

Abstract
The ATP-binding cassette (ABC)-transporter haemolysin (Hly)B, a central element of a Type I secretion machinery, acts in concert with two additional proteins in Escherichia coli to translocate the toxin HlyA directly from the cytoplasm to the exterior. The basic set of crystal structures necessary to describe the catalytic cycle of the isolated HlyB-NBD (nucleotide-binding domain) has now been completed. This allowed a detailed analysis with respect to hinge regions, functionally important key residues and potential energy storage devices that revealed many novel features. These include a structural asymmetry within the ATP dimer that was significantly enhanced in the presence of Mg2+, indicating a possible functional asymmetry in the form of one open and one closed phosphate exit tunnel. Guided by the structural analysis, we identified two amino acids, closing one tunnel by an apparent salt bridge. Mutation of these residues abolished ATP-dependent cooperativity of the NBDs. The implications of these new findings for the coupling of ATP binding and hydrolysis to functional activity are discussed.
Keywords: ABC-transporter, ATPase, catalytic cycle, X-ray crystallography
Introduction
Many Gram-negative bacteria use Type I secretion systems to translocate toxins, hydrolytic enzymes or surface-bound proteins across the cell envelope (Holland et al, 2005). The paradigm of such a transport complex is the haemolysin (Hly)A apparatus, discovered in the early 1980s in uropathogenic Escherichia coli strains (Welch et al, 1981), which like all Type I systems rely on the presence of a C-terminal secretion signal in the transported substrate. The HlyA secretion machinery is composed of three indispensable elements: the ABC-(ATP-binding cassette) transporter HlyB, the membrane fusion protein HlyD, both residing in the inner membrane of E. coli, and the outer membrane protein TolC (Holland et al, 2005).
ABC-transporters, such as HlyB, are ubiquitous ATP-dependent transmembrane proteins (Higgins, 1992; Schmitt and Tampé, 2002) that transport an astonishing variety of transport substrates across membranes, coupled to the consumption of ATP. Despite this transport substrate diversity, all ABC-transporters share the same architecture, comprised of two transmembrane domains or subunits (TMDs) and two nucleotide-binding domains (NBDs) (Schmitt and Tampé, 2002; Davidson and Chen, 2004; Jones and George, 2004). Whereas the α-helical TMDs are divergent in primary structure, the NBDs are rather conserved with respect to sequence and three-dimensional structure. Nevertheless, we could identify a structurally diverse region (SDR) within several different ABC-NBDs (Schmitt et al, 2003) that might be involved in signal transmission to the TMDs. The conservation of the NBDs is also reflected by the fact that all conserved motifs of ABC-transporters, the Walker A and B motif, the C-loop or ABC-signature motif, the Q- and Pro-loop, and the D- and H-loops are located within the NBD.
Over the last decade, we have seen tremendous advances in ABC-transporter research including crystal structures of isolated NBDs (for recent reviews see Ye et al, 2004; Oswald et al, 2006) and full-length transporters (Chang and Roth, 2001; Locher et al, 2002; Chang, 2003; Reyes and Chang, 2005). Based on these structural and biochemical data obtained for isolated NBDs (Chen et al, 2001; Janas et al, 2003), it is now believed that they form a composite dimer in the ATP-bound state. Here, ATP acts as a molecular glue, which is sandwiched between the Walker A motif of one monomer (the cis monomer) and the ABC-signature motif of the other monomer (the trans monomer) (Smith et al, 2002). However, despite rapidly increasing knowledge of the biochemistry and the three-dimensional structure of ABC-transporters, we are still far away from a detailed, molecular understanding of the catalytic cycle of the NBD. Moreover, a comprehensive analysis of the mechanical switches involved in domain movements associated with substrate binding or dissociation is still missing. Importantly, one finds that these features are generally less conserved than the active site geometry. Consequently, a reliable analysis of domain movements, conformational changes and changing patterns of protein–ligand or protein–protein interactions can only be carried out using structures derived from the same protein. So far, this has only been achieved for MalK by solving the structures of the apo-, ATP- and ADP-bound forms (Chen et al, 2003; Lu et al, 2005). In contrast to the HlyB-NBD, MalK fuels transport substrate import and, more importantly, deviates from a canonical NBD by the presence of a large C-terminal domain, required for regulation of the mal operon.
We have now obtained the crystal structures of two mutated forms of the HlyB-NBD, H662A and E631Q, in complex with ATP (dimers) or ADP (monomers), and the crystal structure of the wild-type monomer protein with bound ADP. In combination with the recently published crystal structures of the H662A/ATP-Mg2+ (Zaitseva et al, 2005a) dimer and the structure of the HlyB-NBD in the absence of bound nucleotide (Schmitt et al, 2003), a basic ABC-catalytic cycle can now be described in structural detail. This has revealed important mechanistic insights and several novel features including a structural asymmetry within the dimer in the presence of ATP/Mg2+. We propose that this represents one open and one closed phosphate exit tunnel. Further analysis of this asymmetry enabled us to identify two amino acids of crucial importance for ATP-dependent cooperativity. In combination with a mechanism to store the energy of ATP within the NBD, we now describe the molecular mechanisms that operate to coordinate NBD dimerization, ATP hydrolysis and associated conformational changes in order to fuel transport substrate translocation.
Results and discussion
The NBD of HlyB, comprising residues 467–707, was purified and crystallized in the ATP- and ADP-bound forms (see Materials and methods). For crystallization of the ATP-loaded composite dimer, the H662A and E631Q mutants had to be employed because no suitable crystals could be grown for the wild-type enzyme. ADP-bound forms were obtained for the wild type, the H662A and the E631Q mutant. Data statistics are summarized in Table I.
Table 1.
