

Abstract
Regulation of transcription requires mechanisms to both activate and terminate transcription factor activity. GATA-1 is a key haemopoietic transcription factor whose activity is increased by acetylation. We show here that acetylated GATA-1 is targeted for degradation via the ubiquitin/proteasome pathway. Acetylation positively signals ubiquitination, suggesting that activation by acetylation simultaneously marks GATA-1 for degradation. Promoter-specific MAPK phosphorylation then cooperates with acetylation to execute protein loss. The requirement for both modifications is novel and suggests a way by which degradation of the active protein can be specifically regulated in response to external phosphorylation-mediated signalling. As many transcription factors are activated by acetylation, we suggest that this might be a general mechanism to control transcription factor activity.
Keywords: acetylation, haemopoiesis, phosphorylation, transcription, ubiquitination
Introduction
Many studies investigating the regulation of gene transcription have focused on how transcription is initiated. However, equally important to turning a gene on is the ability to turn transcription off. Indeed, such terminating events are essential to control the level of transcription and thus the amount of gene product generated.
Recently, the ubiquitin/proteasome pathway has been implicated in the destruction of active transcription factors. This limits the half-life of the factor at promoters and thus the level of transcription. Circumstantial evidence for this includes the finding that activation and degradation domains of highly active transcription factors overlap (Thomas and Tyers, 2000) and the presence of components of the 19S proteasome and an E3 ubiquitin protein ligase at promoters (Gonzalez et al, 2002; von der Lehr et al, 2003). More direct evidence came from the finding that phosphorylation of GCN4 by the basal transcription machinery specifically triggers ubiquitination and loss of the promoter-bound factor (Chi et al, 2001). Similarly, liganded and unliganded (inactive) oestrogen receptors are removed from promoters in a cyclical manner via ubiquitin-mediated degradation (Reid et al, 2003).
As removal of transcription factors is likely to play a major role in the regulation of gene activity, an important question is how this degradation is controlled. To date, nothing is known about this regulation. The promoter-specific modification and ubiquitination of GCN4 implies that its degradation is regulated solely by its recruitment to promoters. However, if this is the case, the degradation will not confer any regulation on the level of gene transcription. Instead, it would be advantageous if external signals could regulate transcription factor degradation to thus allow control of transcription in response to stimuli such as the growth rate or cell cycle. Phosphorylation is known to signal the loss of the melanocyte transcription factor, Mi (Wu et al, 2000), but this signalling does not necessarily distinguish between transcriptionally active and inactive protein. Therefore, in addition to external signals, it would be advantageous to have a mechanism to flag the active form of the transcription factor for degradation. As a number of factors are activated by phosphorylation or acetylation, one way to achieve this would be to specifically signal destruction of these modified forms.
We have investigated how transcription factor degradation is regulated using the haemopoietic transcription factor, GATA-1, as a model. GATA-1 activity and levels must be tightly regulated for proper haemopoietic differentiation (Orkin, 2000). For example, experiments in avian myeloblasts demonstrated that the level of GATA-1 activity directly determines the differentiation pathway adopted (Kulessa et al, 1995). Similarly, transgenic experiments have shown that overexpression of GATA-1 normally causes embryonic lethality (Whyatt et al, 2000). One possible explanation for the toxicity of high GATA-1 levels is that a number of GATA-1 target genes are involved in cell cycle regulation (Rylski et al, 2003). Consequently, unless both their initiation and level of transcription are tightly controlled, adverse effects on cell division are likely. Moreover, as GATA-1 auto-regulates its own promoter (Tsai et al, 1991), GATA-1 activity must be strictly regulated to prevent continuous accumulation of GATA-1 in the cell.
An important way by which GATA-1 activity is regulated is by acetylation. This stimulates GATA-1/DNA binding and enhances transcription from a GATA-1-dependent promoter (Boyes et al, 1998). Significantly, acetylation is essential for GATA-1 function during haemopoiesis: mutation of the main sites of acetylation eliminated the ability of GATA-1 to cause erythroid differentiation of GATA-1-deficient G1E cells (Hung et al, 1999). Furthermore, similar mutations considerably reduced differentiation of 416B cells to megakaryocytes (Visvader et al, 1995).
We show here that acetylated GATA-1 is targeted for degradation by the ubiquitin–proteasome pathway. As acetylation activates GATA-1 and also signals its ubiquitination, we suggest that transcription factor activation and degradation are directly linked. We show further that acetylation alone is not enough to cause degradation but rather additional modification by phosphorylation is needed. The requirement for both modifications means that the acetylated, active protein can remain at promoters until its degradation is triggered in response to phosphorylation-mediated signalling. Together, our data suggest a way by which GATA-1 activity can be specifically and continually regulated in response to external cytokine signals to thus control GATA-1-dependent gene expression during haemopoiesis.
Results
Phosphorylation and acetylation affect GATA-1 levels
To investigate how GATA-1 activity is regulated, we transfected expression vectors for wild-type GATA-1 and proteins where the sites of acetylation and phosphorylation had been mutated into Cos 7 and 293T cells. Seven serines have been identified as major sites of GATA-1 phosphorylation (Crossley and Orkin, 1994). In addition, four major sites of acetylation (K245, K246, K252 and K312) and seven more minor sites (K233, K287, K299, K308, K314, K315 and K316) have been mapped (Boyes et al, 1998; Hung et al, 1999). Mutation of these acetylation sites or of all the sites of phosphorylation resulted in considerably higher levels of protein compared to wild-type GATA-1 in transfected Cos 7 and 293T cells (Figure 1A and Supplementary Figure 1A). Similarly, in a haemopoietic cell line (BM-SCF; Cairns et al, 2003), the levels of the FLAG-tagged mutant GATA-1 proteins were higher than FLAG-tagged wild-type protein (Figure 1B). RNase protection analysis showed that expression of each of the GATA-1 constructs is equivalent (Figure 1A and B). These data therefore suggest that acetylation and phosphorylation regulate GATA-1 levels by a post-transcriptional mechanism.
Figure 1.
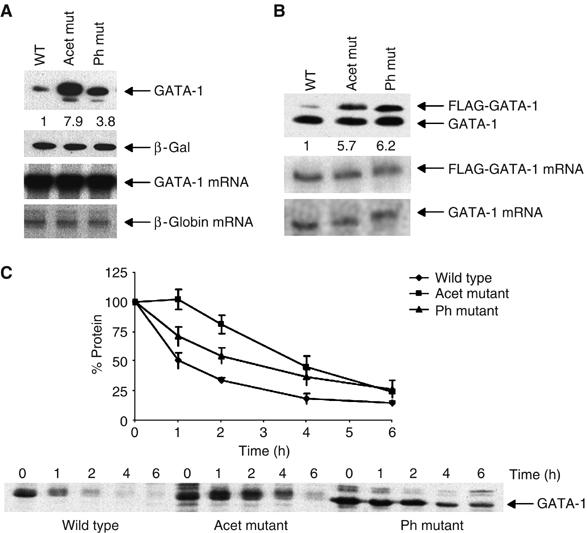
Mutation of phosphorylation and acetylation sites increases GATA-1 stability. (A) Upper panels: Western blot showing GATA-1 levels in Cos 7 cells, transfected with constructs to express wild-type GATA-1 (Wt), phosphorylation (Ph) or acetylation (Acet) mutants. β-Galactosidase is the cotransfection control. Lower panels: RNase protection assay of the corresponding GATA-1 mRNAs; the human β-globin gene is the cotransfection control. (B) Upper panel: Western blot showing the levels of endogenous and FLAG-tagged GATA-1 protein in BM-SCF cells stably infected with retroviruses to express FLAG-tagged versions of GATA-1. Lower panels: RNase protection assay of the FLAG-tagged and endogenous GATA-1 mRNAs. (C) Pulse–chase experiment to determine the half-life of GATA-1. The levels of wild type (⧫), acetylation (Acet ▪) and phosphorylation (Ph ▴) mutants in Cos 7 cells are shown. A value of 100% was given to the amount of protein at time zero; the per cent of protein remaining was calculated relative to this. The plot shows the average of three experiments; error bars show standard error.
