

Abstract
Stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) responds to a variety of stress stimuli and controls cell fates such as cell cycle entrance, apoptosis and senescence. Stimuli such as ultraviolet irradiation and chemical reagents that damage genomic DNA induce the activation of the SAPK/JNK signaling pathway. However, it is unclear how the signal arising in the nucleus owing to DNA damage is transmitted to SAPK/JNK in the cytoplasm. Here, we report that the nuclear components Daxx and Ras-association domain family 1C (RASSF1C) link DNA damage to SAPK/JNK activation in HeLa cells. In response to DNA damage, Daxx localized in promyelocytic leukaemia-nuclear bodies (PML-NBs) undergoes ubiquitination and degradation. RASSF1C, a tumor suppressor and newly identified binding partner of Daxx, is constitutively anchored by Daxx in PML-NBs but is released from the nucleus when Daxx is degraded. This released RASSF1C translocates to cytoplasmic microtubules and participates in the activation of SAPK/JNK. Our data define a novel mechanism by which the Daxx–RASSF1C complex in PML-NBs couples nuclear DNA damage to the cytoplasmic SAPK/JNK signaling pathway.
Keywords: Daxx, DNA damage, JNK, PML body, RASSF1
Introduction
Stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), a member of the mitogen-activated protein kinase family, is activated in response to a wide range of stimuli that impinge on the plasma membrane. These stimuli include certain mitogenic signals, lysophosphatidic acid, inflammatory cytokines and changes in osmolarity induced by sorbitol (Zanke et al, 1996; Davis, 2000; Chang and Karin, 2001; Johnson and Lapadat, 2002; Lin, 2003; Nishina et al, 2004; Wada et al, 2004). Activated SAPK/JNK transduces these signals to the nucleus by phosphorylating target molecules such as c-Jun, JunD and ATF2, which in turn participate in the activation and formation of the AP-1 complex. However, SAPK/JNK can also be activated by a signal(s) that originates in the nucleus in response to genotoxic stress, such as that caused by the DNA-alkylating reagent methyl methanesulfonate (MMS) or ultraviolet (UV) irradiation (Adler et al, 1995; Tournier et al, 2000). Indeed, MMS is one of the most potent triggers of c-jun transcription and c-Jun/ATF-2 hyperphosphorylation mediated by SAPK/JNK activation (van Dam et al, 1995). MMS-mediated c-jun transcription is required for the induction of several AP-1 target genes, including apoptotic factors such as CD95L (Kolbus et al, 2000). Thus, SAPK/JNK activation induced by DNA damage plays a crucial role in the regulation of gene expression leading to apoptosis. However, the molecular mechanism by which DNA damage in the nucleus activates SAPK/JNK in the cytoplasm remains to be elucidated.
Promyelocytic leukaemia-nuclear bodies (PML-NBs) are highly structured protein complexes associated with the nuclear matrix. PML-NBs are composed of a striking variety of proteins, many of which are involved in DNA repair, gene regulation, the antiviral response, proteolysis, tumor suppression or apoptosis (Salomoni and Pandolfi, 2002; Dellaire and Bazett-Jones, 2004). Mice lacking PML-NBs have impaired immune function, exhibit chromosome instability and show increased sensitivity to carcinogens. Functionally, PML-NBs are dynamic sensors of cellular stress that rapidly disassemble following DNA damage.
A key component of PML-NBs is Daxx, a protein involved in apoptosis and transcriptional repression (Ishov et al, 1999; Zhong et al, 2000; Salomoni and Pandolfi, 2002). Daxx has been reported to interact with PML protein (Ishov et al, 1999), apoptosis signaling kinase ASK1 (Chang et al, 1998), and the death receptor Fas (Yang et al, 1997), as well as the transcription factors p53, ETS1, Pax3, NFκB and E2F1 (Michaelson and Leder, 2003). However, the precise function of Daxx in cell death is controversial because conflicting results have been obtained in the transient overexpression and gene knockout studies. Whereas Daxx overexpression potentiates the apoptotic pathways stimulated by Fas (Yang et al, 1997) or TGFβ (Perlman et al, 2001), disruption of the Daxx gene in mice results in early embryonic lethality and extensive apoptosis (Michaelson et al, 1999). Furthermore, small interfering RNA (siRNA)-mediated silencing of Daxx expression strongly sensitizes cells to stress-induced apoptosis that is accompanied by the activation of caspases and SAPK/JNK (Chen and Chen, 2003; Michaelson and Leder, 2003). Thus, evidence exists supporting both an apoptotic and an antiapoptotic roles for Daxx in stressed cells.
The tumor suppressor RASSF1 exists in two major isoforms, RASSF1A and RASSF1C, that are produced from the single RASSF1 gene by alternative splicing (Dammann et al, 2000). RASSF1A contains a Ras-association (RA) domain and an N-terminal cysteine-rich diacylglycerol-binding (C1) domain, whereas RASSF1C contains the RA domain and a unique N-terminal region lacking the C1 domain. RASSF1A, the better-studied isoform, regulates the cell cycle by blocking cell cycle progression and inhibiting cyclin D1 accumulation (Shivakumar et al, 2002; Song et al, 2004; Vos et al, 2004). In addition, RASSF1A localizes to an array of microtubules that extends from the perinuclear region to the periphery of HeLa cells and regulates mitosis through interaction with Cdc20, an activator of the anaphase-promoting complex (Song et al, 2004). By contrast, RASSF1C localizes to the nucleus of HeLa cells but does not influence mitosis. Rather, RASSF1C appears to play an ill-defined role in Ras-mediated cellular activities such as apoptosis (Vos et al, 2000). Thus, the structures and subcellular localizations of RASSF1A and RASSF1C differ and their physiological functions seem to be antagonistic, at least in some contexts.
In this study, we set out to define the nuclear components transmitting signaling initiated by DNA damage to the SAPK/JNK pathway and found that the Daxx–RASSF1C complex in PML-NBs functions upstream of SAPK/JNK. The Daxx–RASSF1C complex acts as a sensor of DNA damage in that the damage triggers the ubiquitination and degradation of Daxx. RASSF1C is released from the PML-NBs and the signal is transmitted to the cytoplasm when RASSF1C translocates to cytoplasmic microtubules and induces SAPK/JNK activation. Thus, our work establishes a new paradigm by which the Daxx–RASSF1C complex couples DNA-damaging stimuli to the SAPK/JNK signaling pathway.
Results
DNA damage induces Daxx degradation by the ubiquitin-proteasome pathway
Previous work had shown that Daxx silencing sensitizes cells to stress-induced apoptosis that is accompanied by SAPK/JNK activation and a correlative decrease in Daxx protein (Chen and Chen, 2003). We therefore analyzed Daxx expression levels in HeLa cells exposed to a variety of genotoxic stresses, including MMS, etoposide and UV-C irradiation. Interestingly, treatment with MMS or UV, but not with etoposide, reduced the level of HA-Daxx protein in a time course-dependent manner (Figure 1A, left and data not shown). Remarkably, we found that pretreatment of the cells with the proteasome inhibitor LLnL inhibited the degradation of HA-Daxx induced by MMS or UV (Figure 1A, right). We next examined the expression levels of endogenous Daxx in MMS- or UV-treated HeLa cells. Western blotting showed that, like exogenous Daxx, endogenous Daxx was degraded after MMS or UV treatment (Figure 1B). Confocal microscopy of these cells revealed that endogenous Daxx, which showed a pattern of nuclear dots and colocalized with PML protein in PML-NBs of untreated cells (Figure 1C), was degraded in response to MMS or UV such that it had almost disappeared from PML-NBs by 3.5 h post-treatment (Figure 1D). These results strongly suggest that DNA damage, such as that caused by MMS or UV, targets Daxx for degradation via the ubiquitin-proteasome pathway.
Figure 1.

DNA damage induces ubiquitination and degradation of Daxx in PML-NBs. (A) Inhibition of HA-Daxx degradation induced by MMS or UV by the proteasome inhibitor LLnL. HeLa cells transfected with pCMV-HA-Daxx were incubated with dimethylsulfoxide or LLnL (100 μM) before treatment with MMS or UV. Cell lysates were prepared at the indicated time points post-treatment and immunoblotted with anti-HA or anti-GAPDH (loading control) antibodies. (B) Degradation of endogenous Daxx in response to MMS or UV. HeLa cells were treated with MMS or UV and lysates prepared at the indicated time points post-treatment were immunoblotted with anti-Daxx antibody. (C) Localization of Daxx in PML-NBs. Untreated HeLa cells were immunostained with anti-Daxx and anti-PML antibodies. All images were obtained by confocal microscopy. Scale bar, 10 μm. (D) Degradation of endogenous Daxx in PML-NBs in response to MMS or UV. Untreated, MMS- or UV-treated HeLa cells were immunostained with anti-Daxx antibody at 3.5 h post-treatment. Scale bar, 10 μm. (E) DNA damage-induced ubiquitination of Daxx. HeLa cells were cotransfected with pCMV-HA-Daxx and pCMV-Flag-ubiquitin, and either left untreated or treated with UV or MMS. HA-Daxx was immunoprecipitated (IP) from lysates of these cells using anti-HA-beads and Daxx ubiquitination was detected by immunoblotting (IB) with anti-ubiquitin antibody. (F) Degradation of full-length Daxx protein only. HeLa cells were transfected with pCMV-HA-Daxx full-length or the deletion mutants HA-Daxx1–501 or HA-DaxxC501. Transfected cells were left untreated or treated with MMS or UV and lysed at the indicated time points post-treatment. Cell lysates were immunoprecipitated with anti-HA beads and immunoblotted with anti-HA antibody.
To determine whether Daxx degradation induced by MMS or UV required ubiquitination, HA-Daxx and Flag-ubiquitin were cotransfected into HeLa cells and the ubiquitination and degradation of HA-Daxx following MMS or UV treatment were examined. As expected, the poly-ubiquitination of HA-Daxx was detected within 1 h of MMS or UV exposure (Figure 1E). To ascertain which portion of the Daxx protein governed its degradation, HA-tagged full-length Daxx, a Daxx deletion mutant representing the Daxx N-terminal domain (Daxx1–501), and a Daxx deletion mutant representing the Daxx C-terminal domain (DaxxC501) were introduced into HeLa cells. When these cells were treated with MMS or UV, we found that neither mutated form of Daxx could be degraded (Figure 1F), indicating that both ends of the Daxx protein are required for its degradation in response to DNA damage. Our findings are the first evidence that DNA damage induces the ubiquitination and degradation of Daxx.
Daxx binds to and colocalizes with RASSF1C in PML-NBs
Although most studies have demonstrated that Daxx localizes primarily in PML-NBs (Ishov et al, 1999), there are some reports that Daxx moves from the nucleus to the cytoplasm in response to certain stresses (Charette and Landry, 2000; Song and Lee, 2003). However, in our study, neither confocal microscopy nor Western blotting revealed a translocation of Daxx to the cytoplasm in response to MMS or UV treatment (Figure 1D and data not shown). We therefore postulated the existence of a molecule(s) that transmits the signal that Daxx has been degraded from the PML-NBs to the cytoplasm. To identify such a molecule(s), we carried out yeast two-hybrid screening using Daxx as bait and determined that RASSF1C bound to Daxx at high frequency (16 out of 3 × 106 transformants). To test whether Daxx could bind to RASSF1C in mammalian cells, we performed co-immunoprecipitation experiments in which cDNAs encoding HA-Daxx, Myc-RASSF1C and Myc-RASSF1A were transfected into COS7 cells. (COS7 cells have a higher efficiency of DNA transfection than HeLa cells.) As was true in yeast, HA-Daxx bound strongly to Myc-RASSF1C in COS7 cells but only weakly to Myc-RASSF1A (Figure 2A). Moreover, the interaction between Daxx and RASSF1C held true for the endogenous proteins, as revealed by immunoprecipitation and immunoblotting in HeLa cells (Figure 2B).
Figure 2.
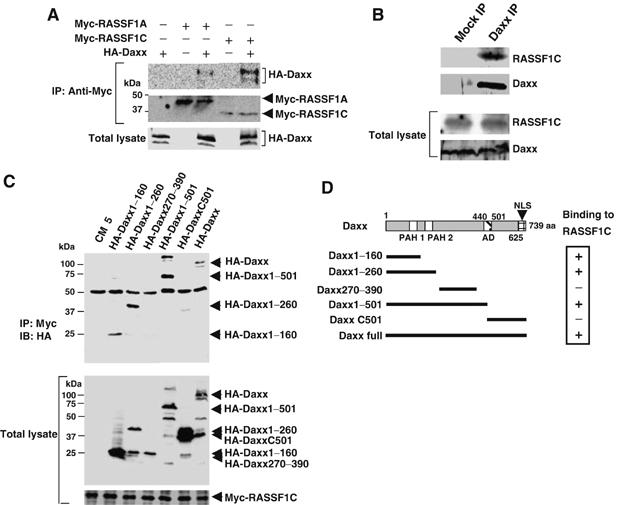
The N-terminal region of Daxx binds to RASSF1C. (A) RASSF1 binds Daxx in COS7 cells. COS7 cells were cotransfected with pCMV-HA-Daxx plus pCMV-Myc-RASSF1A or pCMV-Myc-RASSF1C. Myc-RASSF1A or Myc-RASSF1C was immunoprecipitated using anti-Myc antibody and immunoblotted with anti-HA antibody. (B) Endogenous RASSF1C binds endogenous Daxx. Lysates of untreated HeLa cells were immunoprecipitated with anti-Daxx antibody and immunoblotted with anti-RASSF1C antibody. Mock IP, lysates were immunoprecipitated with control IgG. (C) Mapping of the RASSF1C-interacting domain of Daxx. COS7 cells were cotransfected with HA-Daxx full-length or the indicated HA-Daxx deletion mutants plus Myc-RASSF1C. Immunoprecipitation and immunoblotting were carried out with anti-Myc and anti-HA antibodies, respectively. Numbers indicate amino-acid positions in the full-length Daxx protein. (D) Schematic description of the Daxx deletion mutants used in (C) and their binding to RASSF1C. PAH, paired amphipathic alpha helices domain; AD, acid-rich region; NLS, nuclear localization signal.
To identify the region of Daxx that binds to RASSF1C, we generated a series of Daxx deletion constructs (Figure 2C and D). Full-length HA-Daxx and mutants of HA-Daxx containing the first 260 N-terminal amino acids co-immunoprecipitated with Myc-RASSF1C (Figure 2C and D), suggesting the importance of the Daxx PAH1 and PAH2 domains for binding to RASSF1C. Conversely, the first 220 N-terminal amino acids of Myc-RASSF1C, which contain the RA domain, were sufficient for binding to HA-Daxx (data not shown). We then examined the subcellular localization of RASSF1C and Daxx in HeLa cells using confocal microscopy. In line with the co-immunoprecipitation experiments, GFP-RASSF1C clearly colocalized with DsRed-Daxx in PML-NBs in HeLa cells (Figure 3A–D). However, GFP-RASSF1C could not localize in PML-NBs when coexpressed with DsRed-DaxxC501, a mutant Daxx protein that cannot bind to RASSF1C (Figure 3E–H). Furthermore, using anti-RASSF1C antibody, we were able to demonstrate the colocalization of endogenous RASSF1C with HA-Daxx in the PML-NBs of 293T cells (see Supplementary Figure S1). These results indicate that, in a range of human cells, RASSF1C is localized primarily in the PML-NBs and is anchored in these complexes via binding to the N-terminal region of Daxx.
Figure 3.
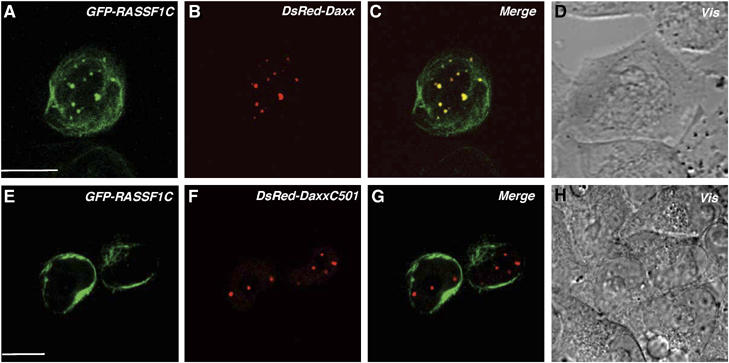
The N-terminus of Daxx is required for the colocalization of RASSF1C with Daxx in PML-NBs. HeLa cells were cotransfected with full-length Daxx (DsRed-Daxx) (A–D) or the C-terminus of Daxx (DsRed-DaxxC501) (E–H) plus GFP-RASSF1C. Transfected cells were cultured for 36 h on a glass-based dish and examined by confocal microscopy. Vis, light microscopic image. Scale bar, 10 μm.
Daxx degradation promotes the translocation of RASSF1C from the nucleus to the cytoplasm
When COS7 cells expressing HA-Daxx and Myc-RASSF1C were treated with MMS or UV, the amount of Daxx bound to RASSF1C was observed to steadily decrease with time (Figure 4A). Although the level of Myc-RASSF1C in total cell lysates of the treated cells remained constant at all time points (Figure 4A), the amount of Myc-RASSF1C in the nuclear fraction gradually declined (Figure 4B). Interestingly, this reduction of Myc-RASSF1C in the nuclear fraction could be rescued by overexpression of HA-Daxx (Figure 4C). These results indicate that, in response to DNA damage, RASSF1C is released from the nucleus in a manner that depends on Daxx degradation.
Figure 4.

The degradation of Daxx promotes the release of RASSF1C from the nucleus. (A) Dissociation of RASSF1C from Daxx in response to DNA damage. COS7 cells expressing HA-Daxx and Myc-RASSF1C were treated with MMS or UV and lysates prepared at the indicated time points were immunoprecipitated and immunoblotted with anti-Myc and anti-HA antibodies, respectively. (B) Reduction of RASSF1C in the nucleus. COS7 cells expressing Myc-RASSF1C were treated with MMS or UV and lysates were prepared at the indicated time points. The nuclear fraction was isolated and immunoblotted with anti-Myc antibody. Nucleolin and β-actin served as a nuclear marker and a cytosolic marker, respectively. (C) Daxx overexpression suppresses RASSF1C release from the nucleus. COS7 cells coexpressing Myc-RASSF1C and HA-Daxx were treated with MMS or UV and the nuclear fractions of lysates were immunoblotted as in (B).
To confirm the release of RASSF1C from the nucleus, we used immunostaining to investigate the subcellular localization of endogenous RASSF1C in HeLa cells subjected to MMS or UV treatment. In untreated cells, endogenous RASSF1C was confined to the nucleoplasm and colocalized with endogenous Daxx in the PML-NBs (Figure 5A, upper panels). Endogenous RASSF1C was not detectable in the cytoplasm of untreated cells (Supplementary Figure S2). However, in MMS- or UV-treated cells, endogenous RASSF1C (but not Daxx) clearly translocated from the nucleus into the cytoplasm (Figure 5A, middle and lower panels, white arrowheads). Intriguingly, RASSF1C appeared to accumulate in the cytoplasm of treated cells as a fiber-like structure (Figure 5A, middle and lower panels). To determine the effect of exogenous Daxx on this translocation of RASSF1C, we analyzed the localization of endogenous RASSF1C at the single-cell level in a mixed culture of control (nontransfected) and HA-Daxx-expressing HeLa cells. In the absence of DNA damage, RASSF1C was detected in the nucleus in both control and HA-Daxx-expressing cells (Figure 5B, upper panels). However, at 1.5 h after MMS or UV treatment, RASSF1C translocated to the cytoplasm in control cells (Figure 5B, white arrowheads) but remained in the nucleus in HA-Daxx-expressing cells (Figure 5B, white arrows). Statistical analysis showed that the frequency of RASSF1C translocation into the cytoplasm after MMS or UV treatment was significantly higher in control cells than in HA-Daxx-expressing cells (Figure 5C). These results indicate that RASSF1C is anchored in the PML-NBs by binding to Daxx and is released from the nucleus into the cytoplasm when Daxx is degraded in response to DNA damage.
Figure 5.
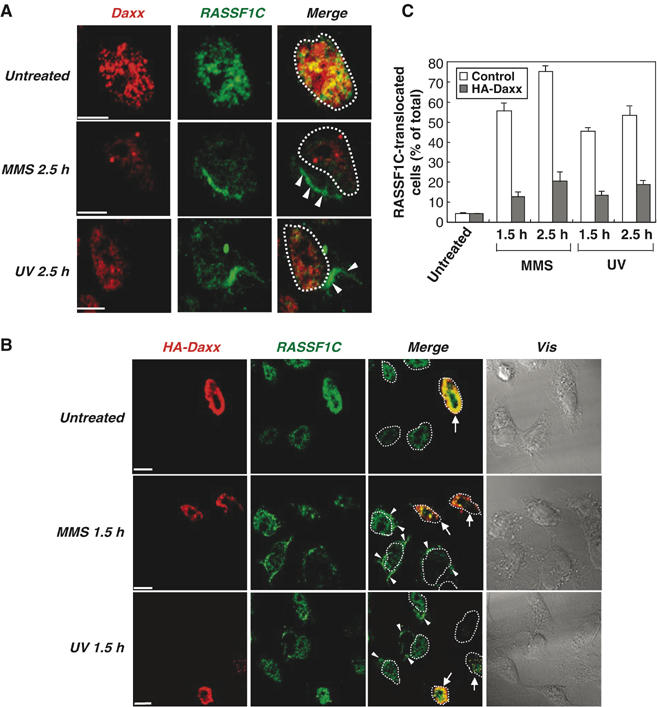
Daxx overexpression inhibits DNA damage-induced translocation of RASSF1C from the nucleus to the cytoplasm. (A) Endogenous RASSF1C translocation to the cytoplasm in response to DNA damage. HeLa cells that were left untreated or treated with MMS or UV were fixed at 2.5 h post-treatment and immunostained with anti-RASSF1C and anti-Daxx antibodies. White arrowheads indicate the accumulation of endogenous RASSF1C in the cytoplasm. White dots outline the nucleus. Scale bar, 5 μm. (B, C) Daxx inhibits RASSF1C translocation to the cytoplasm. (B) Single-cell immunohistochemical analysis. HeLa cells were transfected with pCMV-HA-Daxx and were left untreated or treated with MMS or UV. Cells were fixed at 1.5 h post-treatment and immunostained with anti-HA and anti-RASSF1C antibodies. White arrowheads indicate the accumulation of RASSF1C in the cytoplasm. White arrows indicate cells expressing HA-Daxx. White dots outline the nucleus. Scale bar, 5 μm. (C) Statistical analysis. The number of cells in (B) showing RASSF1C translocation to the cytoplasm at 1.5 and 2.5 h post-treatment was counted in 100 cells/sample and expressed as a percentage of the total cell number. Data shown are the mean percentage±s.e.m. of data obtained from at least three independent experiments.
RASSF1C is translocated to the microtubules in response to MMS treatment
RASSF1C and RASSF1A are known to bind to microtubules and regulate their stabilization (Rong et al, 2004; Vos et al, 2004; Liu et al, 2005). It was therefore not surprising that the cytoplasmic staining pattern of endogenous RASSF1C in DNA-damaged HeLa cells was reminiscent of microtubule staining (Figure 5A and B, white arrowheads). To confirm that the RASSF1C which dissociates from Daxx indeed translocates to the microtubules, we immunostained MMS-treated HeLa cells with anti-RASSF1C and anti-β-tubulin antibodies. The network- and fiber-like pattern of RASSF1C staining largely overlapped that of β-tubulin staining (Figure 6A), clearly indicating that RASSF1C released from the nucleus in response to DNA damage translocates to the microtubules.
Figure 6.
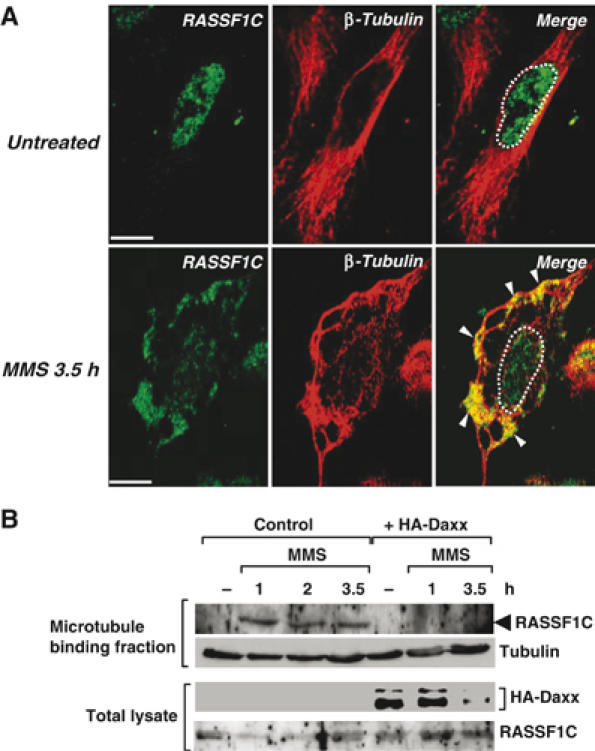
Endogenous RASSF1C is translocated from the nucleus to the cytoplasmic microtubules in response to DNA damage. (A) HeLa cells were left untreated or treated with MMS and immunostained with anti-RASSF1C and anti-β-tubulin antibodies at 3.5 h post-treatment. White arrowheads indicate the accumulation of endogenous RASSF1C at the microtubules. White dots outline the nucleus. Scale bar, 5 μm. (B) Microtubule cosedimentation. HeLa cells transfected with pCMV-HA-Daxx or pCMV empty vector were left untreated or treated with MMS. The cell lysates were incubated with taxol to stabilize microtubules. The microtubule-binding fraction was isolated as described in Materials and methods and immunoblotted with anti-RASSF1C and anti-β-tubulin antibodies.
To demonstrate biochemically that RASSF1C interacts with microtubules following MMS treatment, we performed a microtubule cosedimentation assay (Piddini et al, 2001). Strikingly, endogenous RASSF1C was detected in the microtubule-containing fractions of MMS-treated cells but not in that of untreated cells (Figure 6B, left). When the assay was repeated in MMS-treated HA-Daxx-overexpressing cells, the cosedimentation of RASSF1C with the microtubules was dramatically inhibited (Figure 6B, right). Treatment of HeLa cells with UV resulted in partial colocalization of endogenous nuclear RASSF1C with β-tubulin, suggesting that the binding of RASSF1C to microtubules differs somewhat between MMS- and UV-treated cells (Supplementary Figure S3). Nevertheless, our data are the first demonstration that endogenous RASSF1C is anchored by Daxx in the nucleus and translocates to the cytoplasmic microtubules in response to DNA damage. As the microtubules are involved in the recruitment of signaling molecules and the formation of the so-called signalosome, our findings imply that RASSF1C release owing to Daxx degradation is an important link in the signal transduction pathway leading from the nucleus to the cytoplasm.
Daxx expression influences DNA damage-induced SAPK/JNK activation
To investigate the role of RASSF1C release in DNA damage signal transduction, we first established a time course of SAPK/JNK activation in MMS-treated HeLa cells. Significantly, stimuli that act on the plasma membrane of a cell induce a different pattern of SAPK/JNK activation than stimuli that cause DNA damage. For example, SAPK/JNK activation induced by hyper-osmolarity (sorbitol) is rapid and transient and reaches its maximum within 30 min of stimulus administration (Supplementary Figure S4). In contrast, MMS induces SAPK/JNK activation that is slow to start but is prolonged, reaching at maximum at about 2 h post-stimulus. These results support the hypothesis that MMS-induced DNA damage triggers a signaling mechanism leading to SAPK/JNK activation that is distinct from the mechanism linking cell membrane events to SAPK/JNK activation.
To determine whether the release of RASSF1C from Daxx might be involved in this mechanism, we prepared an siRNA duplex against human Daxx mRNA (Daxx-RNAi) that efficiently knocked down the expression of endogenous Daxx in HeLa cells (Figure 7A). Consistent with the results shown in Figures 4 and 5, this decrease in Daxx was associated with the translocation of much of the nuclear RASSF1C to the cytoplasm (Figure 7B). Statistical analysis confirmed that the proportion of HeLa cells showing RASSF1C accumulation in the cytoplasm was much greater in Daxx-RNAi-treated cells than in control cells. These data clearly show that Daxx serves as an anchor protein for RASSF1C in the nucleus. However, it should be noted that some other component(s) may contribute to the nuclear retention of RASSF1C, since a small amount of RASSF1C could still be detected in the nuclei of Daxx-RNAi-treated cells.
Figure 7.
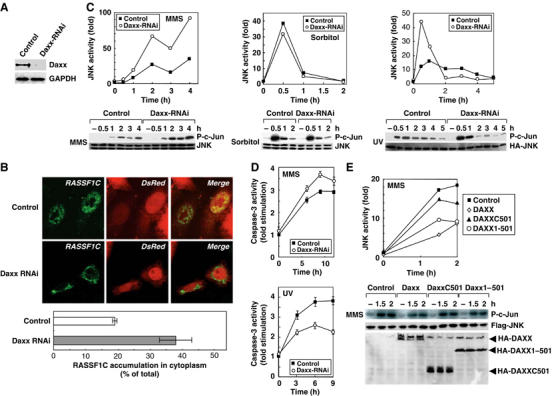
Daxx influences SAPK/JNK activation induced by DNA damage. (A) siRNA-induced reduction of Daxx protein. HeLa cells were transfected with pSuper-control-RNAi or pSuper-Daxx-RNAi vector. Lysates prepared at 65 h post-transfection were immunoprecipitated with anti-Daxx antibody and the immunoprecipitates were immunoblotted with anti-Daxx antibody. (B) Daxx silencing enhances cytoplasmic RASSF1C accumulation. HeLa cells were cotransfected with pSuper-control-RNAi or pSuper-Daxx-RNAi vector plus pCMV-DsRed (to indicate pSuper vector-transfected cells) and immunostained with anti-RASSF1C antibody (upper panels). The percentage of total cells that showed RASSF1C accumulation in the cytoplasm was determined by examination of 100 DsRed-positive cells/sample (lower panel). Data shown are the mean percentage±s.e.m. of data obtained from at least three independent experiments. (C) Daxx silencing promotes MMS- or UV-induced SAPK/JNK activation. HeLa cells transfected with pSuper-control-RNAi or pSuper-Daxx-RNAi vector were treated with MMS, sorbitol or UV and lysates were prepared at the indicated time points post-treatment. SAPK/JNK activity was measured by in vitro kinase assay using the GST-c-Jun substrate (lower panels). Data shown in the upper panels are the number of fold increase in P-c-Jun production in each sample compared to untreated-control. (D) The effects of Daxx silencing on MMS- or UV-induced caspase-3 activation. HeLa cells transfected with pSuper-control-RNAi or pSuper-Daxx-RNAi were treated with MMS or UV and lysates were prepared at the indicated time points post-treatment. Caspase-3 activity was measured as described in Materials and methods. Results shown are the mean fold stimulation±s.e.m. of caspase-3 activation in each sample compared to untreated-control from four independent experiments. (E) The N-terminus of Daxx inhibits MMS-induced SAPK/JNK activation. HeLa cells transfected with control, pCMV-HA-Daxx full-length, HA-Daxx1–501 or HA-DaxxC501 vector were treated with MMS and lysates were prepared at the indicated time points post-treatment. SAPK/JNK actitvity was measured as for (C) except that JNK was immunoprecipitated with anti-Flag antibody (lower panel). Data shown in upper panel are the number of fold increase in P-c-Jun production in each sample compared to untreated control.
We next examined the effect of Daxx-RNAi on SAPK/JNK activation induced by MMS, sorbitol or UV treatment. Although MMS-induced SAPK/JNK activation was enhanced in Daxx-RNAi-treated HeLa cells compared to control cells (Figure 7C, left), there was no difference in sorbitol-induced SAPK/JNK activation in these cultures (Figure 7C, center). UV-induced SAPK/JNK activation in control cells peaked at 1 h and then gradually declined. Curiously, however, in UV-irradiated Daxx-RNAi-treated cells, the early phase of SAPK/JNK activation was enhanced whereas the late phase was suppressed compared to controls (Figure 7C, right). These intriguing results show that the expression level of Daxx specifically influences SAPK/JNK activation in HeLa cells in response to DNA-damaging stimuli but does not play a role in SAPK/JNK activation induced by agents (such as sorbitol) that affect the plasma membrane.
A recent report has shown that the late phase of SAPK/JNK activation is involved in proapoptotic processes, whereas the early phase can play an antiapoptotic role (Ventura et al, 2006). We therefore examined the effect of Daxx silencing on caspase-3 activation in HeLa cells subjected to MMS or UV. Daxx silencing enhanced caspase-3 activity in MMS-treated cells (Figure 7D, top), consistent with the late-phase pattern of SAPK/JNK activation (proapoptotic) in MMS-treated Daxx-silenced cells. On the other hand, Daxx silencing inhibited caspase-3 activity in UV-treated cells (Figure 7D, bottom), consistent with the early phase pattern of SAPK/JNK activity (antiapoptotic) in UV-treated Daxx-silenced cells. These results support two separate physiological roles for the early and late phases of SAPK/JNK activation in promoting cell survival or cell death.
To determine which region of Daxx was required for MMS-induced SAPK/JNK activation, we transfected HeLa cells with vectors expressing HA-Daxx full-length, HA-DaxxC501 or HA-Daxx1–501 (refer to Figure 2D). We found that transfection of HA-Daxx full-length or HA-Daxx1–501, both of which can bind to RASSF1C, reduced MMS-induced SAPK/JNK activation, whereas transfection of HA-DaxxC501, which cannot bind to RASSF1C, only slightly decreased SAPK/JNK activation (Figure 7E). HA-Daxx full-length also attenuated UV-induced SAPK/JNK activation in HeLa cells (Supplementary Figure S5). These results indicate that Daxx negatively regulates DNA damage-induced SAPK/JNK activation in HeLa cells, and that the RASSF1C-binding N-terminal region of Daxx is essential for this regulation.
RASSF1C is required for DNA damage-induced SAPK/JNK activation
To investigate the role of RASSF1C in DNA damage-induced SAPK/JNK activation, we transfected HeLa cells with an siRNA duplex effective against all human isoforms of RASSF1 (RASSF1-RNAi). As expected, RASSF1-RNAi decreased the expression levels of both endogenous RASSF1C mRNA and RASSF1C protein (Figure 8A). We next tested whether RASSF1 silencing affected the localization of Daxx and its degradation in response to DNA damage. In the absence of DNA damage, there was no difference between untreated control cells and RASSF1-RNAi-treated cells in Daxx subcellular localization (Figure 8B). Interestingly, RASSF1 silencing did not affect Daxx degradation induced by MMS or UV treatment either (Figure 8C). However, we found that RASSF1-RNAi almost completely abrogated MMS-induced SAPK/JNK activation and partially inhibited UV-induced SAPK/JNK activation (Figure 8D). There was no effect of RASSF1-RNAi on sorbitol-induced SAPK/JNK activation (Figure 8E). These data suggest that RASSF1 (and most likely RASSF1C) is crucial for transducing the signal that DNA damage has occurred to the SAPK/JNK pathway. Furthermore, caspase-3 activation in response to MMS or UV treatment was inhibited by RASSF1 silencing (Figure 8F). Thus, our data clearly demonstrate that RASSF1 is involved in the late-phase SAPK/JNK activation and associated caspase-3 activation that promotes the apoptosis of DNA-damaged HeLa cells.
Figure 8.
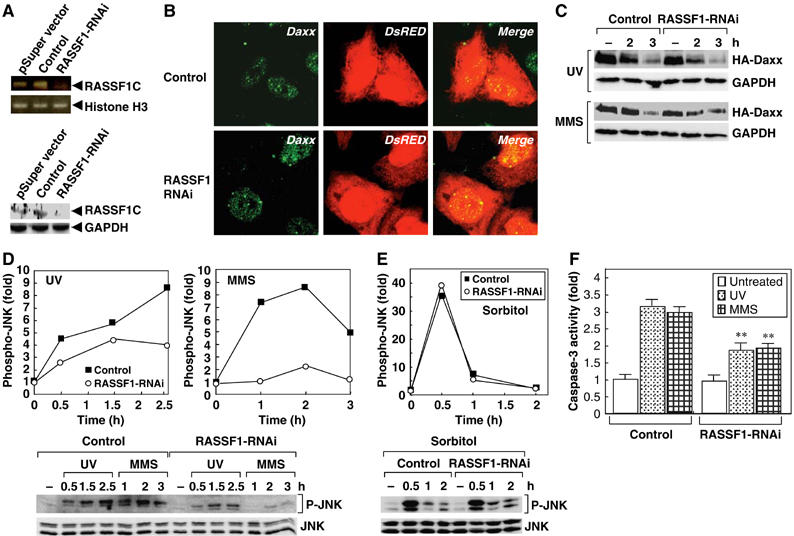
RASSF1C is required for DNA damage-induced SAPK/JNK activation. (A) RNAi-induced reduction of RASSF1C mRNA and protein. HeLa cells were transfected with pSuper-empty, pSuper-control-RNAi or pSuper-RASSF1-RNAi vector. Lysates were prepared at 65 h post-treatment and amounts of RASSF1C mRNA and protein were estimated by PCR analysis (top) and immunoblotting (bottom), respectively. GADPH and Histone H3, loading controls. (B) RASSF1 silencing does not affect Daxx localization. HeLa cells cotransfected with pSuper-control-RNAi or pSuper-RASSF1-RNAi vector plus pCMV-DsRed were immunostained with anti-Daxx antibody. (C) RASSF1 silencing does not affect Daxx degradation in response to DNA damage. HeLa cells cotransfected with pSuper-control-RNAi or pSuper-RASSF1-RNAi vector plus HA-Daxx were treated with MMS or UV and lysates were prepared at the indicated time points. Lysates were immunoblotted with anti-HA antibody. (D, E) The effects of RASSF1 silencing on SAPK/JNK activation. HeLa cells transfected with pSuper-control-RNAi or pSuper-RASSF1-RNAi vector were treated with MMS, UV or sorbitol and lysates were prepared at the indicated time points post-treatment. Activation of SAPK/JNK was determined by immunoblotting with anti-phospho SAPK/JNK (P-JNK) antibody (lower panel). Data shown in upper panel are the number of fold increase in P-JNK production in each sample compared to untreated-control. (F) The effects of RASSF1 silencing on MMS- or UV-induced caspase-3 activation. HeLa cells transfected with pSuper-control-RNAi or pSuper-RASSF1-RNAi vector were treated with UV or MMS and lysates were prepared at 10 h post-treatment. Caspase-3 activity was measured as described in Materials and methods. Results shown are the mean fold increase±s.e.m. in caspase-3 activity in each sample compared to untreated control. **P<0.01 comparing control-RNAi- and RASSF1-RNAi-treated cells (Student's t-test).
Both RASSF1C and RASSF1A bind to the proapoptotic, Ste20-like protein kinase MST1 (Khokhlatchev et al, 2002). SAPK/JNK is a downstream target of MST1, and overexpression of MST1 in a variety of cell types results in apoptosis (Graves et al, 1998). MST1 can form a complex with the Ras-GTP-binding protein, Nore1, and this complex can act as a Ras effector unit that mediates the apoptotic effects of Ras (Ortiz-Vega et al, 2002; Vos et al, 2003). These observations have led us to hypothesize that RASSF1C released from the nucleus in response to DNA damage may positively regulate Ras-Nore1-MST1 signaling and thus activate SAPK/JNK (Supplementary Figures S6 and S7). Indeed, we have found that the inactive forms of H-Ras or MST1 block MMS- and UV-induced SAPK/JNK activation (Supplementary Figures S6C and S7). The inactivation of other small GTPases, such as Rac1, Cdc42 or RhoA, did not affect SAPK/JNK activation (Supplementary Figure S6B). Furthermore, RASSF1C and the active form of H-Ras can bind and cooperate to activate the SAPK/JNK pathway (Supplementary Figure S6A and D). Interestingly, recent reports have shown that RASSF1A can inhibit SAPK/JNK activation (Whang et al, 2005) and that RASSF1A-Nore1 binding exhibits different properties from RASSF1C-Nore1 binding (Ortiz-Vega et al, 2002). These differences may explain the apparently antagonistic effects of RASSF1C and RASSF1A on SAPK/JNK activation.
Discussion
Although it is clear that SAPK/JNK is stimulated by DNA-damaging agents such as MMS and UV, the molecular mechanism linking DNA damage in the nucleus to SAPK/JNK activation in the cytoplasm has been obscure. Based on our results, we propose that the characteristically delayed activation of SAPK/JNK that follows DNA damage is due to degradation of Daxx in PML-NBs that leads to the release of RASSF1C and the translocation of this molecule from the nucleus to microtubules in the cytoplasm (Figures 1 and 7). Our data suggest that Daxx degradation may be the rate-limiting step for late-phase SAPK/JNK activation, because the gradual disappearance of Daxx correlates with the time course of SAPK/JNK activation following MMS treatment (Figure 1B and Supplementary Figure S4). Indeed, Daxx silencing accelerates MMS-induced SAPK/JNK activation and, conversely, Daxx overexpression inhibits SAPK/JNK activation (Figure 7C and E).
On the other hand, the molecular mechanism of UV-induced SAPK/JNK activation is more complicated. UV triggers signaling both from the plasma membrane and from the nucleus, and both of these pathways can lead to SAPK/JNK activation (Kitagawa et al, 2002). The early phase of UV-induced SAPK/JNK activation is triggered primarily by membrane proximal events such as the engagement of the cell surface receptors for epidermal growth factor (EGF), tumor necrosis factor or interleukin-1 (Rosette and Karin, 1996; Kitagawa et al, 2002). In our study, Daxx silencing altered the subcellular localization of RASSF1C in untreated HeLa cells (Figure 7B). In UV-treated HeLa cells, Daxx silencing significantly enhanced the early phase of SAPK/JNK activation but inhibited its late phase (Figure 7C). We believe that the enhancement of the early phase of SAPK/JNK activation may be due to an interaction between RASSF1C released from the nucleus and H-Ras that has been activated by a signal from the cell membrane. Two lines of evidence support this hypothesis. Firstly, the early phase of UV-induced SAPK/JNK activation can be mediated through the EGFR-Ras pathway (Adler et al, 1996; Kitagawa et al, 2002). Secondly, we found that RASSF1C can form a complex with H-Ras that participates in SAPK/JNK activation (Supplementary Figure S6A and D). However, the precise molecular mechanism underlying SAPK/JNK downregulation by Daxx silencing in UV-treated HeLa cells at later time points is unclear. A clue may lie in our observation that Daxx overexpression inhibited the late phase of UV-induced SAPK/JNK activation (Supplementary Figure S5), suggesting that Daxx degradation is also involved in UV-induced late-phase SAPK/JNK activation.
A previous report has shown that Daxx silencing via siRNA stimulates UV-induced SAPK/JNK activation in HeLa cells (Chen and Chen, 2003). Our work has confirmed this result and extended it to show that Daxx degradation is also required for SAPK/JNK activation following MMS treatment of HeLa cells (Figures 1 and 7 and Supplementary Figure S5). These data collectively indicate that Daxx functions as a negative regulator of DNA damage-induced SAPK/JNK activation. However, Khelifi et al (2005) have recently demonstrated that Daxx is required for stress-induced SAPK/JNK activation in primary fibroblasts, and that Daxx is induced in these cells upon exposure to UV or hydrogen peroxide. Thus, whether Daxx promotes or hinders SAPK/JNK activation remains controversial. One possible explanation for the controversial results might be due to different cell types, for example, the tumor suppressor p53 expresses in primary fibroblasts, but not HeLa cells (Matlashewski et al, 1986). It should be noted that p53 modulates the basal level of SAPK/JNK activity, but also affects the susceptibility of a cell to respond to UV (Ramaswamy and Pelling, 1999). Variation in the genetic backgrounds of the cells used in different studies might affect experimental outcomes, perhaps influencing both Daxx function and SAPK/JNK activation in response to DNA damage.
One intriguing aspect of our results is that the ubiquitination and degradation of Daxx in PML-NBs is the key step that induces the activation of downstream signaling molecules and thus the cellular response to DNA damage (Figure 1). In response to genotoxic stress, PML-NBs rapidly disassemble into large supramolecular complexes, dispersing the DNA repair factors contained in these complexes to sites of DNA damage (Dellaire and Bazett-Jones, 2004). However, the signaling network in the PML-NBs that senses DNA damage and activates downstream signaling cascades is not fully understood. Our findings suggest that the ubiquitination of Daxx within the PML-NBs may be an important event in the triggering of this network. The identification of the molecule(s) that catalyzes ubiquitination of Daxx in PML-NBs is therefore of considerable interest. It is tempting to speculate that either the PML protein or the ATRX syndrome protein (Xue et al, 2003), both of which possess a ring-finger-like motif and can bind to Daxx, may act as E3 ubiquitin ligases that ubiquitinate Daxx in response to DNA damage. Another candidate identified while this paper was under review is SPOP/Cul3-ubiquitin ligase. BTB domain-containing speckle-type POZ protein (SPOP) can function as an adaptor binding to Daxx as Daxx is ubiquitinated by Cul3-based ubiquitin ligase (Kwon et al, 2006).
Our results clearly show that endogenous RASSF1C mainly colocalizes with Daxx in the nucleus in unstimulated HeLa cells, and translocates into the cytoplasm following MMS or UV treatment (Figures 5 and 6). However, we also found that overexpressed GFP-RASSF1C localized to the periphery of the nucleus as well as to the PML-NBs (Figure 3). We speculate that, in this latter situation, there may have been insufficient endogenous Daxx to act as a nuclear anchor protein for the high levels of GFP-RASSF1C present. Similar considerations and differences between cell types and experimental conditions may explain the previous finding that overexpressed RASSF1C was localized mostly in the cytoplasm (Rong et al, 2004). These workers (and others) also found that RASSF1C can associate with tubulin and potentially stabilize microtubules (Rong et al, 2004; Vos et al, 2004). In our study, we showed that MMS treatment caused microtubule fibers to thicken and form bundles, phenomena not observed in untreated cells (Figure 6). This change in microtubule structure may be explained by the DNA damage-induced translocation of endogenous RASSF1C to microtubules.
In conclusion, our study demonstrates that Daxx, the protein that anchors RASSF1C in the PML-NBs, undergoes ubiquitin-dependent degradation that promotes RASSF1C release from the nucleus in response to DNA damage. RASSF1C released from the nucleus translocates to the cytoplasmic microtubules and regulates SAPK/JNK activation. These data provide a new paradigm by which the Daxx–RASSF1C complex couples DNA damage in the nucleus to activation of the SAPK/JNK pathway in the cytoplasm.
Materials and methods
Cell culture and transfections
HeLa and COS7 cell lines were maintained as described previously (Kitagawa et al, 2002). For transient expression in mammalian cells, cDNAs were cloned into pCMV5 (Invitrogen). Vectors were transfected into cells using the LipofectAMINE method (Invitrogen).
DNA-damaging stimuli
Cells were treated with 1 mM MMS (Kanto Kagaku), 100 J/m2 UV-C-type delivered via a 254-nm wavelength DNA Stratalinker (Stratagene), 40 μg/ml etoposide (Sigma) or 500 mM sorbitol (Hani Kagaku) for 30 min.
Antibodies
Antibodies to Daxx (M-112), PML (PG-M3), nucleolin/C23 (MS-3), β-actin (C-11), β-tubulin (H-235), ubiquitin (P4D1) and SAPK/JNK1 (C-17 and FL) were from Santa Cruz Biotechnology; anti-phospho-SAPK/JNK was from Cell Signaling; anti-Myc (9E10), anti-Flag (M2) and anti-Flag M2-Agarose affinity gel were from Sigma; anti-hemagglutinin (HA, 12CA5) and anti-HA-conjugated to beads were from Roche; anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from Chemicon; anti-RASSF1C was from the UTSW CBI Antibody Core. Secondary anti-mouse IgG antibodies conjugated with Alexa 488 and 568 were purchased from Molecular Probes.
Immunoprecipitation and immunoblotting
HeLa and COS7 cells were lysed, incubated with specific antibody and beads, fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and subjected to standard immunoblotting procedures as described previously (Kitagawa et al, 2002).
Confocal microscopy and subcellular localization
HeLa cells were cultured on a glass coverslip and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) at room temperature (RT). After treatment with 0.1% Triton X-100 in PBS, the cells were incubated with blocking solution (2% bovine serum albumin, 2% fetal calf serum in PBS) before incubation with primary antibody for 1 h at RT. Cells were washed with PBS and incubated for 1 h with Alexa 488- or Alexa 568-conjugated secondary antibodies. After PBS washes, the coverslip was mounted and viewed on a Carl Zeiss confocal microscope equipped with LSM510 software. Excitation wavelengths were 488 or 543 nm. To visualize subcellular localization of proteins in HeLa cells, cDNAs encoding human RASSF1C and mouse Daxx were cloned into pEGFP-C2 and pDsRed1-C1 (Clontech), respectively. Transfected cells were cultured for 36 h in a glass-based dish (Iwaki) and examined by confocal microscopy as above.
Yeast two-hybrid assay
A human brain cDNA library (Clontech) and the MATCHMAKER GAL4 Two-Hybrid system 3 were used for the yeast two-hybrid assay in which Daxx served as bait.
Nuclear fractionation
Nuclear fractions of treated cells were obtained using the Nuclear/Cytosol Fractionation kit (Biovision) according to the manufacturer's instructions.
Microtubule cosedimentation
The microtubule cosedimentation assay was performed as described previously with slight modifications (Piddini et al, 2001). Briefly, HeLa cells were lysed in an ice-cold solubilizing buffer. To stabilize microtubules, taxol (40 μM; Sigma) was added to the lysates followed by incubation for 30 min at 37°C. Samples were centrifuged through a 10% sucrose cushion at 100 000 g for 10 min at 25°C. The resulting pellets were resuspended in SDS–PAGE loading buffer.
SAPK/JNK kinase assay
HeLa cells were lysed and immunoprecipitated with anti-SAPK/JNK antibody to detect endogenous SAPK/JNK, or anti-Flag or anti-HA antibodies to detect exogenous SAPK/JNK. SAPK/JNK activity in immunoprecipitates was measured using GST-c-Jun as an in vitro substrate in the presence of 60 μM [γ-32P]ATP, as described previously (Kitagawa et al, 2002).
RNA knockdown
Daxx and RASSF1 siRNA vectors were constructed by inserting oligonucleotides containing Daxx or RASSF1 cDNA sequences (Daxx-siRNA, 5′-catcctctctagggtcctg-3′; RASSF1-siRNA, 5′-ggagtacaatgcccagatcaacagcaacc-3′) into pSuper vector. pSuper vectors in which four nucleotides of each inserted cDNA were replaced with random nucleotides were used as controls. To assess the efficacy of RASSF1-RNAi, total RNA of HeLa cells was isolated with Trizol reagent (Invitrogen) and first-strand cDNA was synthesized using SuperScript™ III RNase H− Reverse Transcriptase (Invitrogen). The following paired primers were used for the polymerase chain reaction (PCR): RASSF1C (5′-atgggcgaggcggaggc-3′ and 5′-tcacccaagggggcagg-3′), Histone H3 (5′-cggaattcatggctcgcactaagcaaactg-3′ and 5′-aggtcgactcacgccctctctccg).
Caspase 3 protease activity
Caspase 3 activity was assayed as previously described (Kitagawa et al, 2002). Briefly, HeLa cell lysates were incubated with DEVD-MCA (20 μM; Peptide Institute). The reaction was terminated with cold water, and the release of free MCA (excitation 355 nm, emission 460 nm) was monitored at RT.
Supplementary Material
Supplementary Information Figure S1
Supplementary Information Figure S2
Supplementary Information Figure S3
Supplementary Information Figure S4
Supplementary Information Figure S5
Supplementary Information Figure S6
Supplementary Information Figure S7
Acknowledgments
We thank Drs JM Penninger and W-C Yeh for valuable comments. This work was partly supported by a Grant-in-Aid for Scientific Research on a Priority Area from the Ministry of Education, Culture, Sport, Science and Technology of Japan and the Ministry of Health, Labor and Welfare of Japan.
Competing financial interests The authors declare that they have no competing financial interests.
References
- Adler V, Fuchs SY, Kim J, Kraft A, King MP, Pelling J, Ronai Z (1995) jun-NH2-terminal kinase activation mediated by UV-induced DNA lesions in melanoma and fibroblast cells. Cell Growth Differ 6: 1437–1446 [PubMed] [Google Scholar]
- Adler V, Pincus MR, Polotskaya A, Montano X, Friedman FK, Ronai Z (1996) Activation of c-Jun-NH2-kinase by UV irradiation is dependent on p21ras. J Biol Chem 271: 23304–23309 [DOI] [PubMed] [Google Scholar]
- Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D (1998) Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281: 1860–1863 [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40 [DOI] [PubMed] [Google Scholar]
- Charette SJ, Landry J (2000) The interaction of HSP27 with Daxx identifies a potential regulatory role of HSP27 in Fas-induced apoptosis. Ann N Y Acad Sci 926: 126–131 [DOI] [PubMed] [Google Scholar]
- Chen LY, Chen JD (2003) Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol Cell Biol 23: 7108–7121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP (2000) Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25: 315–319 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Dellaire G, Bazett-Jones DP (2004) PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays 26: 963–977 [DOI] [PubMed] [Google Scholar]
- Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, Chernoff J, Clark EA, Krebs EG (1998) Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J 17: 2224–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG (1999) PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147: 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: 1911–1912 [DOI] [PubMed] [Google Scholar]
- Khelifi AF, D'Alcontres MS, Salomoni P (2005) Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ 12: 724–733 [DOI] [PubMed] [Google Scholar]
- Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J (2002) Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol 12: 253–265 [DOI] [PubMed] [Google Scholar]
- Kitagawa D, Tanemura S, Ohata S, Shimizu N, Seo J, Nishitai G, Watanabe T, Nakagawa K, Kishimoto H, Wada T, Tezuka T, Yamamoto T, Nishina H, Katada T (2002) Activation of extracellular signal-regulated kinase by ultraviolet is mediated through Src-dependent epidermal growth factor receptor phosphorylation. Its implication in an anti-apoptotic function. J Biol Chem 277: 366–371 [DOI] [PubMed] [Google Scholar]
- Kolbus A, Herr I, Schreiber M, Debatin KM, Wagner EF, Angel P (2000) c-Jun-dependent CD95-L expression is a rate-limiting step in the induction of apoptosis by alkylating agents. Mol Cell Biol 20: 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH, Baek SH, Chiba T, Tanaka K, Bang OS, Joe CO, Chung CH (2006) BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of DAXX for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem 281: 12664–12672 [DOI] [PubMed] [Google Scholar]
- Lin A (2003) Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25: 17–24 [DOI] [PubMed] [Google Scholar]
- Liu L, Vo A, McKeehan WL (2005) Specificity of the methylation-suppressed A isoform of candidate tumor suppressor RASSF1 for microtubule hyperstabilization is determined by cell death inducer C19ORF5. Cancer Res 65: 1830–1838 [DOI] [PubMed] [Google Scholar]
- Matlashewski G, Banks L, Pim D, Crawford L (1986) Analysis of human p53 proteins and mRNA levels in normal and transformed cells. Eur J Biochem 154: 665–672 [DOI] [PubMed] [Google Scholar]
- Michaelson JS, Bader D, Kuo F, Kozak C, Leder P (1999) Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev 13: 1918–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JS, Leder P (2003) RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J Cell Sci 116: 345–352 [DOI] [PubMed] [Google Scholar]
- Nishina H, Wada T, Katada T (2004) Physiological roles of SAPK/JNK signaling pathway. J Biochem (Tokyo) 136: 123–126 [DOI] [PubMed] [Google Scholar]
- Ortiz-Vega S, Khokhlatchev A, Nedwidek M, Zhang XF, Dammann R, Pfeifer GP, Avruch J (2002) The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene 21: 1381–1390 [DOI] [PubMed] [Google Scholar]
- Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA (2001) TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol 3: 708–714 [DOI] [PubMed] [Google Scholar]
- Piddini E, Schmid JA, de Martin R, Dotti CG (2001) The Ras-like GTPase Gem is involved in cell shape remodelling and interacts with the novel kinesin-like protein KIF9. EMBO J 20: 4076–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy NT, Pelling JC (1999) Mutational status of the p53 gene modulates the basal level of jun N-terminal kinase and its inducibility by ultraviolet irradiation in A1-5 rat fibroblasts. Mol Carcinog 25: 262–272 [DOI] [PubMed] [Google Scholar]
- Rong R, Jin W, Zhang J, Sheikh MS, Huang Y (2004) Tumor suppressor RASSF1A is a microtubule-binding protein that stabilizes microtubules and induces G2/M arrest. Oncogene 23: 8216–8230 [DOI] [PubMed] [Google Scholar]
- Rosette C, Karin M (1996) Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 274: 1194–1197 [DOI] [PubMed] [Google Scholar]
- Salomoni P, Pandolfi PP (2002) The role of PML in tumor suppression. Cell 108: 165–170 [DOI] [PubMed] [Google Scholar]
- Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA (2002) The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol 22: 4309–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Lee YJ (2003) Catalase, but not MnSOD, inhibits glucose deprivation-activated ASK1-MEK-MAPK signal transduction pathway and prevents relocalization of Daxx: hydrogen peroxide as a major second messenger of metabolic oxidative stress. J Cell Biochem 90: 304–314 [DOI] [PubMed] [Google Scholar]
- Song MS, Song SJ, Ayad NG, Chang JS, Lee JH, Hong HK, Lee H, Choi N, Kim J, Kim H, Kim JW, Choi EJ, Kirschner MW, Lim DS (2004) The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC–Cdc20 complex. Nat Cell Biol 6: 129–137 [DOI] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870–874 [DOI] [PubMed] [Google Scholar]
- van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P (1995) ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J 14: 1798–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ (2006) Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell 21: 701–710 [DOI] [PubMed] [Google Scholar]
- Vos MD, Ellis CA, Bell A, Birrer MJ, Clark GJ (2000) Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J Biol Chem 275: 35669–35672 [DOI] [PubMed] [Google Scholar]
- Vos MD, Martinez A, Elam C, Dallol A, Taylor BJ, Latif F, Clark GJ (2004) A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res 64: 4244–4250 [DOI] [PubMed] [Google Scholar]
- Vos MD, Martinez A, Ellis CA, Vallecorsa T, Clark GJ (2003) The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J Biol Chem 278: 21938–21943 [DOI] [PubMed] [Google Scholar]
- Wada T, Joza N, Cheng HY, Sasaki T, Kozieradzki I, Bachmaier K, Katada T, Schreiber M, Wagner EF, Nishina H, Penninger JM (2004) MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat Cell Biol 6: 215–226 [DOI] [PubMed] [Google Scholar]
- Whang YM, Kim YH, Kim JS, Yoo YD (2005) RASSF1A suppresses the c-Jun-NH2-kinase pathway and inhibits cell cycle progression. Cancer Res 65: 3682–3690 [DOI] [PubMed] [Google Scholar]
- Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W (2003) The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA 100: 10635–10640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Khosravi-Far R, Chang HY, Baltimore D (1997) Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89: 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, Kyriakis J, Liu FF, Woodgett JR (1996) The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr Biol 6: 606–613 [DOI] [PubMed] [Google Scholar]
- Zhong S, Salomoni P, Ronchetti S, Guo A, Ruggero D, Pandolfi PP (2000) Promyelocytic leukemia protein (PML) and Daxx participate in a novel nuclear pathway for apoptosis. J Exp Med 191: 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure S1
Supplementary Information Figure S2
Supplementary Information Figure S3
Supplementary Information Figure S4
Supplementary Information Figure S5
Supplementary Information Figure S6
Supplementary Information Figure S7