripheral blood stem cell support for pulmonary metastases and attained a complete response. K030 remained well until May 1996 when she developed metastatic melanoma in s.c. tissues, lymph nodes, and intestine. Several soft-tissue lesions were excised, introduced into short-term culture, and transduced with pMFG-GM-CSF retroviral supernatants to manufacture autologous, GM-CSF-secreting melanoma vaccines. K030 received 12 intradermal/s.c. immunizations on a weekly basis from October 1996 to January 1997 and achieved a partial regression of the s.c. metastases. A persistent colonic lesion was resected in February 1997, and pathologic examination revealed extensive tumor necrosis with brisk CD4+ and CD8+ T lymphocyte and CD20+ B cell infiltrates (Fig. 1 A–C). Twelve additional immunizations were administered through November 1997, and the patient did well until September 1998, when a new cystic calf metastasis became painful. The lesion was excised and pathologic evaluation again demonstrated extensive tumor necrosis with CD4+ and CD8+ T lymphocyte and CD20+ B cell infiltrates (Fig. 1 D–F). The metastasis was processed to single-cell suspension and transduced with an adenoviral vector encoding GM-CSF to prepare a second set of autologous, GM-CSF-secreting melanoma vaccines. K030 received 11 immunizations with this material through February 1999, but the calf mass recurred and gradually became indurated. Although tissue could not be obtained, the lesion was assessed clinically as progressive tumor. The patient underwent isolated limb perfusion with tumor necrosis factor-α and melphalan followed by removal of a residual mass (that showed extensive necrosis) and radiation therapy. Two subsequent s.c. metastases were resected in November and December 1999, and both displayed extensive necrosis with lymphocyte infiltrates. Adenoviral-mediated gene transfer was used to manufacture a third batch of GM-CSF-secreting vaccines from this material. K030 received 18 more immunizations from March through September 2000, but she then developed a small bowel obstruction that required surgical intervention. In contrast to the previous lesions, pathologic examination of this metastasis disclosed the absence of tumor necrosis and the absence of tumor-infiltrating lymphocytes. A fourth round of vaccines was prepared from this sample, but after four immunizations, rapid disease progression ensued, and the patient died shortly thereafter in March 2001.
Figure 1.

Infiltrated metastases with extensive tumor destruction after vaccination. (A–C) Colon metastasis. (D–F) Calf metastasis. CD4+ T cells (A), CD8+ T cells (B), CD20+ B cells (C), CD4+ T cells (D), CD8+ T cells (E), and CD20+ B cells (F) are shown.
Serologic Response to ML-IAP.
Because pathologic evaluation of multiple necrotic metastases revealed the accumulation of B and T lymphocytes, we hypothesized that at least some melanoma antigens provoked a coordinated humoral and cellular response. To characterize these potential targets, we used a serology-based expression cloning strategy (25). As an autologous tumor cell line was not available when these studies were initiated, we used a melanoma cDNA expression library constructed from a heavily infiltrated metastasis of subject K008, a vaccinated patient previously reported who remains disease-free 6.5 years after treatment (16). One of the antigens identified through screening the library with K030 postimmunization sera was ML-IAP, a recently described caspase inhibitor that contains a single baculoviral IAP repeat and RING domain (17–20). Earlier work established that ML-IAP was frequently expressed in melanoma cells, but mRNA transcripts were not detectable in normal melanocytes by RT-PCR (17).
To examine the humoral response to ML-IAP in more detail, we established an ELISA with bacterially produced recombinant protein. Unexpectedly, K030 harbored IgG antibodies to ML-IAP on study enrollment. Because sera from 60 healthy controls failed to recognize the antigen (data not shown), melanoma development likely triggered the humoral reactivity, although a long-lived memory response to the earlier breast carcinoma was also possible. Antibody titers rapidly increased with vaccination, consistent with an amnestic reaction in the setting of preexisting immunity (Fig. 2A). Moreover, prolonged immunization induced isotype switching. Whereas IgG1 antibodies to ML-IAP were measurable before therapy (data not shown), vaccination elicited IgG4 antibodies as well (Fig. 2B). Pan-IgG and IgG4 antibodies peaked in late 1999, but gradually declined thereafter, despite additional immunizations.
Figure 2.

Vaccination-augmented anti-ML-IAP antibody titers and induced isotype switching. (A) Pan-IgG. (B) IgG4. The ELISA was performed with recombinant GST-ML-IAP protein and K030 sera diluted 1:10,000 for the pan-IgG determinations and 1:100 for the IgG4 measurements. Small arrows denote vaccinations. The peak pan-IgG and IgG4 levels were 4-fold and 10-fold higher than the respective values at study entry. K030 sera also recognized GST-ML-IAP in Western analysis. Reactivity to Candida antigens was not consistently affected by therapy (data not shown).
T Cell Response to ML-IAP.
Because isotype switching typically involves helper T cell function (26), we tested whether ML-IAP-specific CD4+ T cells were associated with tumor destruction. CD8− cells were sorted from the infiltrated calf metastasis and expanded for 4 weeks with 10 units/ml IL-2 to yield a 95% pure population of CD4+ T cells. These lymphocytes demonstrated strong proliferative responses to recombinant GST-ML-IAP, but not to GST (Fig. 3A). In contrast, CD4+ T cells from a healthy donor showed strong reactions to CD3 crosslinking, but not to GST-ML-IAP (not shown). These results indicate the presence of ML-IAP-specific CD4+ T cells in the necrotic metastases, but further studies are required to characterize their cytokine profiles.
Figure 3.

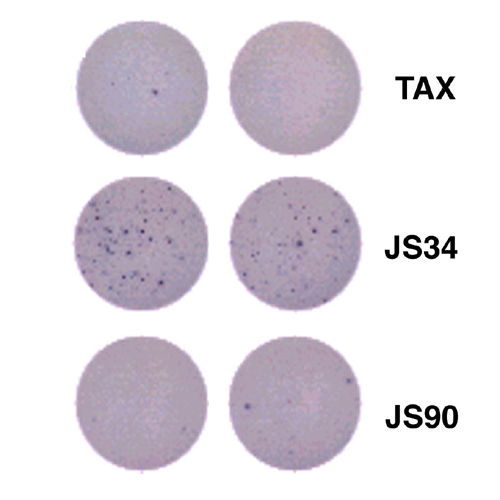
ML-IAP-specific T cells were associated with tumor destruction. (A) Proliferative responses of CD4+ T cells derived from the calf metastasis. (B) JS34- and JS90-tetramer-stained CD8+ T cells from the colon metastasis. CD8+ tetramer+ cells were as follows: JS34, 1.01%; JS90, 0.86%; and Tax, 0.38%. (C) JS34 and JS90 peptides elicited IFN-γ production from CD8+ T cells in the colon metastasis. Spots shown were (no. of replicate wells) as follows: Pol (0, 3); JS34 (20, 4, 10); and JS90 (3, 7, 6). No reactivity was detected with autologous peripheral blood mononuclear cells alone (data not shown). (D) T cells from the calf metastasis showed ML-IAP-specific cytotoxicity (effector-to-target cell ratio of 30:1).
Because dense CD8+ T cell infiltrates were demonstrable in multiple metastases, we investigated whether ML-IAP provoked MHC class I-restricted responses. As K030 was HLA-A201+, we first characterized ML-IAP-derived epitopes that might be presented by this allele. Two peptides identified with computer algorithms (21), JS34 (SLGSPVLGL) and JS90 (RLASFYDWPL), showed strong binding in T2 cell-based assays (data not shown) (22). Soluble HLA-A2 tetramers incorporating these peptides, and a human T-lymphotrophic virus-1 Tax-derived peptide as a control, were synthesized (23) and used to analyze single-cell suspensions from the infiltrated metastases. For these experiments, the infiltrating lymphocytes were studied without any in vitro expansion. Whereas total cell numbers were somewhat limited, JS34-, and to a lesser extent, JS90-tetramer binding CD8+ cells were present in the colon metastasis (Fig. 3B). Minimal reactivity with the Tax tetramer was found. The significance of the CD8− tetramer-reactive cells is unclear. In contrast, the JS34 and JS90 tetramers failed to stain MAGE-3- or telomerase-specific T cell lines derived from the peripheral blood of three healthy HLA-A2 donors (not shown).
To corroborate these findings with a second technique, we tested the tumor infiltrates for ML-IAP reactivity in an ELISPOT assay. Single-cell suspensions from the metastases were again used without in vitro stimulation to analyze their activation status in vivo as closely as possible. Consistent with the tetramer stains, the JS34, and to a lesser extent, JS90 peptides elicited IFN-γ production in the lymphocytes harvested from the colon (Fig. 3C). The JS34 peptide also evoked IFN-γ production in the lymphocytes isolated from the calf metastasis (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org).
To examine whether ML-IAP-specific T cells in the necrotic lesions could mediate cytotoxicity in vitro, we constructed an autologous ML-IAP-expressing melanoma cell line. As ML-IAP was not detectable in M34 cells by Western analysis, we cloned the ML-IAP-coding sequences into the pMFG retroviral vector and generated high-titer stocks of VSV-G pseudotyped viral particles (11, 24). Transduction of M34 cells with the viral preparation resulted in high-level ML-IAP expression (see Fig. 6, which is published as supporting information on the PNAS web site). Cell suspensions from the infiltrated calf metastasis were cultured with irradiated M34-ML-IAP cells and IL-2 for 3 weeks to expand the numbers of effectors. Whereas some cytotoxicity toward the parental line was apparent, increased killing of the ML-IAP-expressing cells was found (Fig. 3D). Because MHC class II molecules were not detectable on resting or IFN-γ-treated M34 cells, the ML-IAP-specific killing was likely to be MHC class I-dependent. The limited numbers of cells available precluded antibody-blocking studies.
ML-IAP Expression in Vivo.
To determine whether immunity to ML-IAP contributed to tumor destruction in vivo, we generated a ML-IAP-specific mAb (3F9) suitable for use in paraffin sections (see Fig. 7, which is published as supporting information on the PNAS web site). The densely infiltrated colon metastasis resected in February 1997 revealed diffuse cytoplasmic expression throughout the tumor, with a minority of cells showing more intense staining (Fig. 4A). Although the calf recurrence in September 1998 primarily revealed necrotic tumor, the few viable malignant cells appeared to demonstrate increased staining relative to the colon metastasis (Fig. 4B). With further vaccination, a population of cells that failed to stain with the antibody began to emerge in the recurrent lesions (Fig. 4C). Indeed, the small-bowel metastasis resected shortly before the patient's demise revealed no ML-IAP expression and the absence of tumor-infiltrating lymphocytes (Fig. 4D).
Figure 4.

Vaccination stimulated the emergence of ML-IAP-negative tumor cells. (A) Colon metastasis. (B) Calf metastasis. (C) s.c./lymph node metastasis. (D) Small-bowel metastasis. The sections were stained with the 3F9 anti-ML-IAP mAb and then were counterstained with hematoxylin.
Discussion
These investigations were undertaken in an effort to learn more about the development of antitumor immunity in a metastatic melanoma patient who achieved a clinical response and long-term survival as a consequence, at least in part, of vaccination with irradiated, autologous GM-CSF-secreting tumor cells. The analysis uncovered a previously unknown cancer-rejection antigen, and yielded insights into both the therapeutic potential and the mechanisms limiting the clinical efficacy of whole-tumor-cell immunizations.
The pathologic evidence of B and T lymphocyte infiltrates in necrotic metastases revealed that GM-CSF-secreting melanoma vaccines stimulated a coordinated humoral and cellular antitumor response. To identify the targets of these reactions, we screened a melanoma cDNA expression library with postimmunization sera. One of the antigens delineated was ML-IAP, a member of the inhibitor of apoptosis protein family (17–20). Although a humoral response to the protein was detectable on study entry, vaccination augmented antibody titers and induced class switching. Moreover, CD4+ and CD8+ T cells specific for ML-IAP were present in the necrotic metastases, as illustrated by proliferation, tetramer, ELISPOT, and cytotoxicity assays.
A key function for ML-IAP in the transformed phenotype suggests that this protein may be an important target for tumor immunity. The ML-IAP gene is localized to chromosome 20q13.3, a region frequently amplified in melanomas and diverse carcinomas (27, 28), and our preliminary immunohistochemical analysis disclosed strong expression in a wide variety of human tumors (data not shown). Previous work demonstrated that ML-IAP antagonizes caspases 3, 7, and 9, thereby blocking death receptor and mitochondrion-based apoptotic pathways (17–20). JNK1 also participates in ML-IAP-mediated suppression of cell death (29), whereas the processed mitochondrial protein SMAC/DIABLO serves as a negative regulator (30). Given these properties, ML-IAP likely endows melanoma cells with a survival advantage during tumor progression and contributes to their resistance to chemotherapy and radiation therapy.
While the increased ML-IAP levels in the calf metastasis of K030 may reflect a selection for melanoma cells that were partially resistant to immune attack, prolonged vaccination was associated with the suppression of these clones and the emergence of antigen-loss variants. These findings raise the interesting issue of how the immune system might control ML-IAP-expressing tumor cells. In this context, convincing evidence indicates that cytotoxic lymphocytes kill their targets in part through caspase-independent mechanisms that involve the intracellular delivery of granzymes (31–35). These proteases directly cleave critical apoptotic substrates including BID, DNA-dependent protein kinase catalytic subunit, nuclear mitotic apparatus protein, inhibitor of caspase-activated DNase, and the nuclear lamins. This cytotoxic pathway might effectively circumvent ML-IAP function in melanoma cells. Further studies are necessary to clarify the roles of CD4+ T cells and antibodies in accomplishing tumor destruction.
In addition to the loss of ML-IAP expression, lethal disease progression was characterized by the absence of tumor-infiltrating lymphocytes and a persistent decline in the titers of anti-ML-IAP antibodies. K030 developed immunity, however, to several other melanoma antigens (F.S.H., J.C.S., and G.D., unpublished data) including ATP6S1, a component of the vacuolar H+-ATPase complex that we previously described as a vaccine target (16). Although the rules governing the integration of responses to multiple cancer antigens remain to be elucidated, our results suggest that immunity to ML-IAP was important for tumor control in this patient. In retrospect, K030 was not vaccinated with a significant number of the antigen-loss variants until her disease was too far advanced. The identification of immunogenic moieties in these resistant cells is, therefore, a high priority, and the ML-IAP-negative M34 cell line should advance these efforts considerably. Understanding the mechanisms by which tumor cells obviate the requirement for ML-IAP is another critical issue for future investigation. One possibility is that other members of the IAP family may subserve a redundant function (36, 37). Nonetheless, our preliminary analysis of the metastases resected shortly before death revealed that X-IAP was expressed in only a minority of the cells and that survivin was undetectable (data not shown).
Overall, the association delineated here between immunity to ML-IAP and tumor destruction should stimulate further work to target this protein therapeutically. ML-IAP is expressed in normal testes, placenta, spinal cord, and lymph node (17–20), but treatment did not elicit significant toxicities in K030. Whereas de novo antibody responses to ML-IAP are uncommon among patients with metastatic melanoma, lung carcinoma, and prostate carcinoma (data not shown), potent antigen-specific immunizations might enhance reactivity. Indeed, a polyvalent vaccine that incorporates ML-IAP, additional antiapoptotic proteins (38, 39), and other gene products crucial for cancer maintenance, such as telomerase (40), may provide a strategy to overcome tumor heterogeneity.
Supplementary Material
Acknowledgments
We thank the Connell-O'Reilly laboratory for excellent processing of patient material; the Kraft Family Blood Bank for help with sera; Christine Sheehan and Esther Brisson (Albany Medical College, Albany, NY) for excellent help with the histological specimens; and Khuong Nguyen and Shannon Bell for excellent help with the mAbs. This work was supported by a Hochschulsonderprogramm-III fellowship of the Deutsche Akademische Austauschdienst (to J.C.S.); Damon Runyon Cancer Research Fund Clinical Investigator Award (to R.H.V.); the Berlex Oncology Foundation; National Institutes of Health (NIH) Grant CA78880 (to F.S.H.); awards to G.D. from the NIH (Grants CA74886, CA39542, and CA66996); the Cancer Research Institute/Partridge Foundation (Clinical Investigator Award); the Cancer Research Institute/Partridge Foundation (Melanoma Initiative); and the Leukemia and Lymphoma Society (Score Award in Acute Myeloid Leukemia). G.D. and R.S. are Clinical Scholars of the Leukemia and Lymphoma Society.
Abbreviations
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- ML-IAP
melanoma inhibitor of apoptosis protein
- ELISPOT
enzyme-linked immunospot
References
- 1.Boon T, van der Bruggen P. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Old L, Chen Y-T. J Exp Med. 1998;187:1163–1167. doi: 10.1084/jem.187.8.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark W, Elder D, Guerry D, Braitman L, Trock B, Schultz D, Synnestvedt M, Halpern A. J Natl Cancer Inst. 1989;81:1893–1904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- 4.Clemente C, Mihm M, Bufalino R, Zurrida S, Collini P, Cascinelli N. Cancer (Philadelphia) 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Mihm M, Clemente C, Cascinelli N. Lab Invest. 1996;74:43–47. [PubMed] [Google Scholar]
- 6.van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 7.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Sakaguchi K, Appella E, Yannelli J R, Adema G J, Miki T, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakker A, Schreurs M, de Boer A, Kawakami Y, Rosenberg S, Adema G, Figdor C. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorn E, Hercend T. Eur J Immunol. 1999;29:592–601. doi: 10.1002/(SICI)1521-4141(199902)29:02<592::AID-IMMU592>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Zorn E, Hercend T. Eur J Immunol. 1999;29:602–607. doi: 10.1002/(SICI)1521-4141(199902)29:02<602::AID-IMMU602>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 11.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan R C. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang A Y, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- 13.Mach N, Gillessen S, Wilson S B, Sheehan C, Mihm M, Dranoff G. Cancer Res. 2000;60:3239–3246. [PubMed] [Google Scholar]
- 14.Reilly R, Machiels J-P, Emens L, Ercolini A, Okoye F, Lei R, Weintraub D, Jaffee E. Cancer Res. 2001;61:880–883. [PubMed] [Google Scholar]
- 15.Soiffer R J, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger J, Hodi F, Liebster L, Lam P, Mentzer S, et al. Proc Natl Acad Sci USA. 1998;95:13141–13146. doi: 10.1073/pnas.95.22.13141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodi F S, Schmollinger J C, Soiffer R J, Salgia R, Lynch T, Ritz J, Alyea E P, Yang J C, Neuberg D, Mihm M, Dranoff G. Proc Natl Acad Sci USA. 2002;99:6919–6924. doi: 10.1073/pnas.102025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vucic D, Stennicke H, Pisabarro M T, Salvesen G, Dixit V M. Curr Biol. 2000;10:1359–1366. doi: 10.1016/s0960-9822(00)00781-8. [DOI] [PubMed] [Google Scholar]
- 18.Kasof G M, Gomes B C. J Biol Chem. 2001;276:3238–3246. doi: 10.1074/jbc.M003670200. [DOI] [PubMed] [Google Scholar]
- 19.Lin J-H, Deng G, Huang Q, Morser J. Biochem Biophys Res Commun. 2000;279:820–831. doi: 10.1006/bbrc.2000.4027. [DOI] [PubMed] [Google Scholar]
- 20.Ashhab Y, Alian A, Polliack A, Panet A, Yehuda D B. FEBS Lett. 2001;495:56–60. doi: 10.1016/s0014-5793(01)02366-3. [DOI] [PubMed] [Google Scholar]
- 21.Parker K C, Bednarek M A, Coligan J E. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 22.Salter R, Cresswell P. EMBO J. 1986;5:943–949. doi: 10.1002/j.1460-2075.1986.tb04307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altman J, Moss P, Goulder P, Barouch D, McHeyzer-Williams M, Bell J, McMichael A, Davis M. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 24.Ory D S, Neugeboren B A, Mulligan R C. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahin U, Tureci O, Schmitt H, Cochlovius B, Johannes T, Schmits R, Stenner F, Luo G R, Schobert I, Pfreundschuh M. Proc Natl Acad Sci USA. 1995;92:11810–11813. doi: 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manis J, Tian M, Alt F. Trends Immunol. 2002;23:31–39. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 27.Barks J H, Thompson F H, Taetle R, Yang J M, Stone J F, Wymer J A, Khavari R, Guan X Y, Trent J M, Pinkel D, Nelson M A. Genes Chromosomes Cancer. 1997;19:278–285. [PubMed] [Google Scholar]
- 28.Korn W M, Yasutake T, Kuo W L, Warren R S, Collins C, Tomita M, Gray J, Waldman F M. Genes Chromosomes Cancer. 1999;25:82–90. doi: 10.1002/(sici)1098-2264(199906)25:2<82::aid-gcc2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Sanna M G, da Silva Correia J, Ducrey O, Lee J, Nomoto K, Schrantz N, Deveraux Q L, Ulevitch R J. Mol Cell Biol. 2002;22:1754–1766. doi: 10.1128/MCB.22.6.1754-1766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vucic D, Deshayes K, Ackerly H, Pisabarro M T, Kadkhodayan S, Fairbrother W J, Dixit V M. J Biol Chem. 2002;277:12275–12279. doi: 10.1074/jbc.M112045200. [DOI] [PubMed] [Google Scholar]
- 31.Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Immunity. 1998;8:451–460. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 32.Thomas D, Du C, Xu M, Wang X, Ley T. Immunity. 2000;12:621–632. doi: 10.1016/s1074-7613(00)80213-7. [DOI] [PubMed] [Google Scholar]
- 33.Sutton V R, Davis J E, Cancilla M, Johnstone R W, Ruefli A A, Sedelies K, Browne K A, Trapani J A. J Exp Med. 2000;192:1403–1414. doi: 10.1084/jem.192.10.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heibein J A, Goping I S, Barry M, Pinkoski M J, Shore G C, Green D R, Bleackley R C. J Exp Med. 2000;192:1391–1402. doi: 10.1084/jem.192.10.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang D, Beresford P J, Greenberg A H, Lieberman J. Proc Natl Acad Sci USA. 2001;98:5746–5751. doi: 10.1073/pnas.101329598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deveraux Q, Reed J. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 37.Miller L. Trends Cell Biol. 1999;9:323–328. doi: 10.1016/s0962-8924(99)01609-8. [DOI] [PubMed] [Google Scholar]
- 38.Andersen M H, Pedersen L O, Becker J C, Straten P T. Cancer Res. 2001;61:869–872. [PubMed] [Google Scholar]
- 39.Hirohashi Y, Torigoe T, Maeda A, Nabeta Y, Kamiguchi K, Sato T, Yoda J, Ikeda H, Hirata K, Yamanaka N, Sato N. Clin Cancer Res. 2002;8:1731–1739. [PubMed] [Google Scholar]
- 40.Vonderheide R, Hahn W, Schultze J, Nadler L. Immunity. 1999;10:673–679. doi: 10.1016/s1074-7613(00)80066-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}