To determine if these modifications affect protein turnover, we examined the half-lives of the wild-type and mutant proteins in Cos 7 cells. The proteins lacking either acetylation or phosphorylation sites had a notably longer half-life than the wild-type protein (Figure 1C), demonstrating that these modifications do indeed affect GATA-1 turnover.
Phosphorylation and acetylation affect GATA-1 ubiquitination
Phosphorylation and acetylation have been implicated in the regulation of protein degradation via the ubiquitin/proteasome pathway (Kouzarides, 2000; Glickman and Ciechanover, 2002). To test if GATA-1 is degraded via this route, we added specific proteasome inhibitors to cells expressing GATA-1. As some caspases are degraded by the ubiquitin/proteasome pathway and GATA-1 is a caspase target (De Maria et al, 1999), we used GATA-1 mutated in a caspase cleavage site in this experiment (Supplementary Figure 1B). We found that all of the inhibitors increase GATA-1 levels (Figure 2A), suggesting that GATA-1 is degraded via the ubiquitin/proteasome pathway.
Figure 2.
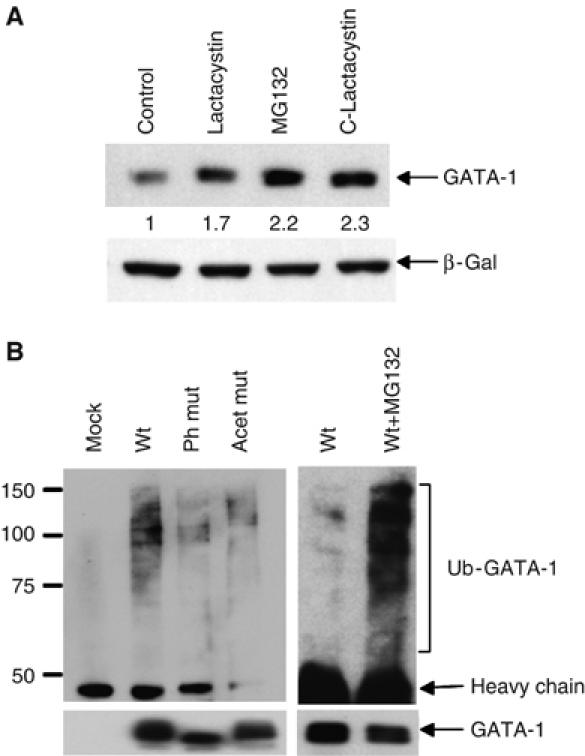
GATA-1 is ubiquitinated. (A) Proteasome inhibitors increase GATA-1 stability. Transfected Cos 7 cells were treated with the proteasome inhibitors shown. Protein levels were analysed by immunoblotting with N6 antibody; β-galactosidase is the cotransfection control. (B) Phosphorylation and acetylation affect GATA-1 ubiquitination. Western blot showing the level of ubiquitination of GATA-1 immunoprecipitated from transfected 293T cells. Normalised levels of GATA-1 were loaded; this is verified in the lower panel where the blot is re-probed with a second anti-GATA-1 antibody; densitometry confirmed GATA-1 ratios as wt 1:Ph mut 0.8:Acet mut 0.95. Lanes 5 and 6 are a short exposure to highlight the difference in GATA-1 ubiquitination ±MG-132. Mock-transfected cells confirm that ubiquitinated proteins do not stick nonspecifically during immunoprecipitation.
To further examine if GATA-1 is ubiquitinated, we used antibodies that detect both monoubiquitin and polyubiquitin chains. Following immunoprecipitation of GATA-1 from transfected 293T cells, higher molecular weight bands corresponding to ubiquitinated GATA-1 were detected in extracts from GATA-1-transfected cells but not from mock-transfected cells (Figure 2B, lanes 1 and 2). Moreover, the proteasome inhibitor, MG-132, increased the amount of ubiquitinated GATA-1 (lanes 5 and 6).
Next, we examined if phosphorylation and acetylation affect GATA-1 ubiquitination by immunoprecipitating wild-type or mutant proteins from transfected 293T cells. To compare the levels of ubiquitination accurately, an equivalent amount of immunoprecipitated GATA-1 was loaded and ubiquitination was analysed as above. Wild-type GATA-1 was consistently found to be more ubiquitinated than either the phosphorylation or acetylation mutant proteins (Figures 2B and 4B). This implies that acetylation and phosphorylation regulate GATA-1 ubiquitination and can account for the altered GATA-1 levels.
Acetylation causes GATA-1 ubiquitination
As acetylation of GATA-1 increases its transcriptional activity and is essential for GATA-1 function during haemopoiesis (Hung et al, 1999), we were keen to determine how acetylation affects GATA-1 degradation. Acetylation could regulate GATA-1 levels in two opposing ways. Lysines are targets for both acetylation and ubiquitination. Therefore, acetylation can stabilise proteins by preventing subsequent ubiquitination (Kouzarides, 2000) as demonstrated for both p53 and Smad7 (Gronroos et al, 2002; Li et al, 2002). However, the opposite possibility also exists: acetylation at some lysines might positively signal ubiquitination at other lysines to thus cause protein loss. To differentiate between these possibilities, we increased acetylation of GATA-1 by using the deacetylase inhibitor trichostatin A (TSA).
Treatment with TSA substantially decreased GATA-1 levels in transfected Cos 7 cells (Figure 3A, upper panel and Supplementary Figure 2A). This suggested that acetylation of GATA-1 triggers its ubiquitination. To test this further, we performed the same experiment but prevented proteasome-mediated degradation by adding the proteasome inhibitor MG-132. This stabilised total GATA-1 levels (Figure 3A, lowest panel) and, as would be expected if acetylation causes ubiquitination, TSA treatment significantly increased the amount of ubiquitinated GATA-1. Likewise, when we examined the changes in acetylated GATA-1 using an antibody that specifically detects acetylated K312 (a major site of GATA-1 acetylation; Supplementary Figure 2B; Boyes et al, 1998; Hung et al, 1999), we found that TSA treatment markedly increased acetylated GATA-1 (Figure 3A, lower panel) concomitant with the increase in GATA-1 ubiquitination. These experiments therefore suggest that increases in GATA-1 acetylation lead to its ubiquitination and degradation. In support of this, we found that coexpression of the acetyltransferase, p300, which is known to modify and increase GATA-1 activity, caused a decrease in GATA-1 levels in transfected NIH 3T3 cells (Figure 3B); p300 mutated in the acetyltransferase domain did not affect GATA-1 levels.
Figure 3.