Data quality and refinement statistics of the crystal structures determined
| Protein | Wild type | H662A | E631Q | H662A | E631Q |
|---|---|---|---|---|---|
| Ligand | ADP | ADP | ADP | ATP | ATP |
| Crystal parameters | |||||
| Space group | C2 | C2 | C2 | P21 | P21 |
| Cell constants at 100 K | |||||
| a, b, c (Å) | 180.37, 34.84, 37.82 | 180.16, 34.77, 38.1 | 178.57, 34.75, 37.52 | 46.56, 195.17, 63.23 | 47.14, 189.26, 63.48 |
| β (deg) | 98.41 | 98.58 | 97.97 | 110.85 | 111.87 |
| Data collection and processing | |||||
| Wavelength (Å) | 1.05 | 1.05 | 1.05 | 1.05 | 1.05 |
| Resolution (Å) | 20–1.6 | 20–1.7 | 20–1.9 | 20–2.6 | 20–2.7 |
| Mean redundancy | 10.9 | 12.2 | 6.9 | 10.2 | 6.8 |
| Completeness (%) | 90.5 (69.5) | 99.2 (88.5) | 94.1 (88.1) | 96.4 (93.2) | 98.9 (98.3) |
| I/σ | 14.7 (2.1) | 26 (3.2) | 10.8 (2.3) | 12.1 (3.2) | 22.5 (5.2) |
| Rsym (%) | 8.8 (25.4) | 6.2 (16.3) | 4.9 (27.7) | 5.7 (19.3) | 9.4 (25.7) |
| Refinement | |||||
| RF (%) | 19.2 (26.8) | 20.5 (24.0) | 20.2 (29.7) | 21.1 (27.3) | 22.6 (29.3) |
| Rfree (%) | 23.1 (36.6) | 24.0 (32.0) | 25.0 (30.1) | 27.9 (37.1) | 28.0 (36.9) |
| R.m.s.d. | |||||
| Bond length (Å) | 0.013 | 0.015 | 0.013 | 0.007 | 0.012 |
| Bond angle (deg) | 1.434 | 1.498 | 1.414 | 1.085 | 1.214 |
| Average B-factor (Å2) | 30.2 | 25.1 | 37.0 | 51.0 | 51.5 |
| Ramachandran plot | |||||
| Most favored (%) | 93.5 | 93.0 | 92.0 | 90.0 | 88.7 |
| Allowed (%) | 5.6 | 6.0 | 7.1 | 8.7 | 10.2 |
| Generously allowed (%) | 0.9 | 0.9 | 0.9 | 0.9 | 0.8 |
| Disallowed (%) | — | — | — | 0.4 | 0.2 |
| Model content | |||||
| Protein residues | 242 | 243 | 243 | 964 | 964 |
| Ligands | ADP | ADP | ADP | 4 ATP | 4 ATP |
| Water molecules | 176 | 246 | 128 | 197 | 152 |
| Values in parentheses correspond to the highest resolution shell (1.65–1.6 Å for the wild-type ADP, 1.76–1.7 Å for the ADP H662A, 1.95–1.9 Å for the E631Q-ADP, 2.65–2.6 Å for the ATP H662A and 2.75–2.7 Å for the ATP E631Q structure). | |||||
The catalytic cycle of the HlyB-NBD
The catalytic cycle, represented by the nucleotide-free and the ATP- and ADP-bound forms of the HlyB-NBD, together with the relevant dissociation constants, is depicted in Figure 1. Crystals of other intermediates of the catalytic cycle such as a vanadate trapped state or an ATP/ADP mixed dimer as proposed to exist for the Mdl1p-NBD (Janas et al, 2003) could not be obtained despite numerous efforts. However, the structures reported here are sufficient to describe important functional states of the basic catalytic cycle of the HlyB-NBD. Whereas the nucleotide-free and ADP states crystallized as monomeric proteins, the ATP and ATP/Mg2+ states formed dimers. As already reported for MJ0796 (Smith et al, 2002) and MalK (Chen et al, 2003), ATP (highlighted in ball-and-stick representation; Figure 1) is sandwiched between the Walker A motif of the cis monomer and the ABC-signature motif of the trans monomer (colored blue and red, respectively, in Figure 1). This composite architecture of the dimer in the ATP- (Smith et al, 2002; Chen et al, 2003) and ATP/Mg2+-bound states (Zaitseva et al, 2005a) shown here is now commonly believed to be the functional, productive catalytic state. It is important to note that in the crystal structures of MalK (Chen et al, 2003; Lu et al, 2005), BtuCD (Locher et al, 2002) and MsbA (Chang, 2003; Reyes and Chang, 2005) lacking ATP, the NBDs are also in an apparent ‘sandwich-like' arrangement in the nucleotide-free state. However, the buried surface area of the NBD:NBD interface in these crystal structures is rather small, 480 Å2 in the case of BtuD (Locher et al, 2002) compared to 1890 Å2 in the case of the ATP/Mg2+-bound state of the HlyB-NBD (Zaitseva et al, 2005a). It is reasonable to suppose that the lateral constraint imposed by the additional C-terminal domain of MalK (Chen et al, 2003; Lu et al, 2005), or the TMDs of BtuCD or MsbA, keeps the NBDs close together in a ‘dimer-like' or open configuration even in the absence of ATP. Importantly, such structures are still readily accessible to nucleotides and we conclude that these ATP-free forms of MsbA and BtuD should be regarded as monomers from a structural point of view. In solution, all other isolated NBDs are also apparently monomers and only in the presence of ATP or in a vanadate-trapped state, can true dimers be identified (Janas et al, 2003; Verdon et al, 2003b; Zaitseva et al, 2005b).
Figure 1.
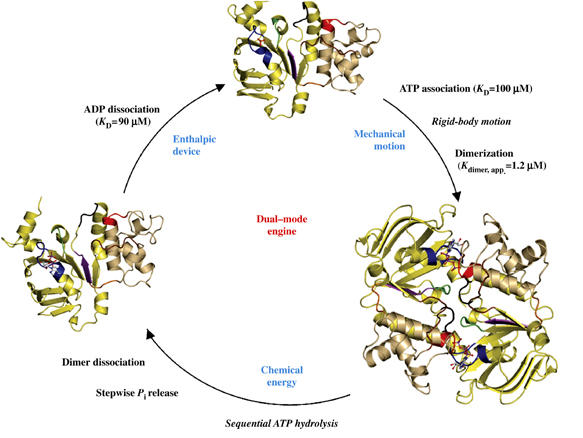
The catalytic cycle of the HlyB-NBD. Crystal structures of the monomeric nucleotide-free (Schmitt et al, 2003), dimeric ATP-bound (H662A and E631Q) and monomeric ADP-bound (wild type, E631Q and H662A) forms (this study) are shown. For simplicity, the structure of the ATP/Mg2+-bound form (Zaitseva et al, 2005a) is not shown. The catalytic domain is colored in light yellow and the helical domain in light tan. Conserved motifs are highlighted and color-coded as follows: Walker A (residues 502–510, blue), Q-loop (residues 549–556, brown), ABC-signature motif (residues 606–610, red), Pro-loop (residues 623–625, orange), Walker B (residues 626–630, magenta), D-loop (residues 634–637, black) and H-loop (residues 661–663, green). Bound ligands are shown in ball-and-stick representation. KD values were taken from Zaitseva et al (2005b).
As shown in Figure 1, the catalytic cycle represents a ‘dual mode' mechanism, combining mechanical movement with the generation of chemical energy in order to ensure function. The catalytic cycle starts with the transition of the NBD from the nucleotide-free to the ATP-bound state, accompanied by a rigid-body motion of the helical domain (shown in light tan in Figure 1) towards the catalytic domain (shown in light yellow), as discussed in more detail below. An additional major change accompanying nucleotide binding in HlyB involves the N-terminus of helix 1, which harbors the last three residues of the Walker A motif (shown in blue). Whereas these three residues adopt a 310-helical conformation in the nucleotide-free state (Schmitt et al, 2003), a regular α-helical structure is observed in all other structures with bound nucleotide. Similarly, a non-canonical conformation of the Walker A motif was observed in the nucleotide-free state of GlcV (Verdon et al, 2003a). This implies that such localized alternative conformations might be important in vivo in controlling ATP binding to particular NBDs. We have also shown that ATP binding promotes dimerization of the HlyB-NBD, not only in the crystal structure but also in solution (Zaitseva et al, 2005b). This is expected as ATP plays an important role in the initiation of the catalytic cycle, engaging both NBDs and driving the formation of the composite dimer.
Comparison of the dimeric ATP- and ATP/Mg2+-bound crystal structures of either the H662A or the E631Q mutant forms revealed a root mean square deviation (r.m.s.d.) of 0.6 Å for 482 Cα-atoms. However, we noted a striking difference, dependent upon Mg2+, in the nature and composition of the water network within the nucleotide-binding site (also see below). Comparison of the three crystal structures of the HlyB-NBD (wild type, E631Q and H662A) in the ADP-bound state revealed an even smaller r.m.s.d., around 0.2 Å over 241 Cα-atoms (wild type/E631Q, 0.209 Å and wild type/H662A, 0.187 Å). This is further supported by an analysis of the B-factors of these ADP-bound structures that are qualitatively identical for all three structures (data not shown). Therefore, we shall only describe in detail the structure of the wild-type HlyB-NBD with bound ADP.
Architecture of the nucleotide-binding site
As shown in Figure 2A, ATP is bound at the dimer interface of the HlyB-NBD via interactions with the Walker A, B and the Q-loop from the cis monomer and interactions with the ABC-signature motif of the trans monomer. Furthermore, the D-loop of the trans monomer interacts with the backbone of S504 (Walker A) of the cis monomer, opening up the possibility to transmit the nature of the functional state of one ATP-binding site to the other site. This canonical arrangement was also observed for MJ0796 (Smith et al, 2002), MalK (Chen et al, 2003) and the HlyB-NBD with bound ATP/Mg2+ (Zaitseva et al, 2005a). Importantly, we note that in contrast to the ATP/Mg2+ structure (Figure 2B), the absence of Mg2+ results in a different arrangement of the water molecules within the ATP-binding site (Figure 2A). As a consequence of this difference, D630 (Walker B) is not in direct contact with the bound nucleotide and the ATP-bound structure has twice as many water-mediated contacts in the active site as the ATP/Mg2+-bound structure, resulting in a larger ligand–protein buried surface.
Figure 2.
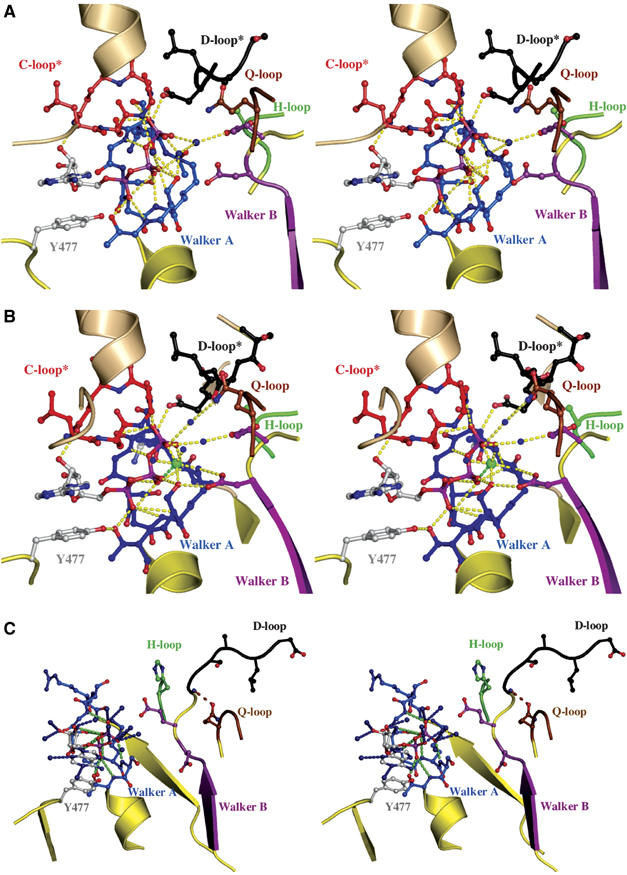
Nucleotide-binding sites. Stereoview of the ATP-binding (A) and ATP/Mg2+-binding(B) sites. Color-coding is identical to Figure 1. Direct and water-mediated protein–ATP interactions are highlighted in yellow. Water molecules are shown as blue spheres and Mg2+ as a green sphere. The interaction between D637 of the D-loop of the trans monomer and S504 of the Walker A motif of the cis monomer is indicated. ATP and amino acids involved in ligand interactions are shown in ball-and-stick representation. * indicates conserved motifs of the trans monomer participating in ATP coordination. (C) Stereoview of the ADP-binding site. ADP and residues involved in ligand interactions are shown in ball-and-stick representation, water molecules are blue spheres, protein–ADP interactions are highlighted in green and ADP–water interactions in blue. Color-coding is identical to Figure 1. The interaction between the side chain of Q550 and the amide backbone of T633 is highlighted by a dashed, brown line.
A closer look into the ADP-binding site is presented in Figure 2C. The adenine ring of ADP interacts with an aromatic amino acid, Y477 in HlyB, via π–π stacking of the two aromatic ring systems. Furthermore, the phosphate moiety of ADP is bound via both side- and main-chain interactions with residues of the Walker A motif. This pattern has been observed in all other crystal structures of NBDs in the ADP-bound state (Gaudet and Wiley, 2001; Karpowich et al, 2001; Yuan et al, 2001; Verdon et al, 2003a) with the exception of the CFTR-NBD1 (Lewis et al, 2004). In contrast to other NBD structures with ADP (Gaudet and Wiley, 2001; Karpowich et al, 2001; Verdon et al, 2003a; Lu et al, 2005), we were not able to obtain a structure for the ADP/Mg2+-bound state, despite the fact that we obtained crystals starting from ATP/Mg2+. This fits with our thermodynamic data (Zaitseva et al, 2005b) and implies that the cofactor Mg2+ is not tightly bound in the ADP state. However, more interesting is the direct interaction of Q550 (Q-loop) with the main chain of T633 (extended D-loop region) when ADP is bound. Thus, after ATP hydrolysis, but only after dissociation of inorganic phosphate (Karpowich et al, 2001), the helical domain rotates outward as a rigid body and is pinned in this position via the Q550–T633 interaction. Strikingly, a hinge axis (see below) covering the Walker B/D-loop region was identified for this particular step in the catalytic cycle. Consequently, it appears that the Walker B/D-loop region is not only involved in NBD–NBD communication and cofactor binding but also serves as an important control point, locking the helical domain into a non-productive position in the absence of ATP.
Structural plasticity of E631
We have recently proposed a ‘linchpin' model for ATP hydrolysis (Zaitseva et al, 2005a, 2005b). A key feature of the model is the catalytic dyad formed by E631 and H662 of HlyB. As a further test of this model, we determined the crystal structure of the E631Q NBD in the presence of ATP, with Mg2+ omitted in view of the residual ATPase activity of the E631Q mutant (Zaitseva et al, 2005a). Data statistics are summarized in Table I. As shown in Figure 3, the substituting Q631 interacts with the side chain of H662 (distance of 3.3 Å; highlighted in yellow in Figure 3) through a single H-bond. This is in contrast to its positioning in the linchpin model where E631 contacts H662 via one main-chain and one side-chain interaction (highlighted in gray in Figure 3). Thus, in the E631Q mutant, H662 is less restricted, resulting in a higher degree of flexibility. Moreover, the geometry of the single hydrogen bond in the E631Q mutant of HlyB-NBD is unfavorable, indicating that the stabilization of the side-chain conformation of the histidine will be minimal. On the other hand, in the ATP-bound dimer of MJ0796 (Smith et al, 2002), the corresponding Q171 forms no interaction at all with the corresponding histidine (H204) (highlighted in cyan in Figure 3). This fits with the strongly reduced ATPase activity of the mutant. Thus, this structure is informative in explaining why the HlyB-NBD E631Q mutant (Zaitseva et al, 2005a), but not the MJ0796 E171Q mutant (Moody et al, 2002), displays residual ATPase activity.
Figure 3.
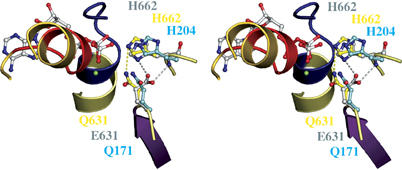
Structural flexibility of the H662–E631 interaction. Stereoview of the ATP-binding site of the HlyB-NBD E631Q mutant, the E171Q mutant of MJ 0796 (PDB entry 1L2T) and the hypothetical model of the wild-type HlyB-NBD in complex with ATP. Side chains of the E631Q mutant of HlyB-NBD (Q631 and H662) are shown in yellow, side chains of the E171Q mutant of MJ0796 (Q171 and H204) in cyan and side chains of the model of wild-type HlyB-NBD (E631 and H662) in gray. The single interaction of Q631 with the side chain of H662 is highlighted in yellow; the bidentate interaction of H662 and E631 proposed in the linchpin model is highlighted in gray. Color-coding is identical to Figure 1.
Hinge regions within the individual structures of the catalytic cycle
The two-domain architecture of ABC-NBDs was first described for HisP (Hung et al, 1998). In the case of HlyB, the catalytic domain corresponds to residues 467–549 and 626–707. This contains the motifs Walker A (residues 502–510) and B (residues 626–630), the D-loop (residues 634–637) and the H-loop (residues 661–663). The helical domain, presumed to act as a communicator between the catalytic domain and the TMDs, corresponds to residues 557–622 of HlyB and contains the ABC-signature motif (residues 606–610) and the SDR (residues 578–605), unique to each type of ABC-transporter (Schmitt et al, 2003). The helical domain is connected to the catalytic domain via the Q-loop (residues 550–556) and the Pro-loop (residues 623–625) (Schmitt et al, 2003). Despite the accumulating structural information however, potential hinges or bending residues within an NBD necessary to facilitate intramolecular movement were not previously identified. An analysis of potential hinges in the HlyB-NBD revealed that in the first step of the catalytic cycle, that is, nucleotide-free to ATP-bound, residues 549–553 (Q-loop) and 617–619 (C-terminus of helix 5) act as mechanical hinges (highlighted in magenta in Figure 4A) allowing inward rigid-body rotation of the helical domain. Thus, the location of the bending residues coincides with the domain definition of the NBD.
Figure 4.
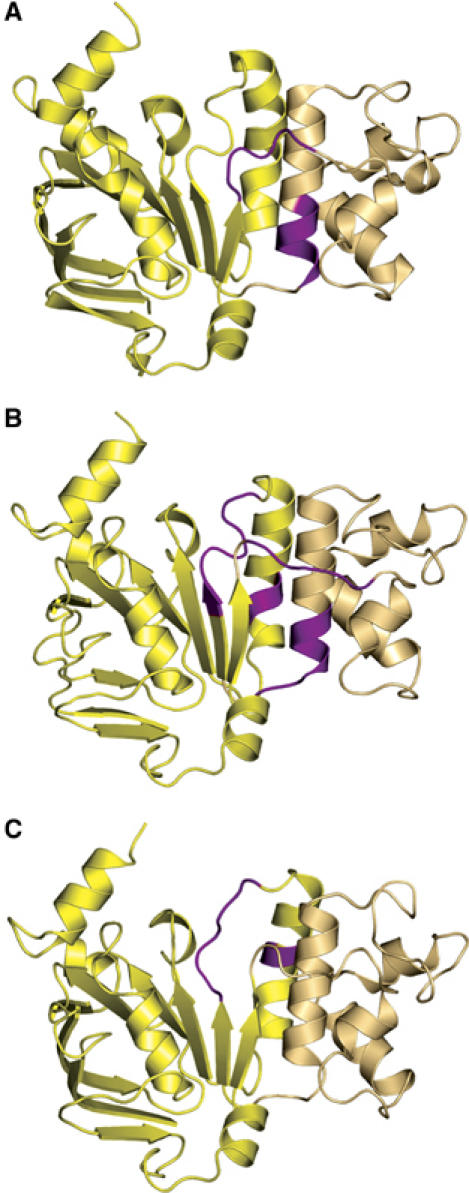
Hinges and bends during functional transitions. Hinges involved in (A) the transition from the nucleotide-free to the ATP-bound state, (B) the transition from the ATP/Mg2+- to the ADP-bound state and (C) the transition from the ADP-bound to the nucleotide-free state. Hinge regions are highlighted in magenta. The catalytic domain is shown in light yellow and the helical domain in light tan. Structures shown correspond to the starting point of the functional transitions.
In the crystal structure of BtuCD (Locher et al, 2002), the Q-loop of BtuD is in contact with the cytoplasmic L-loop of the membrane subunit BtuC. Locher (2004) proposed that this interaction is of importance for TMD–NBD communication. As the Q-loop also serves as a hinge, one can envision that any movement here is transmitted to the TMD in the form of a conformational rearrangement and ultimately the creation of a functional translocation pathway. In such an analysis, however, it is important to be aware of the possible effect of crystal packing on certain conformations under different conditions. In fact, in the apo structure, the Q-loop is partially involved in crystal contacts. Nevertheless, an analysis of the B-factors (Eyal et al, 2005) indicated no stabilization of the observed conformation due to these contacts.
Surprisingly, we find that apparently the requirement for bending residues is more complex in the reverse, outward rotation of the helical domain in the transition ATP/Mg2+-bound to the ADP-bound state (Figure 4B). Thus, in addition to the Q- and the Pro-loop, residues of the Walker B/D-loop region (629–635) and helix 6 (residues 647–648) also act as effective hinges (highlighted in magenta in Figure 4B). In contrast to the apo form, none of these hinges are involved in crystal packing contacts. Finally, in the conversion from the ADP-bound to the nucleotide-free state (Figure 4C), only residues 630–635 (Walker B/D-loop region) and helix 6 (residues 644–645) appear to act as mechanical hinges (highlighted in magenta). This further emphasizes the important role of the extended D-loop region of HlyB, which is located within the dimer interface in a position to play a pivotal role in the architecture of the ATP/Mg2+ dimer and in NBD–NBD communication (Locher et al, 2002; Smith et al, 2002; Chen et al, 2003). In the ADP-bound structure, the C-terminus of helix 6 is involved in crystal packing contacts, but again a B-factor analysis did not suggest that these contacts affect the conclusion that helix 6 acts as a mechanical hinge. Overall, our structural analysis of the HlyB-NBD in various functional states has demonstrated that all steps of the catalytic cycle involve significant conformational changes. In addition, we propose that distinct combinations of mechanical hinges within the protein are used to coordinate the corresponding conformational transitions corresponding to the individual steps of the catalytic cycle. Finally and in summary, whereas the inward rotation of the helical domain upon ATP binding uses the Q- and extended Pro-loops as hinges, the outward rotation in addition uses the Walker B/D-loop region and helix 6. Our analysis also provides a molecular picture of how functional changes during the catalytic cycle might be coupled to the action of mechanical hinges in a way that ultimately ensures efficient NBD–TMD (Q-loop/Pro-loop regions) and NBD–NBD (Walker B/D-loop region) communication.
Detection of asymmetry of the NBD dimer
Starting from the model proposed by Senior et al (1995) for the catalytic cycle of ABC-ATPases, several groups (see for example Senior et al, 1995; Sauna and Ambudkar, 2000; van Veen et al, 2000; van der Does and Tampe, 2004) have suggested that ATP hydrolysis within the NBD dimer is a sequential process leading to models of ‘two cylinder' transporter machines. As most ABC-transporters function as homodimers, this implies some form of asymmetry within the NBD dimer. However, from a structural point of view, this has so far received little attention. To understand better the proposed functional asymmetry, we first analyzed the character of intersubunit protein–protein interactions in both the ATP- and ATP/Mg2+-bound states of the HlyB-NBD (Figure 5). Whereas direct ATP–protein interactions are rather symmetric in both dimer structures (data not shown), symmetric and asymmetric protein–protein interactions do occur, in both the ATP- and the ATP/Mg2+-bound states. As evident from the schematic summary (Figure 5), the pattern of interactions in both states is qualitatively different, with a more prominent asymmetry in the presence of Mg2+. This emphasizes the crucial importance of the cofactor in generating asymmetry and explains why significant asymmetry was not detected in other previously studied ABC dimers, as Mg2+ was absent. The D-loop is heavily involved in asymmetric inter-monomer interactions in the ATP/Mg2+-bound state (Figure 5B) and is therefore well suited to sense changes occurring in the Walker A and ABC-signature motif of the opposing monomers. Nevertheless, owing to the static picture of crystal structures, we cannot yet determine the initial signals and interactions that promote such profound asymmetry.
Figure 5.
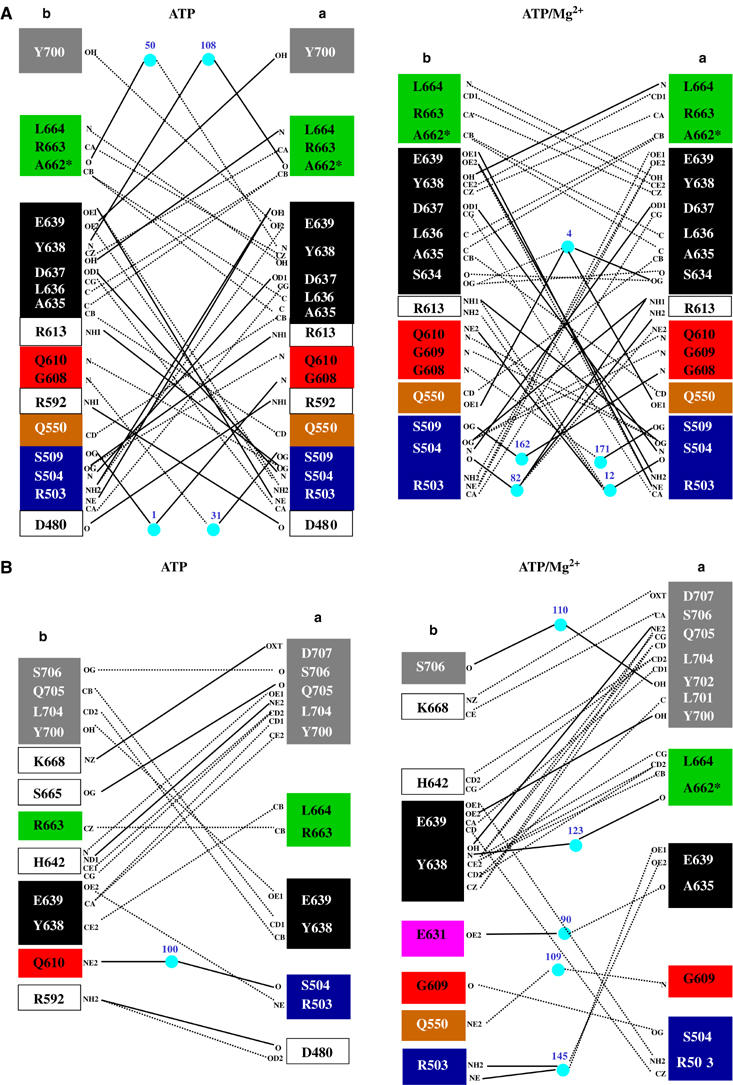
Importance of the cofactor Mg2+ in monomer–monomer interactions in the H662A dimer of HlyB. (A) Symmetric and (B) asymmetric interactions between the monomers of the ATP- (left panel) and ATP/Mg2+- (right panel) bound complexes of the HlyB-NBD H662A. Color-coding is identical to Figure 1. Solid lines represent hydrogen bonds (distance cutoff of 3.2 Å) and dashed line van der Waals interactions (distance cutoff of 4.0 Å). Light blue spheres represent water molecules. Letters indicate the atoms involved in the interactions.
An asymmetric exit tunnel for inorganic phosphate in the presence of Mg2+
The asymmetric pattern of interactions, in both the ATP- and ATP/Mg2+-bound structures, prompted us to perform a detailed cavity analysis of the dimer structures in the hope that this might highlight any functional significance of asymmetry. In Figure 6, the calculated cavities and tunnels in the H662A composite dimer are shown as blue solids. First, in the case of the ATP-bound state with no Mg2+ (Figure 6A), symmetrical tunnels reaching down towards the nucleotide-binding site are detectable. However, these tunnels do not reach the nucleotide and are closed by a salt bridge between D551 and R611, roughly 6 Å above the γ-phosphate group in both ATP-binding sites (indicated by the crossed arrows). The situation is different for the adenine moiety, which is completely solvent accessible in both sites. However, the ribose, α-, and β-phosphate moieties of ATP are also excluded from solvent. Identical results were obtained when we analyzed the ATP-bound dimeric structures of MJ0796 (Smith et al, 2002) and MalK (Chen et al, 2003) (data not shown). Thus, based on a cavity analysis of three structures of dimeric ATP-bound NBDs, inorganic phosphate should not be able to diffuse freely out of the binding site. Rather, a conformational change would have to occur within the protein in order to open an exit path for the cleaved inorganic phosphate.
Figure 6.
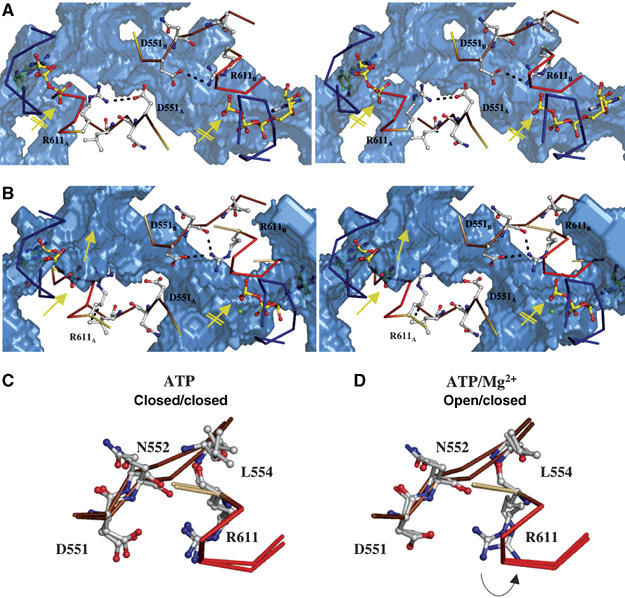
An exit tunnel for inorganic phosphate. Stereoview of the ‘phosphate tunnel' within the two ATP-binding sites of the ATP-bound (A) and the ATP/Mg2+-bound (B) composite dimer of HlyB-NBD H662A. Color-coding is identical to Figure 1. For simplicity only the Walker A, Q-loop and ABC-signature motif are shown in ribbon. Mg2+ is shown as a green sphere and amino acids involved in interactions in ball-and-stick representation. The subscripts indicate the monomer to which the amino acids belong. The accessible solvent area (ASA) of the HlyB-NBD dimer is presented as a transparent, blue solid. The interaction between D551 and R611, which acts as a gate for tunnel opening (no interaction between D551 and R611, closest distance of 4.7 Å) and closing (interaction distance of 3.3 Å), is highlighted. The arrows in panels A and B indicate the open (standard arrow) and closed (crossed arrow) phosphate tunnel. Structural superimposition of the individual monomers of the ATP (C) and ATP/Mg2+ (D) dimer of the HlyB-NBD H662A mutant. The flip of R611 in the ATP/Mg2+ complex, which acts as a tunnel gate, is indicated by an arrow.
We next performed a cavity analysis of the composite ATP dimer with bound Mg2+, calculating the positions of the adenine ring and the α- and β-phosphate moieties of ATP/ Mg2+ with respect to their exposure to solvent. The results showed that these were equivalent to those observed in the ATP-bound dimer without Mg2+. However, quite dramatically, the presence of Mg2+ (absolutely required for hydrolysis) in the composite dimer results in a clear asymmetry with respect to the γ-phosphate region (compare the positions of the standard and crossed arrows in Figure 6B). One site is still closed roughly 6 Å above the γ-phosphate, whereas, in the other site, a ‘phosphate exit tunnel' extends to the γ-phosphate group of the bound ATP. This open tunnel has a smallest diameter of roughly 4.4 Å, which should be sufficient to allow rather unrestricted passage of inorganic phosphate. Superimposition of the individual monomers of the dimeric ATP- and ATP/Mg2+-bound states emphasizes the importance of the salt bridge between D551 and R611 (Figures 6C and D). In the closed state, the salt bridge (distance 3.3 Å) locks the tunnel entrance, whereas in the open state the salt bridge open up owing to a flip of the R611 side chain (Figure 6D). Interestingly, statistical analysis of the conservation of these residues, using more than 10 000 ABC-transporter sequences (derived from www.sanger.ac.uk/cgi-bin/Pfam/getacc?PF0005), revealed conservation of residue 611 (allowing conservative substitutions with respect to the ability to form salt bridges or the alternative to form hydrogen bonds, that is, R (35%), K (31%) and Q (21%)). Position 551 displayed a lower but still significant degree of conservation (E (19%), D (22%), N (11%)). The pairwise distribution of amino acids at positions 551 and 611 further supports the idea of an interacting amino-acid pair. For example, residues capable of forming a salt bridge or a hydrogen bond appear in 66% of the sequences, with a K at position 611 (position 551: D (24%), E (24%), N (10%) and Q (8%)). The same applies if an R is present at position 611 (position 551: D (28%), E (19%), N (10%) and Q (9%)). Thus, we would like to propose that the interaction between R611 and D551 acts as a gate to close or open the phosphate exit tunnel in the HlyB-NBD structure. In the open state, this would allow the free diffusion of the product of ATP hydrolysis, inorganic phosphate. Strong support for the function of R611 and D551 as ‘door-keepers' comes from mutational analysis (Supplementary Figure S1). Mutating R611 or D551 to alanine resulted in proteins that retained ATPase activity although substantially reduced. More importantly, however, substitution of these residues abolished detectable cooperativity with respect to ATP hydrolysis. In contrast, the conservative mutation of R611 to lysine resulted in an enzyme with comparable activity and more importantly that retained positive cooperativity (Supplementary Figure S1). The loss of cooperativity is not simply a nonspecific effect due to the low level of residual activity, as the E631Q mutant of HlyB-NBD still displayed cooperative ATP hydrolysis, despite a reduced ATPase activity comparable to that in the R611A and D551A mutants (Zaitseva et al, 2005a). Thus, these two conserved amino acids may not only be important for asymmetric phosphate release from the HlyB-NBD, but may also be involved in molecular communication between the two ATP-binding sites, which normally results in the so far poorly understood positive cooperativity (Zaitseva et al, 2005b). It is equally important to note here that the presence of the phosphate tunnels could explain biochemical data derived from other isolated NBDs and full-length ABC-transporters, which strongly suggested sequential ATP hydrolysis (see for example (Urbatsch et al, 1995; Janas et al, 2003; Tombline et al, 2005). Janas et al (2003) showed that an NBD dimer could be still detected after single site ATP hydrolysis, but not after the second hydrolysis event. Extrapolated to our structural analysis, this result implies that the release of the first phosphate through the exit tunnel may occur in a fully assembled dimer without significant conformational change, whereas the second release would require a conformational change within the protein. Nevertheless, further investigations are necessary to determine whether this mechanism and its gating through the action of a salt bridge of the precise type discussed above are universal for ABC-ATPase, specific for ABC exporters or unique to HlyB.
The role of helix 6 as an enthalpic device
The F1 motor of the ATP synthase achieves a nearly 100% efficiency by converting chemical energy stored within ATP into elastic strain. Thus, no heat dissipates and all energy can be stored within the protein owing to ‘deformations' of certain structural motifs or secondary structure elements (Wang and Oster, 1998; Sun et al, 2003). A similar situation could arise in ABC-transporters, which ultimately use the energy of ATP to drive transport substrate translocation. We present below one such possibility in relation to the ADP-bound state of the HlyB-NBD and the associated deformation of helix 6. A superimposition of the individual structures of the catalytic cycle indicated that whereas ATP binding results in a rigid-body inward rotation of the helical domain, ATP hydrolysis and NBD dimer disassembly generate the outward rotation of the helical domain together with bending and displacement of helix 6 (residues 637–652), which is located immediately C-terminal to the D-loop. A 15° tilting of helix 6 in the ADP-bound state occurs owing to the breaking of one and the formation of four new hydrogen bonds, when compared with helix 6 in either the ATP-bound or the nucleotide-free state (Figure 7). This tilt is present in all three structures determined for the ADP state (wild type, E631Q and H662A) but not the two ATP dimers (H662A and E631Q). This suggests that helix 6 has an intrinsic conformational flexibility and we would like to suggest that tilting of this helix is used to store energy in the form of elastic strain during the catalytic cycle (Figure 7). It is important to note here that based on the principle of microscopic reversibility (Fersht, 1997), the ADP-bound state is structurally identical whether reached as a result of ATP hydrolysis or conversely from de novo binding of ADP. Thus, based on the observed helix tilting, it is feasible that helix 6 serves as a molecular device to store part of the energy released upon ATP hydrolysis in the form of elastic strain. This strain could be used for functional purposes such as ADP release. This presumably must require an active process of some kind, as the affinity of ADP for the NBD appears to be too high to allow spontaneous dissociation (see below). The utilization of stored energy in this way would provide a route for a programmed release of ADP from monomers, after an outward rotation of the helical domain following ATP hydrolysis and dimer disassembly, with concomitant transmission of conformational changes to the membrane domains.
Figure 7.
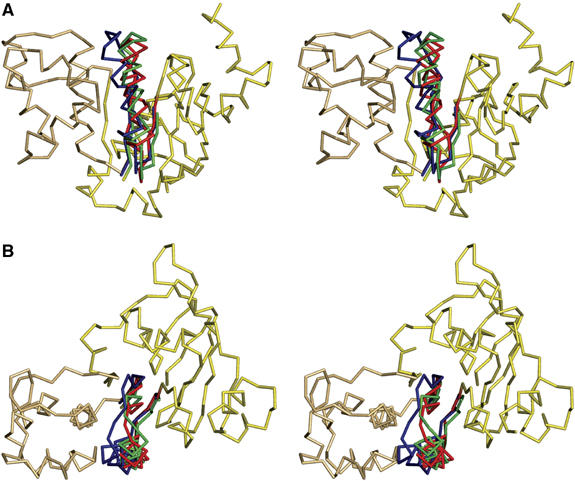
An enthalpic storage device for the chemical energy of ATP. (A) Structural superimposition of the nucleotide-free and ATP- and ADP-bound forms. For simplicity, only the structure of the ATP-bound form is shown as ribbons. Helix 6 of the nucleotide-free state is shown in green, for the ATP-bound state in red and for the ADP-bound state in blue. (B) 90° rotation in the plane with respect to panel A.
Mechano-chemistry associated with the action of ABC-NBDs
In contrast to channels, membrane transport proteins require a source of energy input to translocate the substrate across a membrane and against a concentration gradient. In the case of membrane transporters of the ABC-transporter family, this energy is directly provided by the NBDs. However, as elaborated above, ABC domains not only hydrolyze ATP, but also undergo an ATP-induced dimerization. Consequently, an NBD can use two fundamentally different mechanisms to fuel transport substrate translocation. First, mechanical energy from the rigid-body motion associated with ATP-induced dimerization and second chemical energy obtained from the hydrolysis of ATP.
For a detailed structural and functional analysis of the catalytic cycle, it is important to consider the nature and possible use of all energy generating steps. From the affinity constants for nucleotides and the ATP-induced dimerization constant of the NBD (Zaitseva et al, 2005b), the inherent energies of the individual steps can be calculated based on the Gibbs free energy relation. The ΔG values for ATP binding (KD=87.8±11.2 μM), ADP binding (KD=77.1±13.2 μM) and NBD dimerization (KD=1.2±0.2 μM) were calculated to be −17.3, −17.6 and −33.4 kJ/mol, respectively. From an energetic point of view, as ATP hydrolysis (around −30 to −35 kJ/mol) and NBD dimerization (−33.4 kJ/mol) are very similar, either step could represent the potential ‘power stroke' of the catalytic cycle. However, one has to keep in mind that the above ΔG values are derived from isolated NBDs and the presence of the TMDs of HlyB might change significantly the value of certain steps of the catalytic cycle, shifting the actual energetic distribution. Nevertheless, we emphasize that ABC-ATPases are capable of acting as dual mode engines as a source of both mechanical and chemical energy (Figure 1).
This analysis leads us to propose the following description of the catalytic cycle of the NBD and its functional implications. First, ATP induces dimerization, accompanied by a rigid-body movement of the helical domain, employing the Q- and Pro-loop as hinges. These conformational changes are transmitted to the TMDs and result in a rearrangement of the membrane helices and in initiation of transport. Secondly and equally important is ATP hydrolysis: this is the rate-limiting step of the catalytic cycle and therefore the point of control (Zaitseva et al, 2005a). We now propose that the sequential hydrolysis of two ATPs, reflected by the apparent sequential release of the two inorganic phosphates (see Figure 5), is due to the inherent asymmetry of the dimer. This raises the possibility to couple the hydrolysis steps, accompanied by sequential release of the phosphates for separate purposes. Therefore, overall, one catalytic cycle of the ABC domain would consume two ATPs. This closely resembles, for example, the ATP/transport substrate stoichiometry determined for OpuA from Lactobacillus lactis (Patzlaff et al, 2003). In HlyB, many cycles might be necessary to translocate the large HlyA molecule, although the proton motif force is possibly also involved in HlyA secretion (Koronakis et al, 1991).
An alternative explanation for the molecular events resulting in dimer dissociation has been proposed by Hunt and co-workers (Smith et al, 2002); here, the increased negative charge distribution within the nucleotide-binding site resulting from ATP hydrolysis is suggested to be sufficient for spontaneous dissociation of the dimer. We investigated this idea with respect to HlyB and have calculated the electrostatic surface potential of a monomer of the ATP-bound dimer of H662A. To our surprise and in contrast to the results obtained for MJ0796 (Smith et al, 2002) (Supplementary Figure S2), no significant increase in negative charges accompanying ATP hydrolysis was detected within the ATP-binding site of HlyB. Therefore, it appears unlikely that such a mechanism is sufficient to induce dimer disassembly in HlyB. In fact, superimposition of the hypothetical ADP-bound HlyB-NBD ‘dimer' on the structure of the NBD of BtuCD identical to the ADP-bound form (Locher et al, 2002) strongly suggests that the D-loops are involved in dimer dissociation (Supplementary Figure S3). Here, the resulting steric clashes occurring between opposing D-loops in the modeled HlyB-NBD/ADP dimer clearly indicate that such a ‘dimer' is not stable. As the region of the Walker B/D-loop region acts as one of the four hinges in the ATP–ADP transition (Figure 4), HlyB dimer disassembly could easily be coupled to conformational changes of the ABC domain upon catalysis.
In summary, we propose that the catalytic cycle of the isolated ABC-NBDs is divided into distinct steps that involve different conserved sequence motifs as key players, acting to coordinate intramolecular movements. The energy to drive rearrangement of the TMDs and substrate translocation is provided by two different molecular steps: mechanical energy derived by the rigid-body motion of the helical domain, which is induced by ATP binding ‘to create' the translocation pathway, while sequential ATP hydrolysis provides the chemical energy to complete the movement of the transport substrate across the membrane and to enable ADP dissociation. In this model, one can assign distinct functions to the conserved motifs of NBDs: the Walker A and B motifs, as well as the ABC signature and H-loops are required for ATP binding and hydrolysis, whereas the Pro-, Q- and D-loops act as hinges and are likely central to NBD–NBD and NBD–TMD communication. Finally, we have identified a novel salt bridge in a position capable of controlling sequential release of inorganic phosphate, and essential for cooperative activity.
Materials and methods
Protein purification, ATPase assays, crystallization and data collection
Purification and crystallization of HlyB-NBD and the mutants used were as described previously (Zaitseva et al, 2004). To obtain the E631Q, D551A, R611K and R611A mutants of HlyB-NBD, plasmid pPSG122 was used as a template. The mutation was introduced using the ligase chain reaction according to the protocol of the manufacturer (Epicentre). ATPase activity measurements were performed as described (Zaitseva et al, 2005b). For crystallization of the ADP-bound states, 25 mg/ml of the NBD in 70 mM CAPS pH 10.4, 30% glycerol and 10 mM ADP or ATP/Mg2+ was mixed with an equal volume of 100 mM Tris pH 8.0, 10% PEG 6000 and 5% MPD at 277 K. Crystals of the ATP-bound state of the HlyB-NBD H662A were obtained by mixing equal ratios of 10 mg/ml protein in 1 mM CAPS pH 10.4, 30% glycerol, 2 mM ATP, 1 mM EDTA with 100 mM sodium malonate pH 5.6, 10% PEG 5500-MME and 0.25 mM sodium acetate. Crystals of the ATP-bound state of the HlyB-NBD E631Q were obtained by mixing equal ratios of 10 mg/ml protein in 1 mM CAPS pH 10.4, 30% glycerol, 10 mM ATP, 1 mM EDTA with 100 mM Tris pH 8.5, 10% PEG 6000 and 5% MPD. Crystals were flash-frozen in a nitrogen stream. Data from single crystals were collected at beamline BW-6, DESY, Hamburg. Data were processed using DENZO and SCALEPACK (Otwinowski and Minor, 1997).
Structure determination and refinement
The structures were solved by molecular replacement using AMoRe (Navaza, 1994). The initial solutions were refined using REFMAC5 (Murshudov et al, 1997) employing TLS-grouped refinement, followed by repetitive rounds of manual rebuilding into 1Fo−Fc and 2Fo−Fc maps using O (Jones et al, 1991). In the initial state of refinement of the ATP-bound structure, strict non-crystallographic symmetry (NCS) was applied, which was released in the last four cycles of rebuilding and refinement. In the case of the E631Q mutant structure with bound ATP, NCS of the catalytic domains (residues 467–549 and 625–707) A/C and B/D was kept during all stages of refinement. ARP/wARP (Lamzin and Wilson, 1993) was used to determine water molecules at a conservative threshold of 3σ.
Structural alignments, determination of hinge bending residues and cavity analysis
Structural alignments were performed with LSQMAN (Kleywegt, 1996) using a ‘reduced' catalytic domain (residues 467–540 and 626–707) or the whole NBD (alignments of the different mutants in the same functional state). Bending residues/regions of the HlyB-NBD during the catalytic cycle were determined using the DynDom software (www.cmp.uea.ac.uk/dyndom/main.jsp). Analysis of potential cavities and tunnels in the ATP- and ATP/Mg2+-bound structures of HlyB-NBD H662A was performed using VOIDOO (Kleywegt and Jones, 1994) and MAMA (Kleywegt and Jones, 1999).
Figure preparation
All figures were prepared using Pymol (www.pymol.org).
Protein Data Bank accession code
Coordinates have been deposited under accession numbers 2FF7 (wild-type ADP), 2FFB (ADP E631Q), 2FFA (ADP H662A), 2FGJ (ATP H662A) and 2FGK (ATP E631Q).
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Sander Smits, Eckhard Hoffmann, Robert Ernst and Nils Hanekop for many discussions and support during data collection. We are indebted to Gleb Bourenkov for his excellent assistance. The University of Paris-Sud (IBH) and the DFG (grant Schm1279/2-3 and SFB 628 to LS) supported this work.
References
- Chang G (2003) Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J Mol Biol 330: 419–430 [DOI] [PubMed] [Google Scholar]
- Chang G, Roth CB (2001) Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 293: 1793–1800 [DOI] [PubMed] [Google Scholar]
- Chen J, Lu G, Lin J, Davidson AL, Quiocho FA (2003) A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol Cell 12: 651–661 [DOI] [PubMed] [Google Scholar]
- Chen J, Sharma S, Quiocho FA, Davidson AL (2001) Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc Natl Acad Sci USA 98: 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AL, Chen J (2004) ATP-binding cassette transporters in bacteria. Annu Rev Biochem 73: 241–268 [DOI] [PubMed] [Google Scholar]
- Eyal E, Gerzon S, Potapov V, Edelman M, Sobolev V (2005) The limit of accuracy of protein modeling: influence of crystal packing on protein structure. J Mol Biol 351: 431–442 [DOI] [PubMed] [Google Scholar]
- Fersht A (1997) Enzyme Structure and Mechanism. New York: WH Freeman and Company [Google Scholar]
- Gaudet R, Wiley DC (2001) Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J 20: 4964–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8: 67–113 [DOI] [PubMed] [Google Scholar]
- Holland IB, Schmitt L, Young J (2005) Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway. Mol Memb Biol 22: 29–39 [DOI] [PubMed] [Google Scholar]
- Hung LW, Wang IXY, Nikaido K, Liu PQ, Ames GFL, Kim SH (1998) Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 396: 703–707 [DOI] [PubMed] [Google Scholar]
- Janas E, Hofacker M, Chen M, Gompf S, van der Does C, Tampe R (2003) The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J Biol Chem 278: 26862–26869 [DOI] [PubMed] [Google Scholar]
- Jones PM, George AM (2004) The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci 61: 682–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF (2001) Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure 9: 571–586 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ (1996) Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D 52: 842–857 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D 50: 178–185 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1999) Software for handling macromolecular envelopes. Acta Crystallogr D 55: 941–944 [DOI] [PubMed] [Google Scholar]
- Koronakis V, Hughes C, Koronakis E (1991) Energetically distinct early and late stages of HlyB/HlyD-dependent secretion across both Escherichia coli membranes. EMBO J 10: 3263–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamzin VS, Wilson KS (1993) Automated refinement of protein models. Acta Crystallogr D 49: 129–147 [DOI] [PubMed] [Google Scholar]
- Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23: 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP (2004) Structure and mechanism of ABC transporters. Curr Opin Struct Biol 14: 426–431 [DOI] [PubMed] [Google Scholar]
- Locher KP, Lee AT, Rees DC (2002) The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science 296: 1091–1098 [DOI] [PubMed] [Google Scholar]
- Lu G, Westbrooks JM, Davidson AL, Chen J (2005) ATP hydrolysis is required to reset the ATP-binding cassette dimer into its resting state conformation. Proc Natl Acad Sci USA 102: 17969–17974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J Biol Chem 277: 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr D 50: 157–163 [Google Scholar]
- Oswald C, Holland IB, Schmitt L (2006) The motor domains of ABC-transporters/what can structures tell us? Naunyn Schmiedebergs Arch Pharmacol 372: 385–399 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology, Carter CW, Sweet RM (eds) London: Academic Press [DOI] [PubMed] [Google Scholar]
- Patzlaff JS, van der Heide T, Poolman B (2003) The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J Biol Chem 278: 29546–29551 [DOI] [PubMed] [Google Scholar]
- Reyes CL, Chang G (2005) Structure of the ABC transporter MsbA in complex with ADP.vanadate and lipopolysaccharide. Science 308: 1028–1031 [DOI] [PubMed] [Google Scholar]
- Sauna ZE, Ambudkar SV (2000) Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci USA 97: 2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt L, Benabdelhak H, Blight MA, Holland IB, Stubbs MT (2003) Crystal structure of the nucleotide binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J Mol Biol 330: 333–342 [DOI] [PubMed] [Google Scholar]
- Schmitt L, Tampé R (2002) Structure and mechanism of ABC-transporters. Curr Opin Struct Biol 12: 754–760 [DOI] [PubMed] [Google Scholar]
- Senior AE, al-Shawi MK, Urbatsch IL (1995) The catalytic cycle of P-glycoprotein. FEBS Lett 377: 285–289 [DOI] [PubMed] [Google Scholar]
- Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 10: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Chandler D, Dinner AR, Oster G (2003) Elastic energy storage in beta-sheets with application to F1-ATPase. Eur Biophys J 32: 676–683 [DOI] [PubMed] [Google Scholar]
- Tombline G, Muharemagic A, White LB, Senior AE (2005) Involvement of the ‘occluded nucleotide conformation' of p-glycoprotein in the catalytic pathway. Biochemistry 44: 12879–12886 [DOI] [PubMed] [Google Scholar]
- Urbatsch IL, Sankaran B, Weber J, Senior AE (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem 270: 19383–19390 [DOI] [PubMed] [Google Scholar]
- van der Does C, Tampe R (2004) How do ABC transporters drive transport? Biol Chem 385: 927–933 [DOI] [PubMed] [Google Scholar]
- van Veen HW, Margolles A, Muller M, Higgins CF, Konings WN (2000) The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J 19: 2503–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon G, Albers SV, Dijkstra BW, Driessen AJ, Thunnissen AM (2003a) Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucleotide-bound conformations. J Mol Biol 330: 343–358 [DOI] [PubMed] [Google Scholar]
- Verdon G, Albers SV, van Oosterwijk N, Dijkstra BW, Driessen AJ, Thunnissen AM (2003b) Formation of the productive ATP-Mg2+-bound dimer of GlcV, an ABC-ATPase from Sulfolobus solfataricus. J Mol Biol 334: 255–267 [DOI] [PubMed] [Google Scholar]
- Wang H, Oster G (1998) Energy transduction in the F1 motor of ATP synthase. Nature 396: 279–282 [DOI] [PubMed] [Google Scholar]
- Welch RA, Dellinger EP, Minshew B, Falkow S (1981) Haemolysin contributes to virulence of extra-intestinal E. coli infections. Nature 294: 665–667 [DOI] [PubMed] [Google Scholar]
- Ye J, Osborne AR, Groll M, Rapoport TA (2004) RecA-like motor ATPases—lessons from structures. Biochim Biophys Acta 1659: 1–18 [DOI] [PubMed] [Google Scholar]
- Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF (2001) The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J Biol Chem 276: 32313–32321 [DOI] [PubMed] [Google Scholar]
- Zaitseva J, Holland IB, Schmitt L (2004) The role of CAPS in expanding the crystallisation space of the nucleotide binding domain of the ABC-transporter from E. coli. Acta Crystallogr D 60: 1076–1084 [DOI] [PubMed] [Google Scholar]
- Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L (2005a) H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J 24: 1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva J, Jenewein S, Wiedenmann A, Benabdelhak H, Holland IB, Schmitt L (2005b) Functional characterization and ATP-induced dimerization of the isolated ABC-domain of the haemolysin B transporter. Biochemistry 44: 9680–9690 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures