

Abstract
Injured motor neurons of the adult rat can survive, whereas similar axotomy causes gradual motor neuron death in the adult mouse. We report that the decreased expression of the neuronal glutamate transporter excitatory amino-acid carrier 1 (EAAC1) following nerve injury is associated with motor neuron death in the mouse. Glutamate transporters play a crucial role in prevention of neuronal death by suppressing glutamate toxicity. However, the possible functional role of EAAC1 in preventing neuron death has not been resolved as compared with glial glutamate transporters such as GLT-1. Here, we have revealed a unique ‘rescue' function of EAAC1, which is independent of removal of extracellular glutamate. During apoptotic stimuli, a mitochondrial protein, holocytochrome c synthetase (HCCS), translocates to outside the mitochondria, binds to and suppresses the X-linked inhibitor of apoptosis protein (XIAP), leading to activation of caspase-3. The N-terminus of EAAC1 can bind to HCCS, which interferes with the HCCS–XIAP association, and thereby maintain XIAP activity. This unique anti-apoptotic mechanism of EAAC1 functions in rescuing PC12 cells and motor neurons from NGF deprivation and nerve injury, respectively.
Keywords: axotomy, glutamate transporter, mitochondria, nerve regeneration, neuronal death
Introduction
Peripheral nerve-injured motor neurons in the adult rats can survive and subsequently regenerate; however, those in adult mice undergo progressive neuronal death over a period of several weeks (Kiryu-Seo et al, 2005). The motor neuronal death seen in the adult mice progresses at a much slower rate than that seen in the neonatal animals (Snider et al, 1992), because most of the major anti-apoptotic mechanisms including neurite elongation appear to function in the injured neurons of adult mice as well as in those of adult rats (Kiryu-Seo et al, 2005). The slower motor neuron death seen in adult mice could therefore be an interesting and appropriate model for investigating neurodegenerative diseases such as amyotrophic lateral sclerosis rather than the relatively acute motor neuron death seen in neonates. It seemed likely that an important but slowly acting part of the neuronal survival machinery is missing in the motor neurons of adult mice, but is present in those of adult rats, leading to the gradual death of mouse motor neurons. We therefore looked for the missing part of the survival machinery by screening for nerve injury-associated genes in rats and mice, and focused on the neuronal glutamate transporter excitatory amino-acid carrier 1 (EAAC1) as a candidate gene. The expression of EAAC1 was upregulated in rat motor neurons after axotomy (Kiryu et al, 1995), but downregulated in mouse, suggesting that EAAC1 might have an important role in the prevention of motor neuronal death in the rat.
Of the molecules implicated in glutamate clearance, five members of high-affinity Na+-dependent excitatory amino-acid transporters (EAATs) have so far been discovered: GLAST/EAAT1, GLT-1/EAAT2, EAAC1/EAAT3, EAAT4 and EAAT5 (Danbolt, 2001). Of the glutamate transporters that have been described, the glial type glutamate transporters such as GLAST/EAAT1 and GLT-1/EAAT2 are the major glutamate transporters in the central nervous system (CNS) and are predominantly localized on astrocyte membranes. They are assumed to play a major role in preventing glutamate-induced neuronal death, as well as terminating glutamatergic synaptic transmission (Choi, 1988; Rothstein et al, 1996). In particular, the functional relevance of EAAT2 was clearly demonstrated in a study using GLT1/EAAT2 knockout mice, which developed a phenotype characterized by severe epilepsy (Tanaka et al, 1997). By contrast, EAAC1/EAAT3, which is also widely expressed in CNS and predominantly localized in neuron (Kanai and Hediger, 1992), is unlikely to be a major player in suppression of glutamate toxicity. One reason is that EAAC1-deficient mice develop normally and without apparent neurodegeneration and epilepsy (Peghini et al, 1997). However, a recent study using EAAC1-deficient mice showed that EAAC1 can function as a cysteine transporter and maintain neuronal glutathione homeostasis (Aoyama et al, 2006). It is possible that EAAC1 may have alternative activities distinct from glutamate removal.
After axotomy, motor neuron death is triggered by Fas and proceeds via the established mitochondrial death pathway (Martin and Liu, 2002; Ugolini et al, 2003; Martin et al, 2005). The motor neuron death seen in the mice also progresses gradually via the mitochondrial death pathway in a tumor suppressor protein p53-dependent manner (Kiryu-Seo et al, 2005). The mitochondria-dependent apoptotic machinery has been very well documented (Pettmann and Henderson, 1998; Ferri and Kroemer, 2001; Saelens et al, 2004). In general, the release of cytochrome c from mitochondria into the cytosol is pivotal in the activation of caspases and the ensuing cell death. Concurrent with cytochrome c release, other mitochondrial proteins such as Smac/DIABLO and Omi/HtrA2 are also released from mitochondria into the cytosol. They interact with inhibitor of apoptosis proteins (IAPs) and eliminate their inhibitory effects on caspase activity. However, their IAP suppression activity may be a secondary function occurring only during apoptotic stimuli, as these proteins are essential for maintaining mitochondrial function under normal condition (Jones et al, 2003; Martins et al, 2004). From database searches and knockout studies, there seem to be still many mitochondrial proteins with the potential to act as IAP binding proteins (Vaux and Silke, 2003; Martins et al, 2004).
In this study, we report that EAAC1 has a unique action in preventing mitochondria-mediated neuronal death. This ‘rescue' activity does not depend on glutamate removal, but requires an interaction with a mitochondrial protein, holocytochrome c synthetase (HCCS), which is released from the mitochondria. HCCS is normally localized in the mitochondrial intermembrane space and is essential for holo-cytochrome c synthesis (Schaefer et al, 1996). However, it is now evident that cytosolic HCCS may have a secondary role as an X-linked IAP (XIAP) binding protein under apoptotic stimuli. Here, we consider how EAAC1 may contribute to the survival of injured motor neurons.
Results
EAAC1 shows different expression patterns in rats and mice following axotomy
Following hypoglossal nerve axotomy, injured motor neurons in rats survive and regenerate, whereas those in mice die over a period of several weeks. We found that the signal for EAAC1 mRNA was increased in rat injured motor neurons, but was decreased rapidly in the first days after axotomy in the mouse (Figure 1A and B). Our previous studies using in situ hybridization have demonstrated that most nerve injury-associated genes were regulated similarly in both the rat and the mouse at 7 days after axotomy (Kiryu-Seo et al, 2005). Consistent with this, expression of the other glutamate transporter family members such as GLT-1, GLAST and neutral amino-acid transporters, system ASC transporters (ASCT1 and ASCT2), also responded similarly in rats and mice following hypoglossal axotomy (Figure 2A). RT–PCR analysis using control and injured hypoglossal nuclei confirmed that the expression of all these transporters except for EAAC1 was regulated similarly in both rats and mice (Figure 2B). The upregulation of EAAC1 expression in rats was also observed after alternative peripheral nerve injuries, including facial and sciatic nerve axotomy (Supplementary Figure 1). As EAAC1 expression was unchanged after CNS lesion of rats (Ginsberg et al, 1995, 1996; Lievens et al, 2001), it suggests that this increased EAAC1 expression is critical for the survival of motor neurons in the rat.
Figure 1.

The regulation of EAAC1 mRNA expression is strikingly different in rats and mice following hypoglossal nerve injury. (A) Top panel: In situ hybridization showed that expression of EAAC1 mRNA was upregulated in the injured hypoglossal nucleus of rats, but downregulated in those of mice 7 days following axotomy. Bottom panel: Motor neurons stained with thionine at 7 days after nerve injury. The number of motor neurons did not change at this time point in rats and mice. cc, central canal; XII, hypoglossal nucleus. Scale bars, 100 μm for rat and 50 μm for mouse. (B) Change in EAAC1 mRNA expression after nerve injury in rats (closed circles) and mice (open circles). Each point shows the mean±s.d. of the relative signal intensity. The asterisks indicate statistically significant differences compared with the results found in rats (P<0.01; Student's t-test, n=4).
Figure 2.

The expression of mRNA for glutamate transporter family members after hypoglossal axotomy. (A) Expression of mRNA for glutamate transporter family members was examined by in situ hybridization 7 days after axotomy. EAAC1 mRNA was upregulated (closed arrow) in injured hypoglossal neurons of rat, but downregulated (open arrow) in those of mice. Scale bar, 1.3 mm for rat and 0.8 mm for mouse. (B) RT–PCR using total RNA from control (cont) and injured (inj) hypoglossal nuclei of rats and mice 7 days after axotomy.
The expression of EAAC1 is correlated with the survival of injured motor neurons in mice
To evaluate the relevance of EAAC1 expression to the survival of injured motor neuron, we used EAAC1 transgenic mice (on a C57BL/6 background) and EAAC1-deficient mice (on an ICR background). In EAAC1 transgenic mice, the damage-induced neuronal endopeptidase (DINE) promoter, which we have previously identified as a neuron-specific and injury-associated gene (Kiryu-Seo et al, 2000), was employed. This transgenic mouse was designed to express EAAC1 and GFP simultaneously by inserting an internal ribosome entry site under the control of the DINE promoter. Analysis of mRNA expression in the EAAC1 transgenic mice after axotomy showed that EAAC1 expression was maintained as might be expected because expression of exogenous EAAC1 was increased after nerve injury (Figure 3A and B and Supplementary Figure 2). We then counted the number of surviving motor neurons in wild-type and EAAC1 transgenic mice. As we have described previously, 35 days after nerve injury is a good time point to evaluate neuronal survival and compare the response of the different animals (Kiryu-Seo et al, 2005). At 35 days after axotomy, the wild-type mice had lost approximately 50% of neurons in the injured hypoglossal nuclei. In contrast, the EAAC1 transgenic mice exhibited significantly less neuronal death (Figure 3C and D). This suggests strongly that EAAC1 overexpression in the mouse may mimic the phenotypic response of injured rat motor neurons. In EAAC1-deficient mice (ICR background), there was an accelerated loss of injured neurons as compared with wild-type mice (Figure 3E and F). RT–PCR analysis using hypoglossal nuclei of both EAAC1 transgenic and knockout mice showed that expression of the members of the glutamate transporter family was regulated similarly (Figure 3G). As shown in Figure 3C–F, the survival rate of injured neuron was different between the two mouse strains, C57BL/6 and ICR. Thus, we also examined the effect of genetic background on the motor neuron death after axotomy (Supplementary Figure 3A). At 35 days after axotomy, the survival of neurons in wild-type ICR mice was about 80%, whereas that in wild-type C57BL/6 mice was around 50%. Both strains showed downregulation of EAAC1 mRNA after nerve injury; however, the level of downregulation of EAAC1 mRNA in the two strains correlated well with the survival of axotomized neurons (Supplementary Figure 3B and C). All these data further suggest an important role for EAAC1 in the survival of nerve-injured adult mouse motor neurons.
Figure 3.
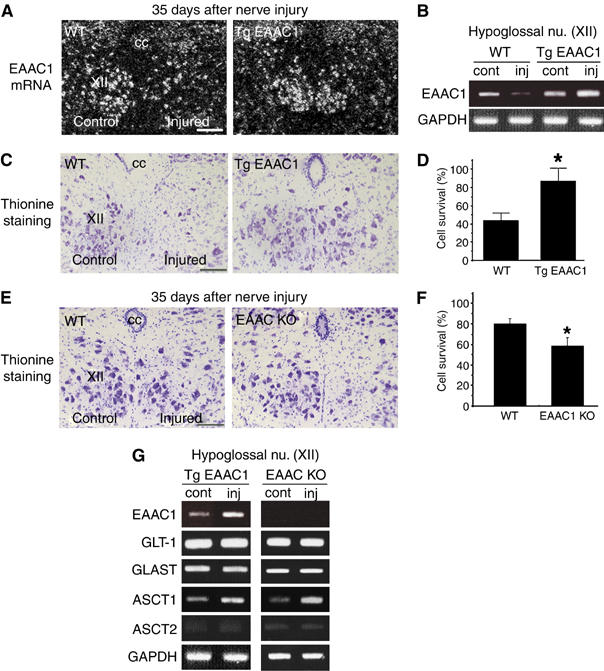
EAAC1 expression influences the survival of injured motor neurons. (A) Sections of hypoglossal motor neurons from wild-type mice (WT) and EAAC1-overexpressing mice (Tg EAAC1) 35 days after axotomy (C57BL/6 background) were subjected to in situ hybridization to examine the expression of EAAC1 mRNA. (B) RT–PCR using hypoglossal nuclei of WT and Tg EAAC1 7 days after axotomy. (C) Thionine staining of hypoglossal nuclei 35 days after axotomy. (D) Percentage of surviving neurons 35 days after axotomy. Results represent the percent ratio of surviving motor neurons on the injured side compared with the contralateral side. Data are mean±s.d. (P<0.01; Student's t-test, n=7). (E) Thionine staining of hypoglossal motor neurons 35 days after hypoglossal nerve injury in WT mice and EAAC1-deficient mice (EAAC1 KO) (ICR background). (F) Percentage of surviving motor neurons in WT and EAAC1 KO 35 days after axotomy. Data are mean±s.d. (P<0.01; Student's t-test, n=11). (G) RT–PCR analysis using hypoglossal nuclei from Tg EAAC1 and EAAC1 KO 7 days after nerve injury. cc, central canal; XII, hypoglossal nucleus; control, contralateral side; injured, injured side; cont, contralateral side; inj, injured side. Scale bar, 50 μm (A,C, E). The asterisks indicate statistically significant differences compared with the results of WT mice (D, F).
EAAC1 prevents neuronal death in differentiated PC12 cells
To examine how EAAC1 may prevent neuronal death, we carried out in vitro studies. We first demonstrated that differentiated PC12 neuronal cells overexpressing EAAC1 took up 14C-labeled glutamate more effectively than control cells (Figure 4A). Expression of EAAC1 in these cells did not alter their level of expression of other glutamate transporters (Supplementary Figure 4A). Although previous reports have indicated that EAAC1 may take up cysteine as well as glutamate (Zerangue and Kavanaugh, 1996; Chen and Swanson, 2003), both EAAC1-overexpressing and control PC12 cells showed similar uptake of cysteine (Supplementary Figure 4B). This may be because PC12 cells express enough endogenous ASCTs (Supplementary Figure 4A).
Figure 4.
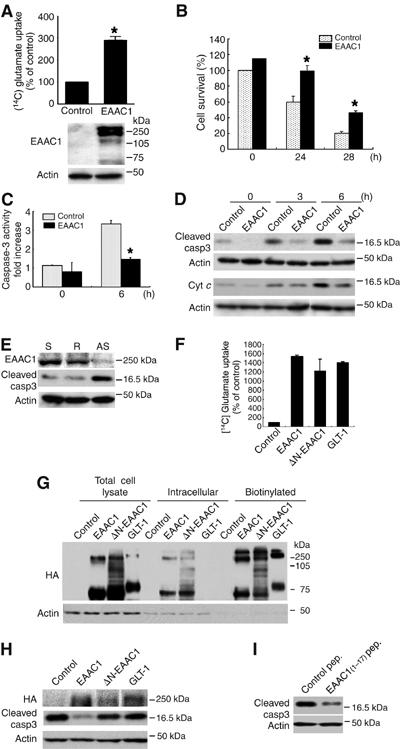
EAAC1 rescues differentiated PC12 cells from cell death after NGF deprivation. (A) Glutamate uptake activity and an immunoblot in control or EAAC1 adenovirus-infected differentiated PC12 cells. Uptake activity is expressed as a percentage of that in control cells (mean±s.d., P<0.01; Student's t-test). (B) Cell viability of differentiated PC12 cells infected with control (dotted bar) or EAAC1 (closed bar) adenoviruses was measured at 0, 24 and 48 h after NGF withdrawal (mean±s.d., P<0.01; Student's t-test). (C) Caspase-3-like activity of differentiated PC12 with control (dotted bar) or EAAC1 (closed bar) adenoviruses was measured at 0 and 9 h after NGF withdrawal. Data are expressed as fold increase compared with nontreated cells (mean±s.d., P<0.01; Student's t-test). (D) Immunoblots of cytoplasmic cleaved caspase-3 and cytochrome c following NGF withdrawal. (E) Immunoblots of suppression of endogenous EAAC1 expression by antisense oligonucleotide and the associated increase in caspase-3 activity. S, sense oligonucleotide; R, random oligonucleotide; AS, antisense oligonucleotide. (F) Glutamate uptake activity in COS-7 cells transfected with indicated plasmids tagged with HA. Data are mean±s.d. (G) Cell surface biotinylation assay using COS-7 cells transfected with the plasmids tagged with HA. (H) Immunoblot of cleaved caspase-3 in differentiated PC12 cells transfected with indicated plasmids tagged with HA. At 48 h after transfection, NGF was withdrawn for 6 h. (I) Immunoblot of cleaved caspase-3. Differentiated PC12 cells were deprived of NGF for 6 h after application of 17-amino-acid peptide (EAAC1 (1–17) peptide).
As the motor neuron death seen in adult mouse is mediated by the mitochondrial death pathway in a p53-dependent manner, it is likely that EAAC1 may act to suppress the mitochondrial death pathway either directly or indirectly. To investigate the possible involvement of EAAC1 in prevention of the mitochondria-mediated death pathway, we examined the effect of EAAC1 overexpression in differentiated PC12 neuronal cells on neuronal survival after NGF withdrawal, which is known to be accompanied by activation of the p53 pathway (Supplementary Figure 4C) (Vaghefi et al, 2004). After NGF withdrawal, EAAC1-expressing PC12 cells exhibited a significant increase in viability as compared to control cells and significantly less activation of caspase-3 (Figure 4B–D). This EAAC1 rescue activity was also observed after p53 adenovirus infection (Supplemental Figure 4D and E). Conversely, knockdown of endogenous EAAC1 expression in PC12 cells by treatment with EAAC1 antisense oligonucleotides significantly induced caspase-3 activity, even in NGF-containing culture medium (Figure 4E). In general, glutamate transporters have been considered to promote neuron survival activity by reducing extracellular/excitotoxic glutamate levels. However, our findings suggest that EAAC1 has an additional anti-apoptotic activity distinct from its ability to remove extracellular glutamate.
N-terminus of EAAC1 is required for survival
To examine whether EAAC1 indeed has another physiological function distinct from glutamate clearance during apoptotic stimuli, we carried out preliminary experiments using some deletion mutants and demonstrated an important role for the N-terminal domain of EAAC1. The function of the N-terminal domain, whose amino-acid sequence diverges most from those of other glutamate transporter family members, is obscure. N-terminal-deleted EAAC1 (ΔN-EAAC1)-expressing cells showed comparable glutamate uptake activity, although the activity was a little lower than that of wild-type EAAC1- and GLT-1-expressing cells, probably because of protein instability (Figure 4F). This result was consistent with protein levels expressed on the cell surface (Figure 4G).
In spite of the similar glutamate uptake activity, a striking difference was that the wild-type EAAC1 effectively inhibited caspase-3 activation after NGF deprivation in differentiated PC12 cells, whereas ΔN-EAAC1-expressing cells did not suppress caspase-3 activity (Figure 4H). GLT-1-expressing cells also failed to suppress caspase-3 activity, although they effectively took up glutamate (Figure 4F and G). When we introduced a 17-amino-acid sequence from the N-terminal domain of EAAC1 (peptide MGKPTSSGCDWRRFLRN) into PC12 cells, this peptide effectively inhibited caspase-3 activity as shown in Figure 4I. These results strongly suggest that the N-terminal domain of EAAC1 may play a crucial role in this ‘rescue' activity, and this ‘rescue' activity is distinct from its activity associated with glutamate clearance. It seemed highly likely that a molecule that might suppress caspase-3 activity either directly or indirectly may be associated with the N-terminus of EAAC1.
EAAC1 binds to a proapoptotic mitochondrial protein, HCCS
To identify possible proteins binding to the N-terminal region of EAAC1, we carried out yeast two-hybrid screening. Of the positive clones, a strongly interacting clone was found to encode HCCS (also known as CCHL or heme lyase) (Schaefer et al, 1996). HCCS is a mitochondrial protein, which converts apo-cytochrome c to holo-cytochrome c by attaching heme in the intermembrane space of mitochondria. First, we showed that endogenous HCCS co-immunoprecipitated with endogenous EAAC1 in differentiated PC12 cells, suggesting that the interaction is physiologically significant (Figure 5A). The next question was how the interaction between EAAC1 and HCCS occurred in spite of their different subcellular localizations. We speculated that HCCS might be released from mitochondria and promote cell death, because many mitochondrial proteins including Smac/DIABLO and Omi/HtrA2 are released into the cytoplasm under apoptotic stimuli and initiate the apoptotic cascade. Therefore, we examined the translocation of endogenous HCCS from the mitochondria into the cytoplasm after NGF deprivation of differentiated PC12 cells, which is the typical neuronal apoptotic paradigm (Figure 5B and C). Immunostaining showed that HCCS was clearly colocalized with cytochrome c in mitochondria in normal PC12 cells. However, after NGF withdrawal, HCCS staining had spread diffusely into the cytoplasm (Figure 5B). In addition, Western blotting using mitochondrial and cytosolic fractions showed that the amount of endogenous HCCS and cytochrome c immunoreactivity in the fractions containing mitochondria were decreased by 3 and 6 h after NGF deprivation (Figure 5C). In contrast, an increased level of HCCS was detected in the cytosolic fraction after NGF deprivation, suggesting that endogenous HCCS may be released from the mitochondria into the cytoplasm as in the classic model of neuronal apoptosis. Furthermore, overexpression of HCCS made the cells more susceptible to death signals such as NGF withdrawal (Figure 5D) and induced caspase-3 activation (Figure 5E). The caspase-3 activity was inhibited by additional expression of EAAC1. EAAC1 expression did not alter the level of HCCS protein (Supplementary Figure 6A).
Figure 5.

HCCS is released from mitochondria and binds to EAAC1. (A) Endogenous interaction between EAAC1 and HCCS in differentiated PC12 cells. Whole-cell lysates (WCL) from differentiated PC12 cells were lysed, subjected to immunoprecipitation (IP) with control IgG and anti-HCCS antibody and blotted (WB) with anti-EAAC1 and anti-HCCS antibodies. (B) Differentiated PC12 cells immunostained with anti-HCCS (red) and anti-cytochrome c (green) antibodies at 0 (NGF+) and 20 h after NGF withdrawal (NGF−). (C) Subcellular localization of endogenous HCCS in differentiated PC12 cells. After NGF deprivation, reduced levels of HCCS and cytochrome c were observed in the heavy membrane/mitochondrial fraction. Elevated levels of these proteins were observed in the cytosolic fraction. (D) Cell viability of differentiated PC12 cells transfected with control (dotted bar) or HCCS (closed bar) plasmids was measured at 0 and 36 h after NGF withdrawal (mean±s.d., asterisk denotes P<0.01; Student's t-test). (E) HCCS-mediated cleaved caspase-3 reaction in differentiated PC12 cells was reversed by cotransfection with EAAC1. The indicated expression vectors were overexpressed in differentiated PC12 cells for 48 h and thereafter collected at 0 (NGF+) and 6 h (NGF−) after NGF withdrawal.
Precise interaction between EAAC1 and HCCS
To verify more precisely the interaction between HCCS and EAAC1, we performed immunoprecipitation analysis using COS-7 cells. Initially, COS-7 cells were transiently transfected with cDNA encoding the C-terminal HA-tagged EAAC1 and GLT-1, together with FLAG-tagged HCCS (Figure 6A). HCCS immunoprecipitated wild-type EAAC1, but did not precipitate GLT-1. When COS-7 cells were transfected with C- and N-terminal HA-tagged EAAC1 together with FLAG-tagged HCCS, HCCS immunoprecipitated both forms of EAAC1 (Supplementary Figure 5). It seems that HCCS can interact with the full-length EAAC1, but not the N-terminus cleaved form of EAAC1. Next, we performed GST pull-down assays with in vitro-translated EAAC1 protein (Figure 6B). Bacterially produced GST-HCCS associated with wild-type EAAC1 protein, but failed to bind to ΔN-EAAC1. Furthermore, the N-terminal EAAC1 peptide competed with EAAC1 protein for binding to GST-HCCS in a dose-dependent manner (Figure 6C). To characterize the association between EAAC1 and HCCS further, we fragmented HCCS, attached FLAG tag and assayed the interaction using immunoprecipitation (Figure 6D). Wild-type EAAC1 strongly interacted with full-length HCCS encoding 272 amino acids and HCCS (148–272) lacking the heme-binding motif. In contrast, EAAC1 exhibited no detectable interaction with HCCS (1–152) lacking a mitochondria-targeting signal. These data showed that the EAAC1 N-terminus can bind to the C-terminal portion of HCCS.
Figure 6.

Precise interaction between EAAC1 and HCCS. (A) COS-7 cells were transfected with cDNA encoding HA-tagged EAAC1 (EAAC1-HA) or HA-tagged GLT-1 (GLT-1-HA) together with FLAG-tagged HCCS (FLAG-HCCS). At 36 h after transfection, cells were solubilized in RIPA buffer. Whole-cell lysates (WCL) from COS-7 cells were immunoprecipitated (IP) using anti-FLAG antibody, followed by Western blotting (WB) with anti-HA antibody. (B) GST pull-down assay with in vitro-translated wild-type and ΔN-EAAC1 protein. Top panel: Autoradiographic analysis showing the interaction between 35S-labeled EAAC1 and bacterially produced GST-tagged HCCS. Bottom panel: Coomassie blue-stained polyacrylamide gel (CBB) of GST fusion protein. (C) EAAC1 peptide competes with in vitro-translated EAAC1 protein for binding to GST-HCCS in a dose-dependent manner. Bound proteins were visualized by immunoblotting with anti-EAAC1 antibody. Bottom panel: Coomassie blue-stained polyacrylamide gel (CBB) of GST fusion protein. (D) COS-7 cells were transfected with EAAC1-HA and the indicated plasmids including deletion mutants of HCCS with attached FLAG tag. The EAAC1–HCCS interaction was examined by immunoprecipitation.
HCCS is a member of a new family of IAP binding proteins
We speculated that HCCS may have a role as IAP binding partner as has been established for Smac/DIABLO and Omi/HtrA2 and we looked at this possibility in more detail. Initially, we demonstrated that endogenous HCCS co-immunoprecipitated with endogenous XIAP in differentiated PC12 cells, suggesting that the interaction is indeed physiologically significant (Figure 7A). We then showed that GST-HCCS successfully interacted with the full-length 35S-labeled XIAP (Figure 7B). Next, expression plasmids for various N-terminal FLAG- or Myc-tagged IAPs, including XIAP, c-IAP1, c-IAP2 and survivin, were transiently transfected in COS-7 cells with a plasmid encoding the N-terminal HA-tagged HCCS. The HCCS was co-immunoprecipitated with XIAP, c-IAP1 and c-IAP2, but did not bind to survivin (Figure 7C). These results indicate that HCCS preferentially binds to some IAP members, such as XIAP, c-IAP1 and c-IAP2, with potent caspase-inhibitory activity.
Figure 7.

HCCS is a new member of IAP antagonists. (A) Endogenous interaction between HCCS and XIAP in differentiated PC12 cells. Whole-cell lysates (WCL) from differentiated PC12 cells were lysed, subjected to immunoprecipitation (IP) with control IgG and anti-HCCS antibody and blotted (WB) with anti-XIAP and anti-HCCS antibodies. (B) Top panel: Autoradiography showing the interaction between 35S-labeled XIAP and GST-tagged HCCS. Bottom panel: Coomassie blue-stained gel (CBB) showing GST and GST-HCCS protein. (C) The interaction of HCCS with IAP family proteins was determined by immunoprecipitation. COS-7 cells were transfected with indicated IAP plasmids with HA-tagged HCCS. (D) Immunoprecipitation using COS-7 cells transfected with mutated forms of HCCS and FLAG-XIAP. MTAV-HCCS or MSAS-HCCS: Alanine-9 or alanine-3 was changed to methionine. (E) COS-7 cells were transfected with FLAG-tagged XIAP together with HA-tagged HCCS truncated forms as indicated. The interaction was examined by immunoprecipitation. (F) Binding between HA-HCCS and fragmented XIAP. Cell lysates of COS-7 cells transfected with HA-HCCS were pulled down with GST-tagged recombinant XIAP proteins. Precipitated samples were subjected to immunoblot analysis using HA antibody. The input lane represents 1/50 of the COS-7 cell lysate used GST pull-down experiment. (G) The HCCS–XIAP or EAAC1–HCCS association was determined by immunoprecipitation. Increasing amounts of transfected EAAC1 or XIAP in COS-7 cells reduced the HCCS–XIAP or EAAC1–HCCS interaction in a dose-dependent manner.
The IAP binding proteins exhibit a common N-terminal sequence, which is required to mediate interaction with IAPs and accelerate cell death. HCCS has an amino-acid sequence containing ATAV or ASAS near the N-terminus, which appears homologous to the consensus sequence of other mammalian IAP antagonists. Therefore, COS-7 cells were transiently transfected with mutated forms of HCCS, in which alanine-9 or alanine-3 was changed to methionine. However, both mutant forms bound as effectively to XIAP as the wild type (Figure 7D). Further immunoprecipitation analysis revealed that the C-terminal portion of HCCS associated with XIAP (Figure 7E). To map the domain of XIAP responsible for HCCS binding, GST fusion proteins corresponding to XIAP fragments were generated. These experiments demonstrated that the BIR2 domain was sufficient for binding to HCCS, and neither BIR1 nor BIR3 bound to HCCS (Figure 7F). Collectively, HCCS may belong to another group of IAP binding proteins distinctly different from Smac/DIABLO and Omi/HtrA2. Furthermore, HCCS associated with XIAP through its C-terminal portion, which also bound to EAAC1. Increasing amounts of transfected EAAC1 or XIAP expression diminished XIAP–HCCS or EAAC1–HCCS co-immunoprecipitation respectively in a dose-dependent manner, suggesting that EAAC1 can compete with XIAP for HCCS (Figure 7G).
The EAAC1–HCCS and HCCS–XIAP interactions in the injured motor neurons
Finally, we carried out immunoprecipitation experiments using injured motor neurons from rats and mice. As the protein expression levels of IAP family members and their neutralizing proteins are not altered after axotomy (Supplementary Figure 6B), interactions between EAAC1/HCCS and XIAP/HCCS in injured neurons of rats and mice, respectively, would be possible. As expected, immunoprecipitation analysis showed that endogenous EAAC1 interacted with endogenous HCCS in injured motor neurons of the rat, but not in those of mice (Figure 8A). On the contrary, endogenous HCCS–XIAP interaction was observed in injured neurons of mice, but not in those of rats (Figure 8B). Collectively, these results suggest that decreased interaction of EAAC1 with HCCS might be responsible for the gradual progressive neuronal death in mice. The total amount of HCCS was unchanged before and after insult. However, immunohistochemistry showed that the intense HCCS staining in the mitochondria became fainter in the injured motor neurons of mice, suggesting that HCCS was being gradually released from mitochondria after axotomy (Supplementary Figure 6C). Interestingly, the expression level of HCCS was much lower in the motor neurons of rats than those of mice, suggesting that motor neurons in mice may be intrinsically more susceptible to nerve injury.
Figure 8.
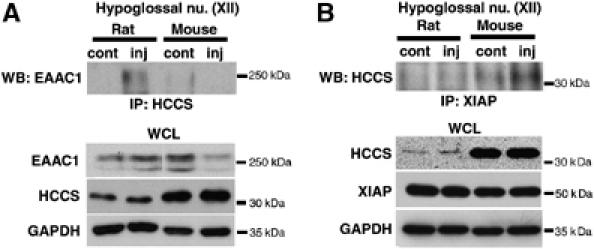
The EAAC1–HCCS and HCCS–XIAP interactions observed in axotomized hypoglossal neurons. (A) Co-immunoprecipitation was performed to assess the interaction between EAAC1 and HCCS in control (cont) and injured (inj) hypoglossal nuclei (XII) of rats and mice 5 days after axotomy. Proteins were precipitated (IP) by anti-HCCS antibody and analyzed on immunoblots (WB) with anti-EAAC1 antibody. Whole-cell lysates (WCL) from hypoglossal nuclei were immunoblotted with anti-GAPDH antibody as an internal control. (B) The HCCS–XIAP interaction was examined by immunoprecipitation using hypoglossal nuclei 5 days after axotomy.
Discussion
In this study, we have shown that EAAC1 is a mild but important survival factor whose expression is downregulated in injured motor neurons of mice, and we have identified a novel role for EAAC1 in preventing mitochondria-mediated neuronal death after nerve injury. For this anti-apoptotic function, the N-terminus of EAAC1 is pivotal, and this domain serves as an inhibitor of interaction between XIAP and its novel antagonistic protein HCCS.
It seemed that EAAC1 expression contributed to the survival of injured motor neurons. The question then arose how EAAC1 might play a role in preventing cell death. In general, the role of the glutamate transporter in the prevention of cell death is well documented, and the transporter-mediated removal of glutamate from the extracellular space would therefore be a very important mechanism for suppressing glutamate-induced neuronal death. However, the functional significance of EAAC1 in this role has remained unclear for the following reasons. The glutamate uptake activity of EAAC1 is relatively low, as compared with GLT-1 (Danbolt, 2001). EAAC1 is located mainly at extrasynaptic sites (He et al, 2000) and EAAC1 knockout mice do not show any apparent phenotypes (Peghini et al, 1997). In our study, it seemed that most established defensive mechanisms involved in resisting excitotoxicity were present in the injured neurons of both rats and mice. For example, glutamate receptors such as NMDAR1 were downregulated in both rats and mice in response to axotomy (data not shown). The expression of other glutamate transporter members (GLT-1 and GLAST) was also regulated in parallel in both rats and mice. Furthermore, EAAC1 was mainly observed in the peri-nuclear region, but not on the cell surface, in regenerating rat motor neurons as well as in neurons in kainate-induced epilepsy and oxidative stress (Supplementary Figure 6D) (Furuta et al, 2003; Yang et al, 2004).
Recently, Aoyama et al (2006) reported an alternative function for EAAC1 in that it prevented age-dependent neurodegeneration by working as a cysteine transporter to maintain intracellular glutathione homeostasis. In our model, the expression of the neutral amino-acid transporter ASCT1 was upregulated in axotomized neurons of rats and mice. In the in vitro experiments, EAAC1 overexpression in PC12 cells prevented mitochondria-mediated neuronal death after NGF withdrawal as well as after p53 adenovirus infection, without any change relative to controls in cysteine uptake (Supplementary Figure 4B). Furthermore, GLT-1 and ΔN-EAAC1 overexpression in PC12 cells failed to prevent cell death in the presence of increased glutamate uptake. The treatment of cells with the EAAC1 N-terminal peptide (17 amino acids) was however able to suppress caspase-3 activity downstream of the mitochondria. These findings imply that EAAC1 has a further additional role under stress conditions, distinct from its established role in glutamate removal and cysteine uptake.
In this study, we have identified HCCS, which resides in the intermembrane space of mitochondria, as an EAAC1 binding protein. Our work shows that HCCS can be released from mitochondria into the cytoplasm and can antagonize the action of XIAP during apoptotic stimuli. EAAC1 could readily interrupt the interaction between HCCS and XIAP and maintain XIAP free from HCCS (Figure 9). We thus speculated that HCCS could behave like other IAP binding proteins such as Smac/DIABLO and Omi/HtrA2, which can be released from mitochondria in response to apoptotic stress, interact with IAPs and accelerate cell death by antagonizing IAP activity (Du et al, 2000; Verhagen et al, 2000; Suzuki et al, 2001a; Danial and Korsmeyer, 2004). However, HCCS seemed to be different from these proteins, because HCCS interacted with only the second BIR domain (BIR2) of XIAP, which was sufficient to inhibit caspase-3 (Takahashi et al, 1998; Suzuki et al, 2001b), and because the interaction was through its C-terminus where previously identified consensus sequences for XIAP interaction are absent. Together with the fact that the mitochondrial targeting signal of HCCS is located at the C-terminus and does not undergo proteolytic processing after mitochondrial entry (Diekert et al, 1999), HCCS may be a new XIAP antagonist like ARTS and XAF (Liston et al, 2001; Gottfried et al, 2004) whose binding also occurs without the consensus sequences. Under normal conditions, HCCS is utilized for synthesis of functional cytochrome c, leading to ATP synthesis in mitochondria. Therefore, loss of this protein leads to severe cell damage, as found for other mitochondrial proteins (Prakash et al, 2002; Jones et al, 2003). Cytosolic HCCS may be rapidly degraded by a mechanism in which XIAP is involved. XIAP has already been established to function as an E3 ubiquitin ligase by regulating the levels of proteins such as MURR1, a protein implicated in copper homeostasis (Burstein et al, 2004). Further study is required to understand the exact physiological roles of HCCS.
Figure 9.

Proposed mechanism for EAAC1-mediated survival. Under stress stimuli, EAAC1 binds to HCCS, which is released into the cytosol from mitochondria. This complex is thought to interfere with the interaction between HCCS and IAP family members and facilitate IAP family proteins to suppress caspase-3 activity, leading to neuronal survival.
The anti-apoptotic activity of EAAC1 observed in vitro presumably also works in vivo as well: EAAC1 sequesters cytosolic HCCS and contributes to maintenance of XIAP free from HCCS (Figure 9). Therefore, lack of the functional interaction between EAAC1 and HCCS gradually decides the fate of the injured motor neurons in the adult mouse. This would provide one possible explanation why decreased expression of EAAC1 influences motor neuron death even though other transporter family members are present and presumably fully functional. Previous work demonstrated that XIAP is considered to be crucial for motor neuron survival (Perrelet et al, 2002, 2004; Inoue et al, 2003). Of the IAP family members, XIAP was the most highly expressed in our model (Supplementary Figure 5B). It seems reasonable then to suggest that EAAC1 contributes to XIAP maintenance after nerve injury.
It may therefore be concluded that EAAC1 has an important anti-apoptotic function, which is independent of removal of extracellular glutamate, and the lack of EAAC1 ‘rescue' activity leads to the gradual progressive motor neuron death seen in the mice. There remain some ambiguities in explaining the precise mechanism of this novel anti-apoptotic function of EAAC1 and how this correlates with glutamate uptake and cysteine uptake. EAAC1 function may change in response to cellular condition. One possibility is that EAAC1 by dint of its uptake activity may sense the increase of extracellular glutamate under stress conditions, which may trigger its ‘rescue' activity to prevent cell death. In addition, the relation between its intracellular trafficking and its novel anti-apoptotic function is also unclear. Most studies have shown that the C-terminal cytoplasmic region of EAAC1 is responsible for intracellular translocation of EAAC1 between the plasma membrane and non-plasma membrane pools such as the endosomes and ER to regulate its glutamate uptake activity (Lin et al, 2001; Cheng et al, 2002; Sheldon et al, 2006). It may be that cooperation with C-terminal binding proteins is necessary to produce enough activity for cell death prevention (Gonzalez and Robinson, 2004). Further studies are warranted to explain the precise mechanism of the novel anti-apoptotic function and it will be particularly interesting to determine whether the defective functioning of EAAC1 may contribute to the gradual progressive neuronal death seen in neurodegenerative disorders in humans.
Materials and methods
Animals
Adult male C57BL/6 mice (7 weeks old), ICR mice (7 weeks old) and Wistar rats (7 weeks old) were obtained from SLC (SLC, Japan). Homozygous EAAC1-deficient mice were purchased from Memorec stoffel gmbh (Koln, Germany). They had an inbred ICR genetic background or had been backcrossed at least 10 times. Age-matched ICR mice were used as controls. EAAC1 transgenic mice were backcrossed with C57BL/6 mice. Age-matched C57BL/6 mice were used as controls. Details of methods for transgenic mice, surgical procedures, histology and RT–PCR are provided in Supplementary data. Studies were performed in compliance with institutional guidelines for animal experiments.
Cell culture, plasmids and adenovirus preparation
Detailed methods are available in Supplementary data.
Glutamate uptake assay
Cells were plated on 24-well plates. After transfection for 48 h, cells were washed once with 0.2 ml transport buffer (125 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.5). The uptake assay was started by addition of 10 mM glutamate (0.1 mM [14C]glutamate inclusive; Amersham) to each well. After incubation for 6 min at 37°C, the solution was removed by suction. Cells were washed twice with 0.2 ml transport buffer and 0.4 ml of 10% SDS was added. Radioactivity was determined by scintillation counting.
Cell surface biotinylation
Details can be found in Supplementary data. The immunoblots in this assay showed that EAAC1 was detected as bands at 66 kDa and around 250 kDa, as glutamate transporters form homomultimers (Haugeto et al, 1996). However, other experiments in this study showed EAAC1 predominantly as the higher molecular weight bands probably because of the different lysis buffer and preparation technique employed. So in the other data EAAC1 is presented as the higher molecular weight bands.
Caspase assays
Detailed methods are available in Supplementary data.
Yeast two-hybrid screening
The MATCHMAKER two-hybrid system was used in accordance with the manufacturer's protocol (Clontech). The plasmid pAS2-1-EAAC1 was constructed by inserting three copies of the N-terminal region of EAAC1 (1–34 amino acids) in-frame with the GAL4 DNA-binding domain and transformed into AH109 cells. Bait-transformed AH109 cells were mated with Y187 cells pretransformed with a mouse brain cDNA library, which was constructed in pACT2 with GAL4 activation domain (Clontech). A total of 3.5 × 106 transformants were screened. Selected colonies were subjected to colony-lifting assays for β-galactosidase expression.
Production of HCCS monoclonal antibody
Detailed methods are available in Supplementary data.
Immunoblotting and immunostaining
Cells were washed with PBS twice. Total protein extracts were resuspended in lysis buffer (50 mM Pipes/NaOH (pH 6.5), 2 mM EDTA, 0.1% CHAPS and 5 mM DTT) containing 20 μg/ml leupeptin, 10 μg/ml pepstatin, 10 μg/ml aprotinin and 1 mM PMSF for cleaved caspase-3 detection, or in lysis buffer (20 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT and 250 mM sucrose) containing 0.1 mM PMSF, 10 μg/ml leupeptin and 10 μg/ml pepstatin for cytochrome c detection, centrifuged to collect the cytosolic fraction and immunoblotted according to the manufacturer's protocol. To examine subcellular localization of HCCS, proteins were extracted using lysis buffer (20 mM HEPES–KOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA and 1 mM DTT) containing 250 mM sucrose and protease inhibitor as described previously (Suzuki et al, 2001a). Primary antibodies included anti-actin (Sigma), anti-caspase-3 antibody recognizing the cleaved form (Cell Signaling Technology), anti-cytochrome c (Pharmingen), anti-EAAC1 (Chemicon), anti-FLAG (M2, Sigma) and anti-HA (3F10, Roche Diagnostics), which were diluted to 1:1000–1:5000.
For immunostaining, differentiated PC12 cells were grown on glass coverslips coated with polyethylenimine and collagen. Cells were fixed in 4% formaldehyde, permeabilized with 0.2% Triton X-100 and incubated with anti-HCCS and anti-holocytochrome c (Pharmingen) antibodies and subsequently a secondary antibody coupled to Alexa Green or Red (Molecular Probes).
Immunoprecipitation and GST pull-down assay
Detailed methods are available in Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Materials and Methods
Acknowledgments
We thank R Takahashi for providing various kinds of the IAP expression plasmids, PC Emson for critical reading and English correction of our manuscript and M Maeda for preparation of facial and sciatic nerve-injured animals. We are grateful to C Kadono, T Ogawa and I Jikihara for their excellent technical assistance, and E Fukui and T Kawai for secretarial assistance. This work was supported by grants from Ministry of Health, Labor and Welfare of Japan, MEXT, the General Insurance Association of Japan and Osaka City University Medical Research Foundation.
References
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA (2006) Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9: 119–126 [DOI] [PubMed] [Google Scholar]
- Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, Wijmenga C, Brewer GJ, Nabel GJ, Duckett CS (2004) A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J 23: 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA (2003) The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem 84: 1332–1339 [DOI] [PubMed] [Google Scholar]
- Cheng C, Glover G, Banker G, Amara SG (2002) A novel sorting motif in the glutamate transporter excitatory amino acid transporter 3 directs its targeting in Madin–Darby canine kidney cells and hippocampal neurons. J Neurosci 22: 10643–10652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1988) Glutamate neurotoxicity and disease of the nervous system. Neuron 8: 623–634 [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1–105 [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219 [DOI] [PubMed] [Google Scholar]
- Diekert K, Kispal G, Guiard B, Lill R (1999) An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc Natl Acad Sci USA 96: 11752–11757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42 [DOI] [PubMed] [Google Scholar]
- Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3: E255–E263 [DOI] [PubMed] [Google Scholar]
- Furuta A, Noda M, Suzuki SO, Goto Y, Kanahori Y, Rothstein JD, Iwaki T (2003) Translocation of glutamate transporter subtype excitatory amino acid carrier 1 protein in kainic acid-induced rat epilepsy. Am J Pathol 163: 779–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Martin LJ, Rothstein JD (1995) Regional deafferentation down-regulates subtypes of glutamate transporter proteins. J Neurochem 65: 2800–2803 [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Rothstein JD, Price DL, Martin LJ (1996) Fimbria–fornix transections selectively down-regulate subtypes of glutamate transporter and glutamate receptor proteins in septum and hippocampus. J Neurochem 67: 1208–1216 [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Robinson MB (2004) Neurotransmitter transporters: why dance with so many partners? Curr Opin Pharmacol 4: 30–35 [DOI] [PubMed] [Google Scholar]
- Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S (2004) The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J 23: 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC (1996) Brain glutamate transporter proteins form homomultimers. J Biol Chem 271: 27715–27722 [DOI] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH (2000) Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol 418: 255–269 [PubMed] [Google Scholar]
- Inoue H, Tsukita K, Iwasato T, Suzuki Y, Tomioka M, Tateno M, Nagao M, Kawata A, Saido TC, Miura M, Misawa H, Itohara S, Takahashi R (2003) The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J 22: 6665–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES (2003) Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425: 721–727 [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA (1992) Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360: 467–471 [DOI] [PubMed] [Google Scholar]
- Kiryu S, Yao GL, Morita N, Kato H, Kiyama H (1995) Nerve injury enhances rat neuronal glutamate transporter expression: identification by differential display PCR. J Neurosci 15: 7872–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Hirayama T, Kato R, Kiyama H (2005) Noxa is a critical mediator of p53-dependent motor neuron death after nerve injury in adult mouse. J Neurosci 25: 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Sasaki M, Yokohama H, Nakagomi S, Hirayama T, Aoki S, Wada K, Kiyama H (2000) Damage-induced neuronal endopeptidase (DINE) is a unique metallopeptidase expressed in response to neuronal damage and activates superoxide scavengers. Proc Natl Acad Sci USA 97: 4345–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens JC, Salin P, Nieoullon A, Kerkerian-Le Goff L (2001) Nigrostriatal denervation does not affect glutamate transporter mRNA expression but subsequent levodopa treatment selectively increases GLT1 mRNA and protein expression in the rat striatum. J Neurochem 79: 893–902 [DOI] [PubMed] [Google Scholar]
- Lin CLG, Orlov I, Ruggiero AM, Dyke-Hoberg M, Lee A, Jackson M, Rothstein J (2001) Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3–18. Nature 410: 84–88 [DOI] [PubMed] [Google Scholar]
- Liston P, Fong WG, Kelly NL, Toji S, Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW, Korneluk RG (2001) Identification of XAF1 as an antagonist of XIAP anti-caspase activity. Nat Cell Biol 3: 128–133 [DOI] [PubMed] [Google Scholar]
- Martin LJ, Chen K, Liu Z (2005) Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci 25: 6449–6459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z (2002) Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J Neurobiol 50: 181–197 [DOI] [PubMed] [Google Scholar]
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J (2004) Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol 24: 9848–9862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peghini P, Janzen J, Stoffel W (1997) Glutamate transporter EAAC1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J 16: 3822–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrelet D, Ferri A, Liston P, Muzzin P, Korneluk RG, Kato AC (2002) IAPs are essential for GDNF-mediated neuroprotective effects in injured motor neurons in vivo. Nat Cell Biol 4: 175–179 [DOI] [PubMed] [Google Scholar]
- Perrelet D, Perrin FE, Liston P, Korneluk RG, MacKenzie A, Ferrer-Alcon M, Kato AC (2004) Motoneuron resistance to apoptotic cell death in vivo correlates with the ratio between X-linked inhibitor of apoptosis proteins (XIAPs) and its inhibitor, XIAP-associated factor 1. J Neurosci 24: 3777–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettmann B, Henderson CE (1998) Neuronal cell death. Neuron 20: 633–647 [DOI] [PubMed] [Google Scholar]
- Prakash SK, Cormier TA, McCall AE, Garcia JJ, Sierra R, Haupt B, Zoghbi HY, Van Den Veyver IB (2002) Loss of holocytochrome c-type synthetase causes the male lethality of X-linked dominant microphthalmia with linear skin defects (MLS) syndrome. Hum Mol Genet 11: 3237–3248 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686 [DOI] [PubMed] [Google Scholar]
- Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23: 2861–2874 [DOI] [PubMed] [Google Scholar]
- Schaefer L, Ballabio A, Zoghbi HY (1996) Cloning and characterization of a putative human holocytochrome c-type synthetase gene (HCCS) isolated from the critical region for microphthalmia with linear skin defects (MLS). Genomics 34: 166–172 [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Gonzalez MI, Robinson MB (2006) A carboxyl-terminal determinant of the neuronal glutamate transporter, EAAC1, is required for platelet-derived growth factor-dependent trafficking. J Biol Chem 281: 4876–4886 [DOI] [PubMed] [Google Scholar]
- Snider WD, Elliott JL, Yan Q (1992) Axotomy-induced neuronal death during development. J Neurobiol 23: 1231–1246 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R (2001a) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 8: 613–621 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Nakata K, Reed JC, Takahashi R (2001b) X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J Biol Chem 276: 27058–27063 [DOI] [PubMed] [Google Scholar]
- Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC (1998) A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 273: 7787–7790 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699–1702 [DOI] [PubMed] [Google Scholar]
- Ugolini G, Raoul C, Ferri A, Haenggeli C, Yamamoto Y, Salaun D, Henderson CE, Kato AC, Pettmann B, Hueber AO (2003) Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J Neurosci 23: 8526–8531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaghefi H, Hughes AL, Neet KE (2004) Nerve growth factor withdrawal-mediated apoptosis in naive and differentiated PC12 cells through p53/caspase-3-dependent and -independent pathways. J Biol Chem 279: 15604–15614 [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J (2003) HtrA2/Omi, a sheep in wolf's clothing. Cell 115: 251–253 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53 [DOI] [PubMed] [Google Scholar]
- Yang Y, Kinney GA, Spain WJ, Breitner JC, Cook DG (2004) Presenilin-1 and intracellular calcium stores regulate neuronal glutamate uptake. J Neurochem 88: 1361–1372 [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP (1996) Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol 493 (Part 2): 419–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Materials and Methods