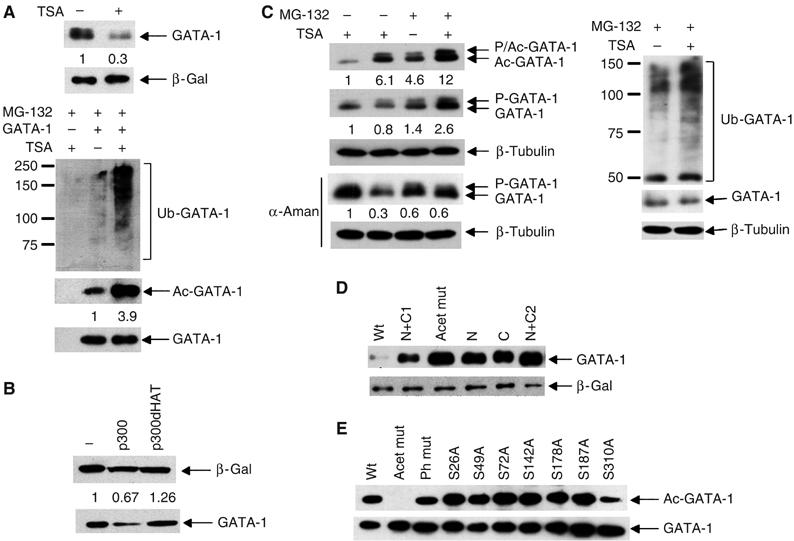
Acetylation of GATA-1 causes ubiquitination. (A) Treatment with TSA increases acetylated and ubiquitinated GATA-1. Upper panel: Western blot showing GATA-1 levels in whole-cell extract from Cos 7 cells that had been transfected with GATA-1 144D (Supplementary Figure 1B) and β-galactosidase with (+) or without (−) TSA treatment. Lower panel: Western blot showing the levels of ubiquitinated GATA-1 (Ub-GATA-1), acetylated GATA-1 (Ac-GATA-1) and total GATA-1 following immunoprecipitation from transfected Cos 7 cells treated with MG-132 with (+) or without (−) TSA. (B) p300 but not p300dHAT decreases GATA-1 levels. NIH3T3 cells were transfected with expression vectors for GATA-1 and p300 or its acetyltransferase mutant, p300dHAT. Whole-cell extracts were used in a Western blot with anti-GATA-1 antibody. β-Galactosidase is the cotransfection control. (C) The decrease in GATA-1 levels upon TSA treatment in haemopoietic (BM-SCF) cells is reversed by MG-132. As for part A except that endogenous GATA-1 was examined. Upper panel: Detection of acetylated GATA-1; second panel: detection of total GATA-1. The right panel shows ubiquitinated and total GATA-1 in the presence of MG-132. Phosphorylated total GATA-1 is indicated by P-GATA-1 and phosphorylated, acetylated GATA-1 by P/Ac-GATA-1. Lambda phosphatase confirmed that the slower mobility form is phosphorylated GATA-1 (Supplementary Figure 2E). Total GATA-1 levels decrease more in Cos 7 cells (A, upper panel) than BM-SCF cells (C, second panel) upon TSA treatment. GATA-1 regulates its own promoter in haemopoietic cells but is controlled by the EF1α promoter in Cos 7 cells. To prevent TSA increasing GATA-1 levels in BM-SCF cells via GATA-1 autoregulation, we added the transcription inhibitor α-amanitin (α-aman). A more substantial decrease is now seen (fourth panel). An anti-β-tubulin Western confirms that equivalent cell numbers were used (lower panel). (D) All acetylation mutants are more stable than the wild-type protein. Groups of acetylation sites were mutated (K to R). N: K245, K246 and K252; C: K308, K312 and K316; N+C1: K245, K246, K252, K312, K314 and K316; N+C2: all N and C mutations. Acet mut is that used in other experiments. The level of GATA-1 protein was determined by Western blotting of whole-cell extracts from transfected 293T cells. β-Galactosidase is the cotransfection control. Mutation of two different sets of three lysines (that are substrates for acetylation; Boyes et al, 1998) resulted in a similar increase in GATA-1 stability, suggesting that it is unlikely that loss of ubiquitination sites caused the increased stability. (E) The phosphorylation mutants are acetylated. GATA-1 phosphorylation mutant proteins (serine to alanine) were immunoprecipitated from transfected 293T cells; a Western blot probed with anti-acetyl-GATA-1 antibody is shown. Equivalent amounts of immunoprecipitated GATA-1 were loaded; this was confirmed by re-probing with a second anti-GATA-1 antibody (lower panel). The anti-acetyl-GATA-1 antibody was raised against amino acids 307–317; the lower signal for S310A and total phosphorylation mutant is probably because some antibodies in the polyclonal mix require S310 to be wild type. A lower signal was not seen with an antibody raised against all acetylated lysines (Supplementary Figure 2C).
We next examined the effects of TSA treatment on endogenous GATA-1 in BM-SCF cells. Consistent with the data in Cos 7 cells, we found that TSA treatment destabilised GATA-1 (Figure 3C, second and fourth panels); in the presence of MG-132, this decrease in GATA-1 levels was prevented and TSA treatment resulted in increased ubiquitination (Figure 3C, right). Importantly, we also found that the levels of acetylated GATA-1 are increased following addition of MG-132 (Figure 3C, upper panel); this implies that acetylated GATA-1 is preferentially degraded. These data are therefore fully consistent with the model that acetylation of GATA-1 positively signals its ubiquitination and leads to its own destruction.
Acetylation is associated with increased GATA-1 activity (Boyes et al, 1998; Hung et al, 1999). Therefore, these data raise the possibility that activation and degradation are linked. Indeed, we found that mutation of two separate sets of lysines ((K245, K246, K252) or (K308, K312, K316)), which cover major acetylation sites, substantially increased GATA-1 stability (Figure 3D). As very similar mutations reduced GATA-1 activity in haemopoietic cells (Hung et al, 1999), these data support the hypothesis that acetylation directly links GATA-1 activation and degradation.
Multiple phosphorylation events are required for GATA-1 ubiquitination
To begin to establish how the degradation of acetylated GATA-1 is regulated, we next asked if phosphorylation is required for acetylation of GATA-1. However, mutation of any single site of phosphorylation did not affect GATA-1 acetylation (Figure 3E and Supplementary Figure 2C). Likewise, GATA-1 that is mutated in all acetylation sites is efficiently phosphorylated (Supplementary Figure 2D and E). Thus, phosphorylation or acetylation of GATA-1 occurs independently of the other modification.
Phosphorylation, nevertheless, is required for GATA-1 ubiquitination (Figure 2B). As this is a potential means of signalling changes in GATA-1 activity, we investigated which of the seven phosphorylation sites regulate GATA-1 ubiquitination. Surprisingly, mutation of every individual serine to alanine generated a protein that was more stable than the wild-type protein (Figure 4A); this suggests that phosphorylation of all seven serines is required for efficient GATA-1 degradation. Consistent with this, all of the mutant proteins showed reduced ubiquitination (Figure 4B). This confirms that all sites of phosphorylation are required for efficient GATA-1 ubiquitination and further suggests that degradation of GATA-1 is very tightly regulated.
Figure 4.

Phosphorylation and acetylation cooperate to cause the degradation of acetylated GATA-1. (A) Each individual phosphorylation mutant is more stable than wild-type protein. Western blot showing GATA-1 phosphorylation mutant proteins immunoprecipitated from transfected Cos 7 cells. Transfection efficiency was normalised to β-galactosidase. (B) Each phosphorylation mutant is less ubiquitinated than wild-type protein. The levels of ubiquitination were determined via immunoprecipitation from transfected 293T cells as described in Figure 2. The same Western blot was re-probed with anti-GATA-1 antibody (lower panel) and confirms that protein is present in all lanes. Higher ubiquitination of the total phosphorylation mutant and S26A (which is similar between the two proteins but notably less than wild type) correlates with their lower protein levels (Figure 4A). (C) Phosphorylation is needed for efficient degradation of acetylated GATA-1. Western blot showing GATA-1 levels in extracts from BM-SCF cells that stably express GATA-1 mutated in serine 26 (Flag-26A). Endogenous GATA-1 in the same cell provides an internal control for degradation upon TSA treatment. β-Tubulin levels verify that equivalent cell numbers were taken (lower panel). (D) MEK inhibitor prevents degradation of acetylated GATA-1. Haemopoietic (BM-SCF) cells were treated with or without TSA in the presence or absence of the MEK inhibitor U0126. Acetylated GATA-1 (Ac-GATA-1; upper panel) and total GATA-1 (GATA-1; lower panel) were detected as in Figure 3. The graph shows quantification of the level of acetylated GATA-1 divided by the amount of total GATA-1 in two further independent experiments where a higher level of acetylated GATA-1 in untreated cells was observed.
Phosphorylation and acetylation cooperate to degrade GATA-1
As both acetylation and phosphorylation induce GATA-1 ubiquitination, we next wanted to determine whether these modifications function independently to cause degradation of distinct pools of GATA-1 or, alternatively, whether both modifications work together. In the latter case, acetylation could flag active GATA-1 for degradation but signalling by phosphorylation would be required to ultimately cause GATA-1 loss. The fact that phosphorylation mutants are poorly ubiquitinated (Figure 4B) but are still efficiently acetylated (Figure 3E) implies that acetylation alone cannot cause substantial ubiquitination. To test whether acetylation and phosphorylation cooperate, and begin to gain a mechanistic insight into the degradation process, we examined whether TSA treatment could cause the degradation of GATA-1 proteins in the absence of phosphorylation. To this end, we used a haemopoietic cell line (BM-SCF) that stably expresses a FLAG-tagged GATA-1 phosphorylation mutant protein (FLAG-26A). Addition of TSA caused only a small decrease in the phosphorylation mutant protein, whereas the endogenous GATA-1 in the same cells was substantially decreased (Figure 4C). Similar results were observed with GATA-1 proteins mutated at serines 142 and 178 (data not shown). These data are therefore consistent with the idea that phosphorylation and acetylation cooperate to mediate GATA-1 degradation. In support of this, addition of the specific MAP kinase inhibitor U0126, which inhibits phosphorylation of GATA-1 at serines 26 and 178 (and growth factor-mediated signalling; Towatari et al, 2004), eliminated the decrease in GATA-1 levels upon TSA treatment (Figure 4D, middle panel, lanes 3 and 4). Furthermore, as would be predicted if phosphorylation causes the degradation of acetylated GATA-1, inhibition of phosphorylation by U0126 caused the preferential stabilisation of acetylated GATA-1 (Figure 4D, upper panel and graph).
Degradation of transcriptionally active GATA-1
As acetylation correlates with GATA-1 transcriptional activity, we next wished to determine if phosphorylation indeed signals the loss of the active protein. If this were the case, we would predict that inhibition of phosphorylation will prevent degradation of acetylated GATA-1 and transcription will be increased. We therefore added MEK inhibitors to transiently transfected cells and analysed the effects on GATA-1-dependent transcription. As can be seen in Figure 5A, MEK inhibitors cause a 10-fold increase in GATA-1 dependent transcription. This effect is specific to GATA-1 that can be acetylated as MEK inhibitors had no effect on transcription by the GATA-1 acetylation mutant. Similarly, no effect was observed with GATA-1 that could not be phosphorylated. These data therefore suggest that phosphorylation signals acetylated, active GATA-1 for degradation.
Figure 5.
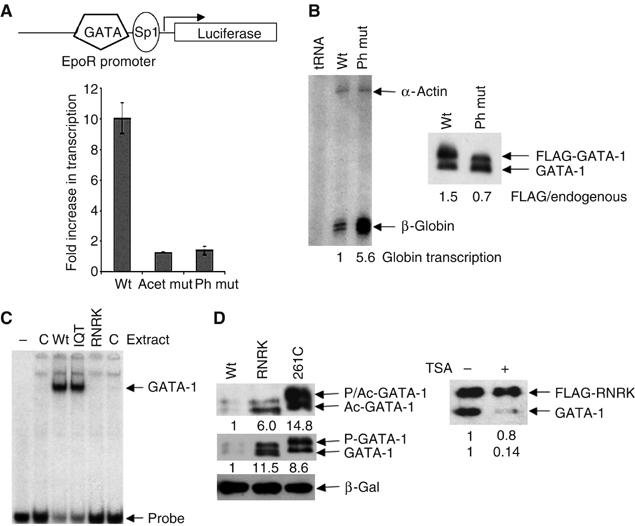
Preferential degradation of transcriptionally active GATA-1. (A) Upper: The reporter construct pGL3EpoR. Lower: Inhibition of phosphorylation increases GATA-1-dependent transcription. NIH3T3 cells, transfected with the constructs shown, were treated with or without the MEK inhibitor U0126. The transcription level in the presence of U0126 divided by that in its absence is plotted. The average of four experiments is shown. (B) The phosphorylation mutant is more transcriptionally active than wild-type GATA-1. Left panel: S1 mapping of β-globin gene transcription in BM-SCF cells that express FLAG-tagged wild-type (wt) or phosphorylation mutant (ph mut) GATA-1. Quantification of β-globin transcription, normalised to α-actin, is shown beneath the gel. Right panel: Western blot showing the level of the FLAG-tagged wild-type or phosphorylation mutant protein and endogenous GATA-1. For this experiment, cell lines were chosen where mutant and wild-type GATA-1 are expressed at similar levels. (C) The RNRK mutant does not bind DNA. Equivalent amounts of whole-cell extract prepared from mock-transfected Cos 7 cells (C) or cells that had been transfected with expression vectors for wild-type (wt) or mutant GATA-1 proteins (IQT and RNRK) were used in a gel mobility shift assay under the conditions described (Boyes et al, 1998). Control experiments confirmed that transfection efficiency was equivalent. (D) The DNA binding mutants are more stable than wild-type GATA-1. Left panel: Western blot of extracts from Cos 7 cells transfected with wild-type GATA-1 and the DNA binding mutants, C261P and RNRK, probed with anti-GATA-1 antibody or anti-acetyl-GATA-1 antibody. β-Galactosidase is the cotransfection control. Right panel: Haemopoietic (BM-SCF) cells, expressing FLAG-tagged RNRK GATA-1, were treated with or without TSA. A Western blot probed with anti-GATA-1 antibody (N-6) is shown.
Next, we examined the activity of GATA-1 proteins mutated in the sites of phosphorylation. As these proteins are acetylated (Figure 3E) but are not degraded by phosphorylation-mediated signalling, we expected that they would be more transcriptionally active than the wild-type protein. Indeed, transcription of a physiologically relevant gene, β-globin, is more than five-fold higher in haemopoietic BM-SCF cells that stably express the phosphorylation mutant compared to wild-type GATA-1 (Figure 5B). Similarly, the phosphorylation mutant is also more active in transfected 293T cells when transcription at similar levels of protein is compared (Supplementary Figure 3A).
DNA-bound GATA-1 is preferentially degraded
These data demonstrate that phosphorylation signals transcriptionally active GATA-1 for degradation but they do not show if it is only the active protein that is lost. To investigate this, we looked at the stability of transcriptionally inert proteins by making use of the fact that GATA-1 activity requires GATA-1/DNA binding (Martin and Orkin, 1990). Two different GATA-1 DNA binding mutants were expressed in Cos 7 cells: C261P (Martin and Orkin, 1990), where the C-terminal zinc-finger is destroyed, and RNRK, which disrupts DNA contacts in the C-terminal basic region (Figure 5C). For both mutations, lack of DNA binding (and lack of GATA-1-dependent transcription; Supplementary Figure 3B) correlates with increased protein stability (Figure 5D and Supplementary Figure 3C). Together, these data suggest that GATA-1 that is competent to bind DNA is preferentially targeted for degradation.
DNA binding is required for MAPK-dependent phosphorylation of GATA-1
We noted that these DNA binding mutant proteins are both acetylated and phosphorylated (Figure 5D). To further test if GATA-1 degradation requires DNA binding in addition to acetylation and phosphorylation, we treated cell lines that stably express a FLAG-tagged version of the RNRK mutant with TSA. Figure 5D (right) shows that the FLAG-tagged RNRK mutant is markedly less degraded than wild-type GATA-1 present in the same cell. This indicates that there is additional regulation that restricts degradation only to the DNA-bound protein. Two explanations seemed likely: the E3 ubiquitin protein ligase could be situated at promoters, thereby restricting GATA-1 ubiquitination to this location. Alternatively, promoter-specific modifications might be required to modify GATA-1 fully, thus restricting GATA-1 degradation to the DNA-bound form.
To further analyse the mechanism of degradation, we decided to test the possibility of promoter-specific modifications. Acetyltransferases interact with GATA-1 in the absence of DNA binding (Blobel et al, 1998; Boyes et al, 1998); acetylation is known to enhance GATA-1/DNA binding (Boyes et al, 1998) and DNA binding prevents subsequent acetylation of GATA-1 (Figure 6A). Together, these data suggest that acetylation of GATA-1 occurs before DNA binding; consequently, we would expect acetylation of the DNA binding mutant and wild-type protein to be similar. By contrast, although the DNA binding mutant appears to be phosphorylated on the basis of slower mobility (Figure 5D), it is possible that not all seven phosphorylation sites are modified. To test if an additional promoter-specific phosphorylation event regulates degradation, we labelled Cos 7 cells that had been transfected with expression vectors for either wild-type GATA-1 or the RNRK DNA binding mutant with inorganic phosphate. The sites of phosphorylation were then determined by two-dimensional phosphopeptide mapping (Crossley and Orkin, 1994). We found that phosphorylation of two serines, S26 and S178, was significantly reduced in the DNA binding mutant (Figure 6B). Importantly, these residues are target sites for MAP kinases (Towatari et al, 2004). This therefore correlates very well with our data showing that inhibition of MAPK signalling increases GATA-1-dependent transcription (Figure 5A) and that inhibition of MAPK signalling stabilises acetylated GATA-1 (Figure 4D). Moreover, it strongly implies a role for MAPK signalling in causing degradation of the active transcription factor. Control experiments confirmed that both the wild-type and RNRK mutant proteins are nuclear (Figure 6C). Furthermore, the RNRK mutation does not destroy known docking sites of MAPK on substrate proteins (Jacobs et al, 1999) and hyper-activation of the MAPK pathway with constitutively active MAPK shows that under these artificial conditions the RNRK mutant can be phosphorylated (Figure 6D). In addition, phosphorylation does not alter GATA-1 DNA binding (Crossley and Orkin, 1994; Supplementary Figure 4) implying that phosphorylation is not required for recruitment to promoters thereby causing default suicide degradation. Together, these data suggest that GATA-1 must be bound to DNA for MAP kinase phosphorylation and highlight this phosphorylation event as a potentially key regulatory step in the degradation process.
Figure 6.
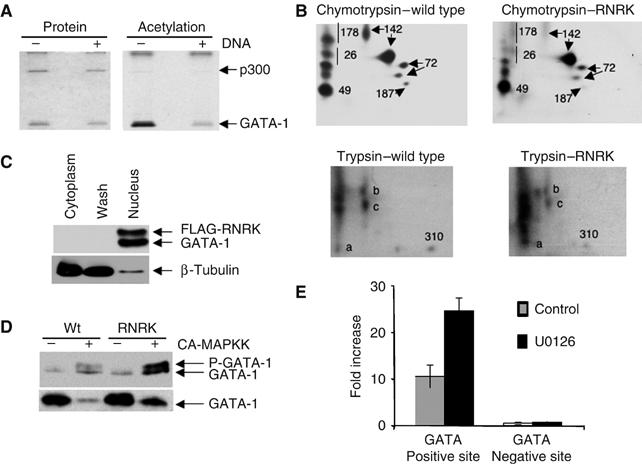
DNA binding is required for MAPK phosphorylation but not acetylation of GATA-1. (A) DNA binding inhibits acetylation of GATA-1. A purified peptide of the double zinc-finger region of GATA-1 was either bound or unbound to an oligonucleotide with a palindromic GATA-1 binding site and an in vitro acetylation reaction with p300 was performed (Boyes et al, 1998). At this binding site, the half-life of GATA-1/DNA binding is 70 min and thus should persist during the acetylation reaction. The stained gel (left) shows that equivalent amounts of protein were loaded. The autoradiograph (right) shows acetylation of GATA-1 and p300. (B) DNA binding is required for phosphorylation of GATA-1 at S26 and S178. Phosphopeptide maps of wild-type GATA-1 and RNRK mutant protein that had been immunoprecipitated from transfected, 32P-labelled Cos 7 cells and then digested with chymotrypsin or trypsin as indicated are shown. The sites of phosphorylation were identified from Crossley and Orkin (1994). One spot showing phosphorylation at S142 is smeared in the mutant protein; as the other spot is present at normal intensity, we suggest that phosphorylation at S142 is unaffected by DNA binding. The spot previously correlated with phosphorylation at S310 is visible in the trypsin digests. For the DNA binding mutant, this spot is shifted slightly to the left, indicative of a higher molecular weight. We suggest that this alteration in tryptic digestion pattern is owing to loss of a cleavage site in the RNRK mutant. (C) The RNRK mutant protein is localised to the nucleus. Nuclear and cytoplasmic extracts were prepared (Supplementary methods) from BM-SCF cells, which stably express FLAG-tagged RNRK mutant. An equivalent volume of each extract was analysed by Western blotting using anti-GATA-1 and anti-β-tubulin antibodies. (D) The RNRK mutant protein can be phosphorylated. Cos 7 cells were transfected with expression vectors for the GATA-1 proteins shown either with or without an expression vector for the constitutively active MAP kinase (CA-MAPKK). Whole-cell extract was used in a Western blot with anti-GATA-1 antibody (N6). Upper panel: The amount of extract used was varied to achieve more equivalent GATA-1 levels for ease of comparison of the phosphorylation shift. Lower panel: An equivalent volume of extract from each transfection was used and indicates that expression of CA-MAPKK causes loss of GATA-1. (E) GATA-1 binding to the EKLF enhancer is increased when MAPK signalling is inhibited. Chromatin immunoprecipitation was performed in BM-SCF cells following treatment with (UO126) or without (control) MEK inhibitor. The DNA was analysed by real-time PCR using primers spanning a GATA-1 site in the EKLF enhancer (positive site) and a negative control site. The fold enrichment compared to input Ct values is shown.
If transcriptionally active GATA-1 is indeed degraded in response to MAPK signalling, we would predict increased GATA-1 occupancy at regulatory elements when MAPK signalling is inhibited. To test this, we carried out chromatin immunoprecipitation and examined GATA-1 occupancy at the EKLF enhancer in the presence and absence of MEK inhibitors. As expected, when MAPK signalling is prevented, GATA-1 occupancy is increased (Figure 6E). This provides further evidence that the transcriptionally active form of GATA-1 is being signalled for degradation by phosphorylation.
Growth factors signal degradation of acetylated GATA-1
Finally, we wished to determine how this phosphorylation is signalled in its normal context of haemopoietic cells. As cytokine signalling is central to haemopoietic differentiation, we investigated if cytokines can modulate GATA-1 loss. To this end, we used two erythroid/myeloid progenitor cell lines: BM-SCF (described above) and BM-Epo (Cairns et al, 2003). The advantage of these cell lines is that they can be grown only in the presence of SCF or erythropoietin, respectively, and thus the effects of these cytokines can be specifically examined.
Cells grown initially in either SCF or erythropoietin were starved of growth factor for 6 h; the relevant factor was then re-administered and the effects on GATA-1 modifications were examined. As shown in Figure 7A, SCF and erythropoietin both increase GATA-1 phosphorylation, concomitant with an increase in ubiquitination and reduction in GATA-1 protein. Mutation of the phosphorylation sites eliminated the phosphorylation shifts and protein turnover (Figure 7B). Importantly, as expected if MAPK signals GATA-1 ubiquitination, use of MEK inhibitors prevented both phosphorylation of GATA-1 and the increase in ubiquitination (Figure 7C). Together, these experiments indicate that SCF and erythropoietin signal via MAPK-dependent phosphorylation to cause GATA-1 ubiquitination and degradation, thus further highlighting the importance of this signalling pathway in regulating the degradation of GATA-1. Furthermore, they verify that GATA-1 levels can be modulated in response to normal growth factor signals. Consistent with the idea that both acetylation and phosphorylation are required for turnover, we find that the acetylation mutant is stable under mitogen stimulatory conditions and, moreover, that mitogen stimulation preferentially leads to the degradation of acetylated GATA-1 (Figure 7D).
Figure 7.
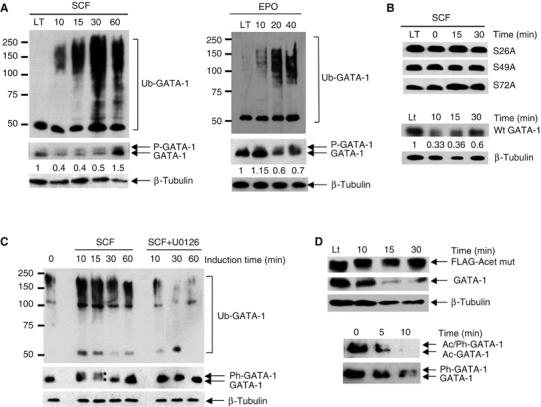
Regulation of GATA-1 ubiquitination by SCF and erythropoietin. (A) GATA-1 ubiquitination is increased in response to signalling by SCF and erythropoietin. BM-SCF or BM-Epo cells were starved of cytokine for 6 h. SCF or erythropoietin was then re-administered and GATA-1 immunoprecipitated at the times shown. GATA-1 ubiquitination was analysed with an anti-ubiquitin antibody and GATA-1 levels with an anti-GATA-1 antibody (M-20; middle panel). Sixty minutes after induction, GATA-1 levels increased, most likely owing to new GATA-1 synthesis. Phosphorylated GATA-1 (P-GATA-1) is indicated. LT indicates cells grown continuously (long term) in SCF. β-Tubulin levels verify that equivalent cell numbers were used (lower panel). (B) Phosphorylation is required for SCF-induced GATA-1 turnover. BM-SCF cells that express FLAG-tagged GATA-1 mutated in individual phosphorylation sites (S26A, S49A and S72A) were induced with SCF. Immunoblotting with anti-FLAG antibody showed that SCF did not cause a phosphorylation shift or loss of GATA-1. FLAG-tagged wild-type GATA-1 is lost like the endogenous protein (compare lower panel and panel A middle panel). (C) Inhibition of phosphorylation prevents GATA-1 ubiquitination. BM-SCF cells were starved of growth factor for 6 h. SCF was then re-administered either in the absence (SCF) or presence (SCF+U0126) of the MEK inhibitor U0126. Samples were taken at the times indicated, immunoprecipitated with anti-GATA-1 antibody and the level of ubiquitination determined via Western blotting using an anti-ubiquitin antibody. The middle panel shows GATA-1 levels following re-probing of the blot with a second anti-GATA-1 antibody (M-20). Phosphorylated and non-phosphorylated GATA-1 are indicated by asterisks. (D) Acetylation is required for GATA-1 turnover. Upper panel: GATA-1 acetylation mutant is not degraded in response to SCF signalling; endogenous GATA-1 in the same cell is lost (second panel from top). Fourth panel: Acetylated GATA-1 (Ac-GATA-1) is degraded faster than bulk GATA-1 (lower panel, where the same blot was re-probed) upon mitogen signalling.
Discussion
Regulation of transcription requires that the activity of key transcription factors is stringently regulated. We show here that acetylated GATA-1 is targeted for degradation, thus demonstrating that an active form of the transcription factor can be selectively removed. Furthermore, as acetylation positively controls ubiquitination, this implies that activation by acetylation simultaneously flags the factor for degradation. Acetylation, however, is not enough to cause protein loss. We find further that phosphorylation and acetylation must cooperate for efficient ubiquitination; this suggests that acetylated GATA-1 can remain active at promoters until phosphorylation-mediated signalling causes its loss. Consistent with this, we find that phosphorylation of GATA-1 by MAP kinases requires that GATA-1 is bound to DNA. Together, these data suggest a novel means by which the specific and regulated degradation of active transcription factors can be achieved in response to external signals.
In principle, the regulated loss of an active transcription factor is likely to be advantageous to the cell. Most importantly, it provides a means to continuously regulate gene transcription. Thus, once transcription is initiated, there is a mechanism to control the output of a gene and, if necessary, to cease transcription. Our data therefore add to recent reports proposing that active transcription factors are degraded, to allow promoter re-setting (Lipford et al, 2005). However, there is only one report that describes a mechanism by which the selective degradation of an active factor can be achieved (Chi et al, 2001). In this case, phosphorylation of GCN4 by the basal transcription machinery targets the active protein for degradation. However, it is unlikely that all active transcription factors will be substrates for phosphorylation by the basal machinery. Instead, we suggest that as many transcription factors are activated by acetylation, by simultaneously activating and marking for degradation, acetylation might provide a more general signal for the degradation of active factors. Consistent with this, HIF1α is also degraded when acetylated (Jeong et al, 2002), although in this case it is not known how acetylation affects its transcriptional activity.
Acetylation alone is not enough to signal degradation, as indicated by the increased stability of the phosphorylation mutants. The cooperation between acetylation and phosphorylation for efficient degradation has a number of advantages. Firstly, it means that degradation of active GATA-1 can be regulated in response to external signals. Thus, acetylated GATA-1 can be marked for degradation but remains active until phosphorylation signals its loss. This contrasts with the other examples of active transcription factor degradation described to date where the mere presence of a factor at a promoter automatically leads to its degradation (Chi et al, 2001; Reid et al, 2003; Sundqvist and Ericsson, 2003). Secondly, it means that phosphorylation will primarily signal degradation of the acetylated, active protein, not the entire pool of GATA-1. Indeed, as less than 2% of GATA-1 is acetylated (Supplementary Figure 5), degradation of GATA-1 is very specific. The requirement for both modifications also permits tight control of the degradation. Consistent with this, we find that all seven phosphorylation sites are required for optimal ubiquitination. Sequence analysis indicates that these phosphorylation sites are targets for different kinases, suggesting that a number of kinase cascades need to converge to trigger degradation of acetylated GATA-1. A similarly large number of phosphorylation events were found to be necessary for ubiquitination of the cell cycle-regulated protein Sic1 (Nash et al, 2001). In this case, phosphorylation was thought to create an ultrasensitive switch so that protein degradation is initiated only at a critical level of cyclin-dependent kinase activity (Harper, 2002). Likewise, the multisite phosphorylation required for GATA-1 degradation might provide a sensitive switch or allow its degradation in response to different signalling pathways. In the experiments described here, MAPK signalling is ultimately responsible for GATA-1 loss. However, in other situations, different kinases might signal GATA-1 degradation. For example, given its role in the cell cycle (Whyatt et al, 1997; Rylski et al, 2003), GATA-1 activity might be regulated in response to cyclin-dependent kinases.
The finding that GATA-1 must be bound to DNA for degradation suggests yet another safeguard to prevent inappropriate loss. Although it remains possible that the specific E3 ubiquitin protein ligase is located at promoters to thus restrict GATA-1 ubiquitination, we find surprisingly that DNA binding is required for phosphorylation of GATA-1 by MAP kinases. These data therefore add weight to our model that degradation of acetylated, active GATA-1 at promoters is regulated by external signals and are fully consistent with our finding that inhibition of MAP kinases increases GATA-1-dependent transcription (Figure 5A). As DNA binding prevents acetylation of GATA-1 (Figure 6A), they also highlight the temporal order of modifications leading to degradation, namely that modification by acetylation must occur first, followed by MAP kinase phosphorylation of the DNA bound form. We further suggest that this promoter-specific modification is likely to provide additional stringency to the regulation of degradation. Indeed, if degradation relied solely on location of the E3 ubiquitin protein ligase, then the mere presence of a factor at promoters would automatically lead to its degradation. How MAPK phosphorylation of GATA-1 is restricted to the DNA binding form is unclear. One possibility is that DNA binding induces a conformational change that makes S26 and S178 simultaneously accessible. Alternatively, scaffold proteins might play a role in restricting MAPK activity to the DNA-bound form.
It is likely that yet further regulatory steps restrict the amount of transcriptionally active GATA-1. We find GATA-1 degradation is accelerated in the presence of the histone deacetylase inhibitor TSA (Figure 3A and C). This suggests that under normal growth conditions deacetylases remove acetylation from GATA-1 to stabilise it. Exactly how the balance between de novo acetylation and activation and deacetylation and stabilisation is achieved is unknown. However, it seems likely that regulators of acetylation and deacetylation are further important players in controlling the level of gene transcription.
Together, our data show a mechanism by which a transcriptionally active form of GATA-1 can be continually and carefully regulated. Moreover, we show a mechanism of achieving this regulation in response to physiological cytokine signalling. Exactly how MAP kinases are specifically directed to acetylated GATA-1 at promoters to signal degradation and how acetylation itself is regulated, however, are the subjects of future studies.
Materials and methods
Plasmids
A FLAG tag was cloned in-frame with GATA-1 cDNA using the NcoI site at the first ATG of GATA-1 and a SmaI site in the vector Bft4; Bft4 has a FLAG tag cloned into the BamHI site of pBluescript II KS. GATA-1 mutants were prepared from Bft4GATA-1 via PCR. The acetylation mutant was prepared by converting all lysines (233, 245, 246, 252, 287, 299, 312, 314, 315 and 316) in the DNA binding domain to arginine except lysine 308 (required for DNA binding); primer sequences are available on request. The phosphorylation mutants (serine to alanine) were a gift from Dr M Crossley (Crossley and Orkin, 1994). GATA-1 expression vectors were made by inserting an XbaI–ClaI fragment from Bft4GATA-1 constructs into pEF-XC (Mizushima and Nagata, 1990). Retroviral expression constructs were made by inserting the same fragment into MSCV-IRES-GFP (a gift from Dr A Nienhuis). PCMVβ-galactosidase was from Clontech. The RNRK DNA binding mutant was prepared by mutating amino acids 305–308 to ANAA. The luciferase reporter plasmid, pGL3EpoR, was prepared by cloning −337 to +121 of the mouse erythropoietin receptor promoter into the KpnI/BglII sites of pGL3 (Promega). CA-MAPKK was a gift from Professor C Marshall.
Cell culture and transfections
Cos 7 cells were grown at 35°C and 293T and NIH3T3 cells at 37°C in DMEM with 10% fetal calf serum and 2 mM L-glutamine. Transfection was via calcium phosphate precipitation, using 15 μg DNA per 10 cm plate.
BM-SCF and BM-Epo cells were grown as described (Cairns et al, 2003). Starvation was for 6 h. MEK inhibitor, U0126 (Promega), was used at a final concentration of 10 μM. Retroviral infection was performed according to http://www.stanford.edu/group/nolan/protocols using MSCV-IRES-GFP-GATA-1 expression vectors. Positive cells were FACS sorted on the basis of GFP expression.
Cells were treated with proteasome inhibitors for 8 h. The inhibitors, used at the concentrations indicated, were from the sources given in parentheses: lactacystin: 10 μM (Professor EJ Corey, Harvard); MG-132: 2.5 μM, clasto-lactacystin: 5 μM (Calbiochem).
TSA was used at a final concentration of 100–200 ng/ml for Cos 7 and 293T cells and 500 ng/ml for BM-SCF cells. α-Amanitin was used at a final concentration of 2.5 μM.
Antibodies
Antibodies were purchased as follows: anti-GATA-1 antibodies N6 (rat monoclonal) and M-20 (goat polyclonal) from Santa Cruz; mouse monoclonal anti-β-galactosidase from Promega; mouse monoclonal anti-ubiquitin (clone FK2) from Affiniti Research; mouse monoclonal anti-tubulin and mouse anti-FLAG (M2) from Sigma. Anti-acetyl-GATA-1 antibody was raised against a GATA-1 peptide acetylated at lysine 312. Serum was affinity purified using acetylated and non-acetylated peptides. Antibodies that reacted only with the acetylated peptide were used (Supplementary Figure 2B). The dilutions were as follows: N6 (1:1000); M-20 (1:400); anti-β-galactosidase (1:3000); anti-ubiquitin (1:250); anti-tubulin (1:2000); anti-FLAG (1:1000) and anti-acetyl-GATA-1 (1:100). All secondary antibodies were from Jackson ImmunoResearch and were used at 1:5000.
Western blotting and immunoprecipitations
Cells were harvested 48–72 h after transfection; GATA-1 was immunoprecipitated as described (Boyes et al, 1998).
To detect ubiquitinated GATA-1, where indicated, cells were treated with10 μM MG-132 for 8 h before harvesting. Samples were immunoprecipitated with N6 antibody covalently coupled to POROS matrix (Applied Biosystems) and analysed by Western blotting with anti-ubiquitin antibody. Where heavy chain is indicated, GATA-1 was immunoprecipitated with uncoupled antibody.
Membranes were blocked for 1 h at room temperature with 5% milk (5% BSA for the anti-ubiquitin antibody). Antibodies were incubated with the membrane for 1 h at room temperature except anti-ubiquitin and anti-acetyl-GATA-1 antibodies, which were incubated overnight at 4°C.
RNase protection, S1 mapping and luciferase assay
The RNase protection assay was as described (Boyes and Bird, 1992); S1 mapping and probes are as described (Cairns et al, 2003). The luciferase assay used the Promega kit according to the manufacturer's instructions.
Pulse–chase experiments
Cos 7 cells, in 5 cm dishes, were transfected with 7.5 μg of GATA-1 expression vectors for 24 h. The cells were then washed with PBS and labelled with 100 μCi of [35S]methionine (Perkin Elmer Life Sciences) per ml of methionine-free DMEM. After 30 min at 37°C, the cells were washed with PBS and chased with complete DMEM. Extracts were made at the times shown, immunoprecipitated with N6 antibody and Western blotted. Band intensities were analysed with a phosphorimager.
Cell labelling and two-dimensional phosphopeptide mapping
Cos 7 cells were transfected with expression vectors for wild-type GATA-1 or the RNRK DNA binding mutant protein. Cell labelling, immunoprecipitation and 2D phosphopeptide mapping were performed as described (Crossley and Orkin, 1994) except that labelling was overnight.
Chromatin immunoprecipitation
Crosslinked chromatin was prepared from 6 × 107 BM-SCF cells (Letting et al, 2003) that had been treated with or without U0126. Immunoprecipitation was with coupled N6 antibody; 0.9 ng of immunoprecipitated or input DNA was analysed by real-time PCR using SYBR green and EKLF positive and negative primers (Rodriguez et al, 2005). The fold increase compared to input DNA was calculated as in Rodriguez et al. All immunoprecipitations were performed in duplicate and real-time PCR in triplicate. Isotype-matched IgG was used as a control.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank M Crossley for the phosphorylation mutant constructs, A Nienhuis for the MCSV-IRES-GFP vector and C Marshall for the CA-MAPKK vector. We also thank D Barford, J Mermoud and P Rigby for helpful comments on the manuscript, A Ashworth, R Marais and S Mittnacht for helpful discussions and I Titley for FACs sorting. SO was supported by Telethon, AIRC, COFIN and Cariplo. AH was supported by a Marie Curie fellowship (HPMF-CT-2001-01317). We thank the Wellcome Trust for supporting this work (grant 059602). JB is grateful for support from the Lister Institute of Preventive Medicine and a University of Leeds Research Fellowship.
References
- Blobel GA, Nakajima T, Eckner R, Montminy M, Orkin SH (1998) CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci USA 95: 2061–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J, Bird A (1992) Repression of gene by DNA methylation depends on CpG density and promoter strength:evidence for involvement of a methyl-CpG binding protein. EMBO J 11: 327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J, Byfield P, Nakatani Y, Ogryzko V (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396: 594–598 [DOI] [PubMed] [Google Scholar]
- Cairns L, Ciro M, Minuzzo M, Morle F, Starck J, Ottolenghi S, Ronchi A (2003) Induction of globin mRNA expression by interleukin-3 in a stem cell factor-dependent SV-40 T-antigen-immortalized multipotent hematopoietic cell line. J Cell Physiol 195: 38–49 [DOI] [PubMed] [Google Scholar]
- Chi Y, Huddleston MJ, Zhang X, Young RA, Annan RS, Carr SA, Deshaies RJ (2001) Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev 15: 1078–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley M, Orkin SH (1994) Phosphorylation of the erythroid transcription factor GATA-1. J Biol Chem 269: 16589–16596 [PubMed] [Google Scholar]
- De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, Srinivasula SM, Alnemri ES, Testa U, Peschle C (1999) Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 401: 489–493 [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Delahodde A, Kodadek T, Johnston SA (2002) Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296: 548–550 [DOI] [PubMed] [Google Scholar]
- Gronroos E, Hellman U, Heldin CH, Ericsson J (2002) Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 10: 483–493 [DOI] [PubMed] [Google Scholar]
- Harper JW (2002) A phosphorylation-driven ubiquitination switch for cell cycle control. Trends Cell Biol 12: 104–107 [DOI] [PubMed] [Google Scholar]
- Hung HL, Lau J, Kim AY, Weiss MJ, Blobel GA (1999) CREB-binding protein acetylates hematopoietic transcription factor GATA-1 at functionally important sites. Mol Cell Biol 19: 3496–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev 13: 163–175 [PMC free article] [PubMed] [Google Scholar]
- Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW (2002) Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111: 709–720 [DOI] [PubMed] [Google Scholar]
- Kouzarides T (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J 19: 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulessa H, Frampton J, Graf T (1995) GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev 9: 1250–1262 [DOI] [PubMed] [Google Scholar]
- Letting DL, Rakowski C, Weiss MJ, Blobel GA (2003) Formation of a tissue-specific histone acetylation pattern by the hematopoietic transcription factor GATA-1. Mol Cell Biol 23: 1334–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Luo J, Brooks CL, Gu W (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 277: 50607–50611 [DOI] [PubMed] [Google Scholar]
- Lipford JR, Smith GT, Chi Y, Deshaies RJ (2005) A putative stimulatory role for activator turnover in gene expression. Nature 438: 113–116 [DOI] [PubMed] [Google Scholar]
- Martin DI, Orkin SH (1990) Transcriptional activation and DNA binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes Dev 4: 1886–1898 [DOI] [PubMed] [Google Scholar]
- Mizushima S, Nagata S (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res 18: 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M (2001) Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414: 514–521 [DOI] [PubMed] [Google Scholar]
- Orkin SH (2000) Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet 1: 57–64 [DOI] [PubMed] [Google Scholar]
- Reid G, Hubner MR, MetivierR, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F (2003) Cyclic, proteasome-mediated of liganded and unliganded ERalpha on responsive promoters is an integral feature of estrogen signalling. Mol Cell 11: 695–707 [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Bonte E, Krijgsveld J, Kolodziej KE, Guyot B, Heck AJ, Vyas P, de Boer E, Grosveld F, Strouboulis J. (2005) GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J 24: 2354–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylski M, Welch JJ, Chen YY, Letting DL, Diehl JA, Chodosh LA, Blobel GA, Weiss MJ (2003) GATA-1-mediated proliferation arrest during erythroid maturation. Mol Cell Biol 23: 5031–5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundqvist A, Ericsson J (2003) Transcripion-dependent degradation controls the stability of the SREBP family of transcription factors. Proc Natl Acad Sci USA 100: 13833–13838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Tyers M (2000) Transcriptional regulation: Kamikaze activators. Curr Biol 10: R341–R343 [DOI] [PubMed] [Google Scholar]
- Towatari M, Ciro M, Ottolenghi S, Tsuzuki S, Enver T (2004) Involvement of mitogen activated protein kinase in the cytokine-regulated phosphorylation of transcription factor GATA-1. Hematol J 5: 262–272 [DOI] [PubMed] [Google Scholar]
- Tsai SF, Strauss E, Orkin SH (1991) Functional analysis and in vivo footprinting implicate the erythroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes Dev 5: 919–931 [DOI] [PubMed] [Google Scholar]
- Visvader JE, Crossley M, Hill J, Orkin SH, Adams JM (1995) The C-terminal zinc finger of GATA-1 or GATA-2 is sufficient to induce megakaryocytic differentiation of an early myeloid cell line. Mol Cell Biol 15: 634–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG (2003) The F-Box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 11: 1189–1200 [DOI] [PubMed] [Google Scholar]
- Whyatt D, Lindeboom F, Karis A, Ferreira R, Milot E, Hendriks R, de Bruijn M, Langeveld A, Gribnau J, Grosveld F, Philipsen S (2000) An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature 406: 519–524 [DOI] [PubMed] [Google Scholar]
- Whyatt DJ, Karis A, Harkes IC, Verkerk A, Gillemans N, Elefanty AG, Vairo G, Ploemacher R, Grosveld F, Philipsen S (1997) The level of the tissue-specific factor GATA-1 affects the cell-cycle machinery. Genes Funct 1: 11–24 [DOI] [PubMed] [Google Scholar]
- Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, Fisher DE (2000) c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev 14: 301–312 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures