

Abstract
Suv39h1 is a histone H3 lysine-9 (H3-K9) specific methyltransferase (HMT) that is associated with gene silencing through chromatin modification. The transition from proliferation into differentiation of muscle cell is accompanied by transcriptional activation of previously silent muscle genes. I report Suv39h1 interaction with myogenic regulator MyoD in proliferating muscle cells and its HMT activity, which is associated with MyoD, diminishes as differentiation proceeds. The Suv39h1–MyoD complex was detected on the chromatin regulatory regions of a silent differentiation signal muscle gene myogenin and that Suv39h1 presence correlated with H3-K9 methylation. Increased Suv39h1 expression repressed MyoD-dependent muscle gene expression and this property required its HMT activity. This repression required Suv39h1 association with MyoD as well as sustained methylation of H3-K9 on myogenin promoter. Suv39h1 was required for muscle gene repression because its abrogation by siRNA activates these gene expressions by MyoD. These findings suggest that Suv39h1 presence in association with MyoD on the promoter of muscle genes silences gene transcription, providing a necessary checkpoint between proliferation and differentiation.
Keywords: chromatin, histone lysine-9 methylation, muscle differentiation, muscle gene repression, myogenic regulator MyoD
Introduction
Skeletal muscle differentiation is a genetic program regulated at the transcriptional level. The myogenic basic helix–loop–helix (bHLH) transcription factor MyoD acts as a regulator in muscle cell determination (myoblast) and differentiation (Sabourin and Rudnicki, 2000). In initiating differentiation, MyoD activates transcription by binding to the E-box located in enhancer/promoter regions of muscle-specific genes. The initial event in the transition of proliferative muscle cell (myoblast) to differentiate involves the activation of the key early muscle gene myogenin followed by growth arrest and late muscle genes (Andres and Walsh, 1996). Although MyoD is expressed in myoblasts prior to the onset of differentiation, it fails to activate gene expression in these cells without differentiation promoting cues. MyoD failure in myoblasts may be to prevent premature differentiation for the maintenance of cell identity. There are several well-described mechanisms by which the inability of MyoD in myoblasts to initiate differentiation program is impeded (Berkes and Tapscott, 2005; Forcales and Puri, 2005; Sartorelli and Caretti, 2005); however, the precise mechanism is still unclear.
Among the diverse mechanisms, the role of chromatin modifiers such as histone acetylases (HATs), deacetylases (HDACs) and SWI/SNF chromatin remodeling factor in controlling MyoD activity has recently been expanded considerably. Evidences established that HDACs negatively and HATs with added subsequent recruitment of SWI/SNF complexes positively regulates MyoD activity in the induction of muscle differentiation (Berkes and Tapscott, 2005; Forcales and Puri, 2005; Sartorelli and Caretti, 2005). The HDACs and HATs regulates MyoD activity by balancing the acetylation levels of both MyoD and histone on chromatin through post-translational modification (Sartorelli et al, 1999; Mal et al, 2001; Polesskaya et al, 2001). A recent work demonstrated that the polycomb Ezh2 histone methyltransferase (HMT), which methylates histone H3 at lysine-27 (H3-K27) in gene silencing (Silva et al, 2003), also represses skeletal muscle gene expression and differentiation (Caretti et al, 2004). This repression by Ezh2 required its recruitment by nonmyogenic transcriptional regulator YY1 and subsequent H3-K27 methylation on the chromatin region of muscle gene. Moreover, the data also demonstrated that Ezh2 targets late muscle MCK and MHCIIb genes but not the early gene myogenin. Like methylation on H3-K27, methylation on H3-K9 is also associated with transcriptional silencing (Kouzarides, 2002; Sims et al, 2003). Moreover, H3-K9 methylation by Suv39hHMT stimulates the binding of heterochromatin proteins (HPs) in gene silencing (Lachner et al, 2003). Thus, it appears that methylation on H3-K9 triggers a sequence of events that ultimately lead to transcriptional silencing. Previous studies have demonstrated the presence of methylated-H3-K9 at the chromatin region of myogenin gene in undifferentiated but not differentiated muscle cells (Zhang et al, 2002b; Mal and Harter, 2003).
In this work, I have tested the hypothesis that Suv39h1 in cooperation with MyoD may be engaged in the control of muscle gene expression and differentiation. The data demonstrate that Suv39h1 represses muscle gene activation via a mechanism that is dependent on both its presence in association with MyoD on the promoter of muscle gene and HMT activity. Suv39h1 represses muscle gene expression and differentiation in an epigenetic manner that sustains H3-K9 methylation at the promoter region of the differentiation initiating critical muscle gene myogenin. This is the first demonstration of a functional and direct interaction between Suv39h1 and MyoD in the control of muscle gene expression. Thus, the data uncover a novel role of Suv39h1 in MyoD-dependent transcriptional control at the regulatory checkpoint between proliferation and differentiation.
Results
Suv39h1 represses MyoD-responsive muscle gene transcription
Since myogenin activation correlates with the loss of methylation on H3-K9 from its regulatory regions (Zhang et al, 2002a; Mal and Harter, 2003), I tested whether Suv39h1 regulates muscle gene transcription by MyoD. Hence, C3H10T1/2 fibroblasts were cotransfected with a vector encoding MyoD and one of a MyoD-responsive reporter plasmid (myogenin promoter, Myogenin-Luc; mutimurized E-boxes, 4RE-Luc) or control SV40 reporter (SV40-Luc) in the presence and absence of vector encoding Flag-tagged Suv39H1 (Figure 1A). Suv39h1 inhibited specifically the activation of MyoD-responsive reporters. Moreover, this inhibition by Suv39h1 occurred in a dose-dependent manner (Figure 1B). An identical pattern of gene repression by Suv39h1 was also observed in C2C12 myoblasts, which expresses endogenous MyoD (Figure 1C). Gene repression by Suv39h1 was also observed when MyoD-responsive MCK-reporter replaces 4RE reporter (right E-box from muscle creatine kinase MCK enhancer was multimerized) (Supplementary Figure 1S). To determine whether Suv39h1HMT activity was responsible for this effect, both C3H10T1/2 and C2C12 cells were cotransfected with a vector encoding either wild-type Suv39h1 (wt) or HMT inactive mutant Suv39h1-H324K along with MyoD expression plasmid (only in C3H10T1/2 cells) and indicated reporters. The results showed that Suv39h1 required its HMT activity for repressing reporter gene activation (Figure 1D and E). Since a transiently transfected plasmid does not recapitulate the endogenous chromatin structure, I assessed whether Suv39h1 was capable of inhibiting MyoD-responsive reporter in native chromatin environment. Hence, I generated polyclonal population of C2C12 cells with (C2-4RE-LacZ) or without (C2-Em-LacZ) chromatin integrated reporter 4RE-LacZ. Results (Figure 1F) showed MyoD responsiveness to the integrated reporter gene activation. However, when C2-4RE-LacZ cells were transfected with vector encoding Suv39h1 (wt) or Suv39h1-H324K, I found that enzyme-active Suv39h1 also represses chromatin integrated MyoD-responsive reporter activity (Figure 1F). The functional difference between Suv39h1 (wt) and Suv39h1-H324K was not due to their differential expression (Figure 1G). Together, these results demonstrate that Suv39h1 inhibits MyoD-dependent gene transcription and that its HMT activity is essential for this inhibition.
Figure 1.
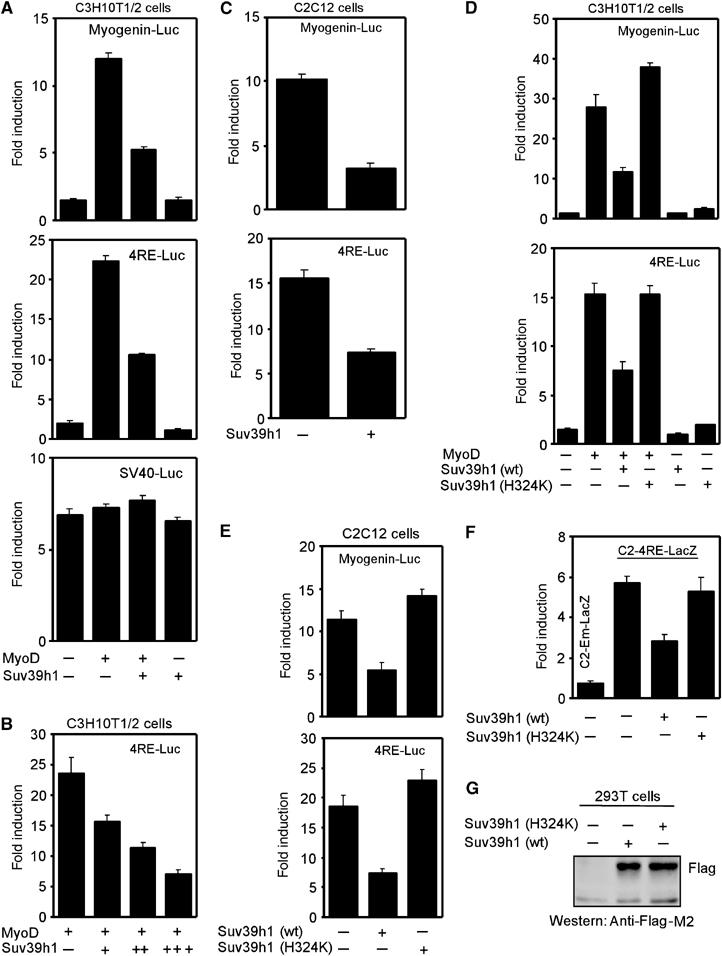
Suv39h1 inhibits MyoD-mediated transcription in an HMT activity dependent manner. (A) C3H10T1/2 cells were cotransfected with indicated reporters and vectors encoding MyoD and Suv39h1. (B) C3H10T1/2 cells were cotransfected with indicated reporter, vectors encoding MyoD (0.2 μg) and an increased concentration of Suv39h1 (0.4, 0.6 and 0.8 μg). (C) C2C12 cells were cotransfected with the indicated reporters and Suv39h1 expression vector. (D, E) C3H10T1/2 or C2C12 cells were cotransfected with indicated reporters plus vector encoding enzymatic active (wt) or inactive H324K version of Suv39h1 in the presence of vector expressing MyoD. (F) Chromatin integrated control (C2-Em-LacZ) or 4RE-LacZ (C2-4RE-LacZ) cells were transfeted with active (wt) and inactive H324K version of Suv39h1. Normalized reporter activity was expressed as fold activation. (G) 293T cells were transfected with equal amount of plasmid encoding Flag-tagged wt or mutant version (H324K) of Suv39h1. Their expression levels were monitored by immunoblot analysis of equal amount of cell extracts using Flag antibodies.
Suv39h1 physically interacts with MyoD
Since Suv39h1 inhibited MyoD-dependent gene transcription, I tested whether Suv39h1HMT can associate with MyoD. Therefore, glutathione-beads containing either bacterially produced GST-MyoD or control GST were incubated with the nuclear extracts of HeLa cells. Subsequently, bound proteins associated with the beads were subjected to an HMT activity assay using radiolabel S-adenosym methionine (SAM) as a methyl donor and full-length histone H3 as a substrate. I found that GST-MyoD was associated with H3HMT activity but not GST (Figure 2A). To determine whether MyoD-associated H3HMT is an H3-K9-specific HMT, I repeated the above experiment, except unlabelled SAM and N-terminus (1–57 amino acids) of H3 fusion protein GST-H3 (N) were used in this case (Tachibana et al, 2001). Immunoblot analysis using trimethylated-H3-K9 antibodies indicated that MyoD is associated with an H3-K9 trimethylase activity (Figure 2B). Since Suv39h is an H3-K9 trimethylating HMT (Peters et al, 2003), I asked whether MyoD-associated H3HMT activity was indeed due to Suv39h. Hence, MyoD immunoprecipitates retrieved from the extracts of wt and Suv39h knockout (Suv39h−/−) mEF cells stably expressing MyoD were subjected to H3HMT activity assay. The results indicated that MyoD recovered an H3HMT activity from wt but not in Suv39h−/− mEFs (Figure 2C). Immunoblot analysis of cell extracts showed that MyoD's inability to associate with H3HMT activity in Suv39h−/− mEFs was not due to lack of its expression in these cells (Figure 2D). Together, these results demonstrated that MyoD-associated H3HMT was due to its association with Suv39h.
Figure 2.
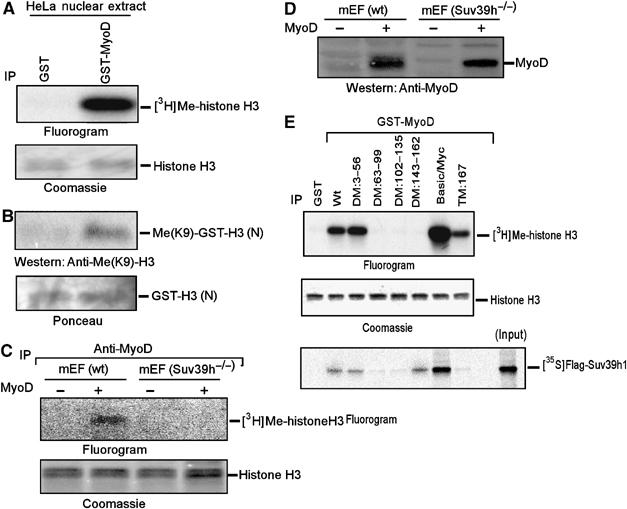
HMT activity of Suv39h1 associates with MyoD and Suv39h1 binds directly to MyoD in vitro. (A) Glutathione-beads containing 5 μg of GST-MyoD or control GST were used to purify HMT activity from HeLa nuclear extracts. Fluorography and Coomassie detected methylated H3 and total input H3, respectively. (B) Similar to (A), except GST-H3(N) was used as substrate instead of H3. Methylation on H3-K9 was detected by immunoblot analysis using trimethylated H3-K9 antibodies and substrate H3 by ponceau. (C) MyoD immunoprecipitates retrieved from nuclear extracts of growing wt and Suv39h knockout (Suv39h−/−) mEFs expressing with or without MyoD were subjected to HMT activity assay. Methylated H3 and total input H3 were detected by fluorography and Coomassie, respectively. (D) The expression of MyoD in extracts used in C was determined by immunoblot analysis with MyoD antibodies. (E) Equal amount of GST, wt or mutant GST-MyoD proteins were prebound to glutathione-beads. This was carried out in two sets. In one set, beads with attached proteins were used to purify HMT activity as in the case of (A). In the second set, beads with attached proteins were incubated with purified 35S-Flag-Suv39h1, and associated proteins along with input control were analyzed by fluorography.
In order to determine the sites on MyoD with which Suv39h interacts, I repeated the experiment as described in Figure 2A with GST, full-length GST-MyoD (wt) and a series of GST-MyoD deletion and truncation mutants (Mal et al, 2001). The results of this experiment (Figure 2E, upper panel) showed that the deleted acidic region (DM:3–56) and N-terminal fragment of MyoD were efficient as MyoD (wt) in associating with Suv39h. However, the deleted cysteine–histidine (C/H) (DM:63–99), highly basic (DM:102–135) or HLH (DM:143–162) domains of MyoD failed to associate with Suv39h. Surprisingly, a MyoD fragment spanning the highly basic and HLH regions (Basic/Myc) was bound robustly with Suv39h than MyoD (wt). The observed difference in MyoD interaction with Suv39h1 was not due to the unequal amount of substrate H3 (Figure 2E, middle panel). I next performed an in vitro binding experiment to determine whether MyoD directly interacts with Suv39h. After incubating a purified 35S-labelled Flag-Suv39h1 with preceding GST-MyoD proteins, I was able to establish the MyoD-Suv39h1 direct interaction (Figure 2E, lower panel). However, I cannot entirely exclude the possibility that there may be an intermediary protein co-purified with 35S-labelled Flag-Suv39h1 mediating the interaction with MyoD. Moreover, I found that mutant MyoD proteins lacking acidic or HLH region interacted with Suv39h1; however, mutants devoid of either C/H or the basic or the C-terminal region failed in this interaction. Furthermore, MyoD(Basic/Myc) protein showed profound interaction with Suv39h1. I conclude that instead of a single region, the C/H, basic and C-terminal regions on MyoD all were involved in its interaction with Suv39h1.
Suv39h1 represses trans-activation by MyoD once MyoD tethers it to DNA
The above findings underscore the importance of Suv39h1 in MyoD-dependent transcriptional repression. However, it raises the possibility that Suv39h1 interaction with MyoD may perturb critical DNA binding function of MyoD, since its DNA binding basic region interacted with Suv39h1 (Figure 2E). To address this, I evaluated Suv39h1's ability to repress trans-activation by MyoD using a Gal4-reporter assay (diagrammed in Figure 3A). This system enables me to separate the trans-activation and DNA-binding functions of MyoD. Hence, C3H10T1/2 cells were cotransfected with plasmids for Gal4-luciferase and MyoD fused to Gal4 DNA binding domain in the presence and absence of vector encoding Suv39h1.The results are shown in Figure 3B, which demonstrates that the trans-activation of reporter gene by Gal4-MyoD was inhibited by Suv39h1 (wt). Conversely, cotransfection of Suv39h1-H324K failed to inhibit this trans-activation by MyoD. The specificity of this assay was also verified by the fact that Suv39h1 (wt) failed to inhibit the trans-activation of reporter gene by the activation domain of ATF2 fused to Gal4. To evaluate whether repression of MyoD trans-activation by Suv39h1 was due to their physical association, I repeated the above assay with Gal4-fused mutants of MyoD deleted in the trans-activation (ΔN), basic (ΔB) or C-terminal portion (ΔC). Note that the latter two mutants of MyoD have impaired Suv39h1 interactions (Figure 2E). I found that MyoD mutants (ΔB) or (ΔC) activated the reporter gene (Figure 3C) (Weintraub et al, 1991), and Suv39h1 (wt) failed to repress this activation. Together, the data suggest that Suv39h1 directly interacting with MyoD represses MyoD-dependent transcription in an HMT activity dependent manner.
Figure 3.
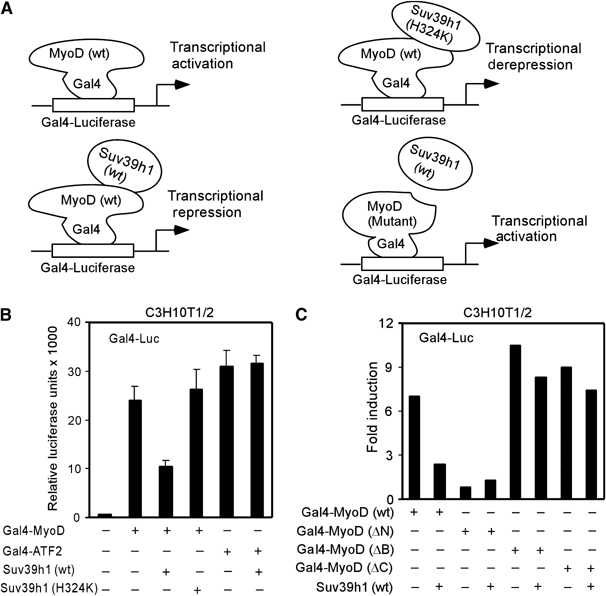
Suv39h1 requires its HMT activity and MyoD interaction to inhibit trans-activation function of MyoD. (A) Schematics indicating that HMT activity of Suv39h1 as well as it interaction with MyoD blocks the trans-activation function of MyoD. (B) C3H10T1/2 cells were cotransfected with a Gal4-luciferase reporter plus vectors encoding Gal4 fusion full-length MyoD or ATF2 trans-activation domain and expression vector for Suv39h1 (wt) or Suv39h1-H324K. (C) Similar to (B), except plasmids encoding Gal4 fusion MyoD wt and mutants were cotransfected in the presence and absence of Suv39h1. Values after normalization were expressed as relative luciferase unit in (B) and fold induction in (C).
Suv39h1 associates with MyoD in undifferentiated muscle cells
Since MyoD is expressed solely in skeletal muscle cells, I tested whether MyoD is associated with an Suv39h1 HMT activity in these cells. Hence, MyoD immunoprecipitates recovered from nuclear extracts of C2C12 cells, grown in growth (GM) or differentiation (DM) media, were subjected to H3HMT activity assay. The MyoD-associated H3HMT activity was observed in undifferentiated but not in 2 or 4 day differentiated muscle cells (Figure 4A). Immunoblot analysis confirmed that the regression of MyoD-associated this activity in differentiated cells was not due to the absence of MyoD in its immunoprecipitate. MyoD was also equally efficient in associating H3HMT activity in a subconfluent or confluent culture of C2C12 cells (0 h in DM), and its associated activity declined as differentiation proceeds (Supplementary Figure S2A). Since MyoD-associated H3HMT activity was observed in myoblasts, I evaluated MyoD-associated activity on H3-K9 methylation. Hence, MyoD immunoprecipitates retrieved from the nuclear extracts of undifferentiated and differentiated C2C12 cells were analyzed as in the experiment that has been described in Figure 2B, except dimethylated-H3-K9 antibodies were used in this case. The results showed that MyoD-associated H3HMT activity was able to methylate H3-K9 and its associated this H3-K9HMT activity diminished upon differentiation (Figure 4B). The declined MyoD-associated H3-K9HMT activity during the differentiation of C2C12 cells suggests the possibility that Suv39h1 protein level may decline during this process. In order to test this, nuclear extracts prepared from C2C12 cells at different stages of differentiation were immunobloted with Suv39h1 antibodies. In parallel, nuclear extracts of wt and Suv39h−/− mEFs were analyzed to confirm the specificity of the Suv39h1 antibodies. The results showed the decline of Suv39h1 protein levels along with the appearance of a slower migrating form of it as C2C12 cells undergo differentiation (Figure 4C). However, this slower migrating form disappeared after treatment with phosphatase (data not shown), suggesting its phosphorylation during differentiation.
Figure 4.
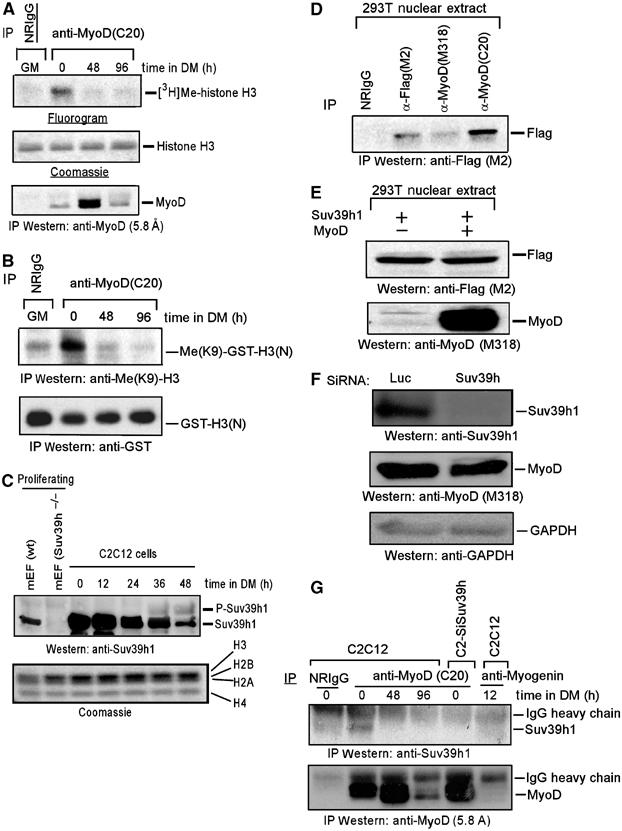
Suv39h1 associates with MyoD in undifferentiated muscle cells. (A) MyoD or control NRIgG immunoprecipitates retrieved from nuclear extracts of C2C12 at different stages of differentiation (confluent in GM (0); 48, 96 h in DM) were subjected to HMT activity assay. Commassie or fluorography detected histone H3. In a separate set of experiment, the above immunoprecipitates were immunoblotted using MyoD antibodies. (B) Similar to (A), except GST-H3 (N) was used as substrate for HMT activity assay instead of H3. Immunoblot analysis using dimethylated H3-K9 and GST antibodies determined methylated H3-K9 and substrate input, respectively. (C) Immunoblot of nuclear extracts derived from growing wt and Suv39h−/− mEFs and C2C12 myoblasts at different stages of differentiation probed with Suv39h1 antibodies. Coomassie staining of core histone determined the loading control. (D) 293T cells were transfected with vectors encoding Flag-Suv39h1 alone or in combination with MyoD as shown in (E). Nuclear extracts derived from combined or single transfected cells were immunoprecipitated separately with two different antibodies against MyoD or Flag-M2 antibodies, respectively, and the precipitated proteins were revealed with Flag antibodies. Nonspecific NRIgG was used as the control. (E) Immunoblot of extracts (50 ug) used in (D) probed with antibodies against Flag and MyoD. (F) C2C12 myoblasts were infected with lentivirus expressing siRNA against control (Luc) or Suv39h, and their extracts were immunoblotted with antibodies to Suv39h1, MyoD and GAPDH. (G) Nuclear extracts derived from growing C2C12 myoblasts infected with lentivirus expressing siRNA against Suv39h and C2C12 myoblasts at different stages of differentiation were immunoprecipitated with MyoD antibodies. Similarly, myogenin was immunoprecipitated from C2C12 cells 12 h in DM using antibodies against it. The immunoprecipitated proteins were then immunoblotted with Suv39h1 antibodies. NRIgG was used as a negative control. The membrane was re-probed with MyoD antibodies.
Although prior in vitro binding assay establishes MyoD-Suv39h1 interaction (Figure 2E), I asked whether MyoD could also interact with Suv39h1 in vivo. Hence, nuclear extracts obtained from 293T cells co-expressing exogenous MyoD and Flag-Suv39h1 were immunorecipitated with MyoD or control NRIgG antibodies, and the immunoprecipitated proteins were immunoblotted with Flag antibodies. In parallel, Flag-Suv39h1 was immunoprecipitated from 293T cells expressing it alone and processed. The results showed MyoD association with Suv39h1 when both proteins were coexpressed (Figure 4D). The level of the exogenously expressed proteins was also verified by immunoblot analysis of the equivalent amount of the extracts (Figure 4E). When a similar binding experiment was performed in 293T cells coexpressing both MyoD and Flag-Suv39h1-H324K, I found it association with MyoD (data not shown). Hence, the inability of Suv39h1-H324K to inhibit MyoD-dependent gene transcription (Figure 1D and E) was not due to the lack of its association with MyoD. Next, I wished to determine whether Suv39h1 interacts with MyoD in muscle cells. To assess Suv39h1–MyoD interaction in muscle cells, first I depleted endogenous Suv39h from C2C12 muscle cells through siRNA against Suv39h (Suv39h siRNA) using lentiviral delivery system. Others have already used this siRNA sequence to knockdown endogenous Suv39h (both Suv39h1 and Suv39h2) in these C2C12 cells (Ait-Si-Ali et al, 2004). As a control, C2C12 cells were infected with lentivirus expressing siRNA against luciferase (Luc siRNA). Immunoblot analysis of the cell extracts shows that Suv39h was depleted in C2C12 cells that have received Suv39h siRNA, and this siRNA has no effect on endogenous MyoD protein level (Figure 5F). When MyoD immunoprecipitates retrieved from nuclear extracts of Suv39h-depleted C2C12 cells were tested for MyoD-associated H3HMT activity, its associated this activity was abolished in these cells (Supplementary Figure S2B). Having established that Suv39h is solely contributing to the MyoD-associated H3-K9HMT activity in C2C12 cells, I assessed Suv39h1–MyoD interaction in C2C12 cells. Hence, nuclear extracts obtained from C2C12 cells at different stages of differentiation and Suv39h-depelted undifferentiated C2C12 cells were immunoprecipitated with MyoD antibodies. In parallel, nuclear extracts derived from C2C12 cells, grown in GM or DM for 12 h, were immunoprecipipitated with NRIgG and myogenin antibodies, respectively. In C2C12 cells, myogenin expression was detected as early as 12 h in DM (Mal and Harter, 2003). The immunoprecipitated proteins were then immunoblotted with Suv39h1 antibodies (Figure 4G). The results showed that Suv39h1 was coimmunoprecipitated with MyoD from undifferentiated but not with MyoD either from differentiated or Suv39h-depleted extracts or with myogenin from 12 h differentiated extracts. Together, the data suggest Suv39h1 specific interaction with MyoD in undifferentiated muscle cells; however, it failed to interact with MyoD or myogenin in differentiated muscle cells.
Figure 5.
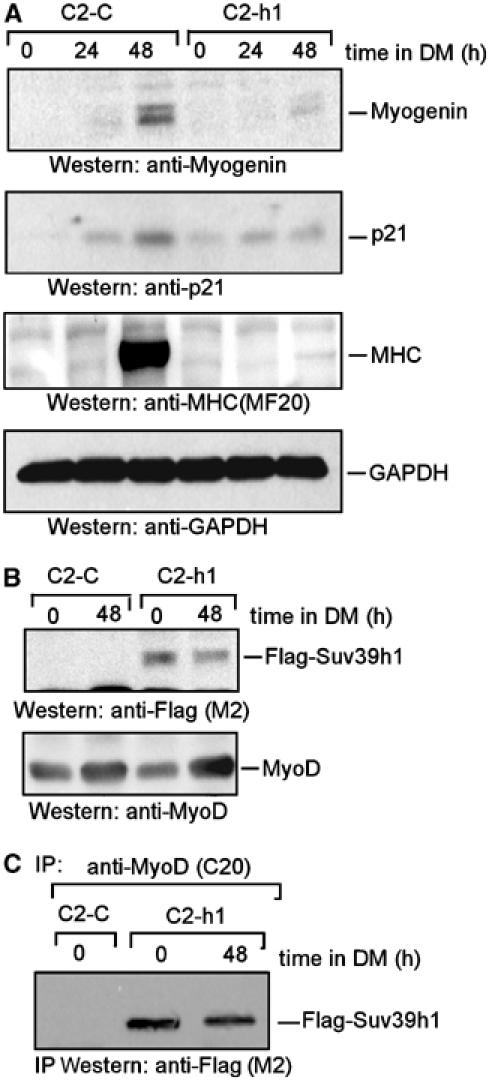
Suv39h1 represses muscle differentiation-responsive gene expression. (A) Immunoblot of cell extracts derived from C2C12 cells expressing with (C2-h1) or without (C2-C) exogenous Suv39h1, grown in GM (0 h) or DM for indicated times, probed with antibodies against myogenin, p21, MHC and GAPDH. (B) Immunoblot of the same extracts as depicted in (A) probed with antibodies against Flag and MyoD. (C) Cell extracts derived from C2-C and C2-h1 cells grown in DM for indicated times were immunoprecipitated with MyoD antibodies, and the precipitated proteins were immunoblotted with Flag antibodies.
Suv39h1 inhibits muscle gene expression and differentiation
Since Suv39h1 represses MyoD-dependent gene transcription, I evaluated whether it can repress differentiation-responsible muscle (such as myogenin and myosin heavy chain, MHC) and non-muscle (growth arrest p21) gene expression. Hence, cell extracts derived from stable C2C12 cells expressing with (C2-h1) or without (C2-C) Flag-Suv39h1 were subjected to immunoblot analysis (Figure 5A). The results showed that under differentiation conditions, while both early myogenin, p21 and late MHC were expressed in C2-C cells; however, expression of these genes was severely impaired in C2-h1 cells. Immunoblot analysis of these cell extracts further indicated that both C2-C and C2-h1 cells expressed comparable levels of Flag-Suv39h1 and MyoD (Figure 5B). Since MyoD interaction with Suv39h1 was detected in C2C12 cells under condition of growth but not differentiation (Figure 4G), I investigated the possibility that it may be associated with Flag-Suv39h1 in C2-h1 cells under both situation. Hence, MyoD immunoprecipitates retrieved from nuclear extracts of C2-h1 cells grown in GM or DM media were analyzed by immunoblotting using Flag antibodies. The results showed MyoD interaction with Flag-Suv39h1 in C2-h1 under both cultured conditions (Figure 5C). When MyoD immunoprecipitates were further tested for its associated H3-K9HMT activity, it was detected under both situations (data not shown). Together, the data suggest that Flag-Suv39h1 interaction with MyoD restores MyoD-associated H3-K9HMT activity and the repression of differentiation-responsible gene expression in C2-h1 cells even under differentiation conditions. To further verify that Suv93h1 indeed affected MyoD ability in this gene repression phenomenon, I evaluated its effect on MyoD-mediated myogenin and p21 expression in C3H10T1/2 cells by immunoblot analysis. The results of this experiment (Supplementary Figures S3A and B) indicated that Suv39h1HMT activity represses MyoD function on gene activation. Moreover, C2-h1 cells not only failed in muscle gene activation but these cells also failed to form myotubes in the presence of cytosine-arabinoside (Ara-C) (Supplementary Figure 4S), which eliminates nondifferentiating myoblasts from the culture (Mal et al, 2000). Altogether, these findings indicate that Suv39h1 interaction with MyoD prevents gene expressions that are necessary for differentiation.
Suv39h1 induces H3-K9 methylation at the regulatory regions of myogenin gene
Myogenin activation correlated with the loss of promoter-associated H3-K9 methylation from its silent state (Zhang et al, 2002b; Mal and Harter, 2003). Since C2-h1 cells failed to activate myogenin expression but not the C2-C cells (Figure 5A), I evaluated the status of dimethylated-H3-K9 at this gene promoter before and after inducing these cells to differentiate. Therefore, I performed chromatin immunoprecipitation (ChIP) with dimethylated-H3-K9 antibodies and analyzed the immunoprecipitated DNA fragments by PCR using primers for the myogenin promoter region. The results of this experiment are shown in Figure 6A. Dimethylated-H3-K9 was found at the promoter region of myogenin gene derived from both C2-h1 and C2-C cells under growth conditions. However, under differentiation conditions, dimethylated-H3-K9 was detected at this chromatin region derived from C2-h1 but not C2-C cells. These data suggest that ectopic Flag-Suv39h1 in C2-h1 cells sustained the dimethylatable H3-K9 at the myogenin promoter. Suv39h1 can convert unmodified and also monomethyalted-K9 of H3 to its ultimate trimethylated-K9 form (Peters et al, 2003). I assessed by ChIP using antibodies against dimethylated- or trimethyaled H3-K9 that it is the trimethylated form of H3-K9 enriched at the regulatory regions of silent myogenin gene (Supplementary Figure S5). Together, the data establish that Suv39h1 maintained the trimethylation on H3-K9 at the myogenin promoter.
Figure 6.
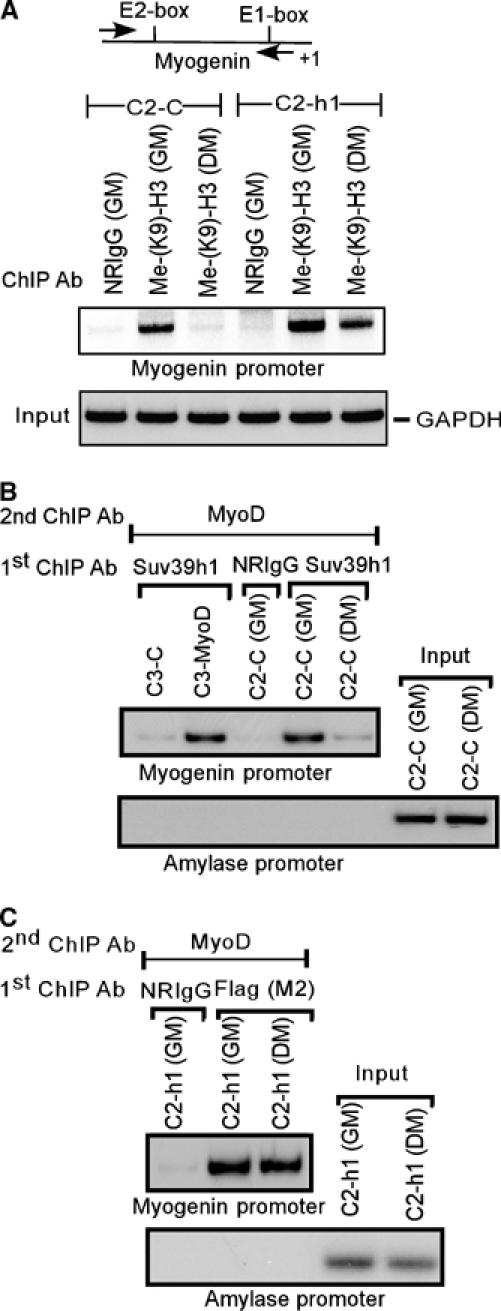
Chromatin recruitment of Suv39h1 in association with MyoD induces methylation of H3-K9 at the regulatory regions of muscle myogenin gene. (A) ChIP with control NRIgG and anti-dimethylated antibodies on chromatin obtained from C2-C and C2-h1 cells, cultured either in GM or DM for 48 h. The precipitated DNA was amplified by PCR using primers for the myogenin promoter. As an input control, 0.04% of total chromatin was amplified by PCR using primers for GAPDH coding region. (B) Re-ChIP analysis of muscle myogenin and nonmuscle amylase promoters. Chromatin derived from growing C3H10T1/2 cells stably expressing with (C3-MyoD) or without MyoD (C3-C), and C2-C cells (C2C12 cells expressing without exogenous Suv39h1) grown either in GM or DM for 48 h were initially ChIP with indicated antibodies. The released associated proteins from the initial ChIP were re-immunoprecipitated with MyoD antibodies (Re-ChIP). Finally, the precipitated DNA was amplified by PCR using specific primers for the myogenin and amylase promoter. As an input control, 0.04% total chromatin of C2-C cells was PCR amplified using primers for amylase promoter. (C) Re-ChIP was performed as described in B except that chromatin was obtained from C2-h1 cells (C2C12 cells expressing exogenous Suv39h1) and antibodies against Suv39h1 was replaced by Flag antibodies. Here, chromatin of C2-h1 cells was used for input control.
Endogenous Suv39h1 interaction with MyoD in undifferentiated muscle cells is perfectly correlated with the presence of trimethylated-H3-K9 at the myogenin promoter. These results suggest that MyoD may recruit Suv39h1 on the myogenin promoter and reside in association with MyoD at this gene promoter in myoblasts. To test this hypothesis, I utilized chromatin reimmunoprecipitaion (Re-ChIP) assay in C3H10T1/2 cells after retroviral transduction with (C3-MyoD) or without (C3-C) MyoD and in C2C12-derived control C2-C cells. The Re-ChIP assay was performed sequentially with Suv39h1 followed by MyoD antibodies, and the immunoprecipitated DNA fragment was analyzed by PCR using specific primers for myogenin and nonmuscle amylase promoter regions. Suv39h1 alongside with MyoD was detected specifically at the chromatin region of myogenin derived from growing C3-MyoD and undifferentiated C2-C cells but not from growing C3-C cells or differentiated C2-C cells (Figure 6B). This result also revealed that MyoD tethered Suv39h1 to this gene chromatin region because Suv39h1 was detected only in C3H10T1/2 cells that express exogenous MyoD. Moreover, ChIP analysis with MyoD antibodies showed that MyoD was bound on the myogenin promoter derived from both undifferentiated and differentiated C2-C cells (data not shown). At variance with others (Simone et al, 2004; Blais et al, 2005; de la Serna et al, 2005), MyoD binding to the chromatin region of myogenin in undifferentiated C2-C cells is consistent with our earlier work on in parent C2C12 cells (Mal and Harter, 2003). Since Suv39h1 association with MyoD correlated their presence at the chromatin region of myogenin, I would then expect that Flag-Suv39h1-MyoD complex was associated with this chromatin region in C2-h1 cells irrespective of the cultured conditions. Therefore, an identical Re-ChIP assay as described above was performed, except Flag antibodies replaced Suv39h1 antibodies. As expected, Flag-Suv39h1 alongside with MyoD was detected at the chromatin region of myogenin derived from C2-h1 cultured under both growth and differentiation conditions (Figure 6C). Collectively, the data establishes that Suv39h1 in association with MyoD at the myogenin promoter region induces methylation on H3-K9 at this region.
Suv39h1 depletion in muscle cells induces differentiation promoting gene expression
The results presented so far suggest that Suv39h1 positioning on the chromatin regulatory domain through interaction with MyoD induces H3-K9 methylation in the repression of critical myogenin expression. If this were true, I would expect that delineating the Suv39h protein would result in activation of myogenin and also other differentiation-dependent gene expression. To test this hypothesis, immunoblot analysis was performed on cell extracts obtained from Suv39h depleted (Suv39h-siRNA) or control (Luc-siRNA) C2C12 cells (as described in Figure 4F). The result of this experiment is shown in Figure 7. Myogenin was expressed in similar levels like MyoD both in control and Suv39h-depleted C2C12 cells grown in differentiation conditions. Surprisingly, myogenin expression was detected only in Suv39h-depleted C2C12 cells under growth conditions, although MyoD is expressed more or less same level compared to control cells in this situation. The expression pattern of MHC was identical to that of myogenin expression in Suv39h-depleted versus control C2C12 cells. On the other hand, p21 was expressed in much higher levels in Suv39h-depleted C2C12 cells under both cultured conditions. These findings are completely opposite to a recent report stating that Suv39h depletion inhibits muscle (myogenin and MHC) gene expression in these cells (Ait-Si-Ali et al, 2004). However, consistent with this group, the S-phase cyclin A2 gene expression was downregulated in control but not in Suv39h-depleted C2C12 cells grown in differentiation conditions (Supplementary Figure S6). Together, these findings suggest that Suv39h1 constrains muscle gene expression in undifferentiated muscle cells.
Figure 7.
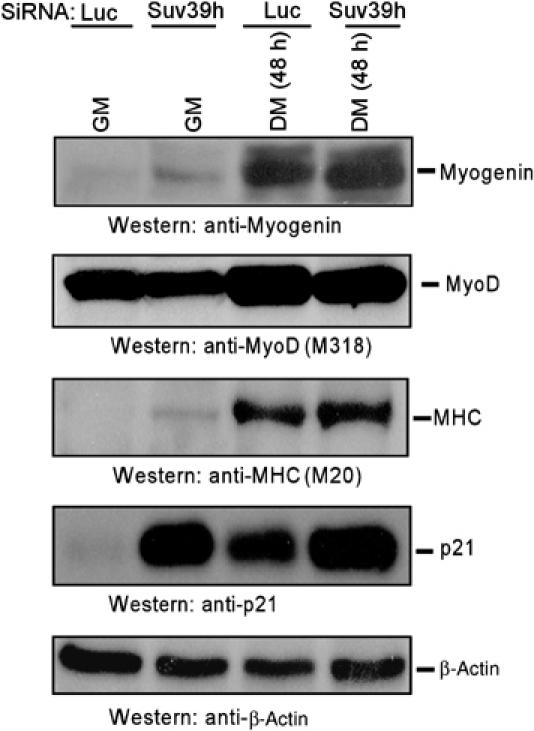
Suv39h depletion induces muscle differentiation-responsible gene expression in myoblast cells. C2C12 myoblasts were infected either with lentivirus expressing siRNA against control (Luc) or Suv39h and then incubated in GM or in DM for 48 h, and their extracts were immunoblotted with antibodies to myogenin, MyoD, MHC, p21 and β-actin.
Discussion
This study has uncovered a functional and biochemical link between Suv39h1 and MyoD for the repression of muscle gene activation prior to differentiation. The results revealed that Suv39h1 inhibited muscle differentiation by binding to and inhibiting MyoD targets differentiation-responsible gene expression. In particular, Suv39h1 associating with MyoD was recruited at the chromatin regulatory regions of myogenin gene, which is induced by MyoD at the onset of differentiation and a critical determinant in myogenic program. Once recruited, Suv39h1 repressed myogenin expression by methyting H3-K9 on its promoter. These data support a model in which Suv39h1 modulates MyoD in making the decision whether muscle cells undergo differentiation or continue proliferates.
The identification of Suv39h as the H3-K9-specific HMT provided the first direct link that stable methylation modification on H3-K9 was associated with permanent gene repression (Rea et al, 2000). Its involvement in reversible repression of euchromatin genes was also established (Sims et al, 2003). Recent discovery of lysine-specific demethylase (Shi et al, 2004; Metzger et al, 2005) further suggests that stable histone methylation does not last forever. In muscle cells, activation of myogenin coincides with the loss of methylated H3-K9 from the chromatin region of its transactional repressed state upon differentiation (Zhang et al, 2002b; Mal and Harter, 2003). Recently, we have demonstrated that MyoD occupied the myogenin promoter before myoblasts (transcriptionally repressed) are induced to differentiate (Mal and Harter, 2003). However, others have shown that MyoD is absent from this gene promoter in myoblasts (Simone et al, 2004; Blais et al, 2005; de la Serna et al, 2005). Simon et al used ethidium bromide dye as a detection method whereas we have used radioactive 32P. Since MyoD binding to the myogenin promoter in myoblasts is lower than in differentiated cells (Mal and Harter, 2003), their low sensitivity detection assay may account for the differences in our findings. Blias et al also suggested that MyoD does not bind to the myogenin promoter. However, their data presented in their Supplementary Table S1 indicated that there was no significant difference in the values for MyoD binding in myoblasts between the myogenin and actin alpha-1 genes, which they claim to be bound by MyoD. Since the results for both genes fall close to their arbitrary cutoff, it is difficult to conclude that MyoD binds to one gene but not the other. While de la Serna et al also failed to show MyoD binding to the myogenin promoter, they used murine NIH3T3 fibroblast cells expressing exogenous MyoD in their study not the mouse myoblast C2C12 cell line of our experiments, which represents a well-established in vitro muscle differentiation model that recapitulates this process in vivo. These different cell systems not only differ in MyoD binding, they also differ in their histone H4 status (i.e. hyperacetylated versus hypoacetylated (Lu et al, 2000), respectively). Thus, a direct comparison between the two sets of experiments cannot be made since the underlying properties of the cell systems vary. Moreover, MyoD binding to the myogenin promoter in myoblasts may also be explained by a recent report by Berkes et al (2004). According to this report, MyoD would be recruited to this gene promoter indirectly through homeodomain protein Pbx. Together, it would strongly argue against MyoD binding to myogenin promoter in myoblasts. The presence of MyoD on the chromatin of myogenin promoter enriched with methylated H3-K9 in myoblasts (Mal and Harter, 2003) suggests that Suv39h may be inhibiting MyoD activity in order to repress myogenin expression under this situation. I have found that Suv39h1HMT activity blocks MyoD-dependent gene activation. It is also possible that Suv39h1 may be inhibited MyoD gene activation function indirectly through MEF2. This is because MEF2 cooperates with MyoD in the activation of muscle genes (Naya and Olson, 1999) and its activity is repressed by HP1α through the formation of a complex consisting of MEF2 interacting protein MITR, HP1α and Suv39h1 (Zhang et al, 2002b). However, this is unlikely, since MEF2 expression is initiated after the onset of differentiation (Naya and Olson, 1999) and MyoD directly activates MEF2C, the first member of MEF2 family expressed in skeletal muscle (Dodou et al, 2003).
The results also indicate that MyoD can associate with an H3-K9 methylase activity of Suv39h and it directly interacts with Suv39h1. The data further demonstrate that the Suv39h1 interacting regions on MyoD are those that harbor lysine residues for acetylation-mediated execution of muscle differentiation and its chromatin remodeling activity (Gerber et al, 1997; Sartorelli et al, 1999). There was also an incongruity in the HLH region on MyoD with respect to its direct interaction with Suv39h1 and associated H3 HMT activity. This discrepancy might occur due to the failure of this mutant MyoD to associate with HDAC1, since it directly interacts with MyoD through HLH region (Mal et al, 2001). The current experiment used commercially purchased calf thymus histone H3, which was highly acetylated, as a substrate for H3HMT activity assay. Since incorporation of a methyl group on histone would require prior removal of the acetyl group from it by deacetylase; failure to methylate H3 may be due to the absence of HDAC1 in this mutant MyoD (without HLH) complex. I am currently addressing this hypothesis by investigating whether the use of deacetylated histone H3 as a substrate would restore HMTase activity to this mutant MyoD complex. From the in vitro binding data, one can hypothesize that Suv39h1 exerts its repressive effect on MyoD gene activation function by affecting the DNA binding ability of MyoD. This is because Suv39h1 interacts with MyoD through DNA binding basic domain. However, the data from DNA tethering experiment indicate that Suv39h1 represses trans-activation by MyoD only when MyoD retains its ability to associate with Suv39h1. This suggests that Suv39h1 is quite capable of inhibiting MyoD gene activation function concurrently associating with the MyoD. In this scenario, Suv39h1 might target H3-K9 methylation on the regulatory region of MyoD-responsive genes.
The data also demonstrated that in muscle cells, MyoD is associated with Suv39h1 and this interaction is correlated with MyoD-associated H3-K9 methylase activity in undifferentiated but not in differentiated muscle cells. In differentiated muscle cell, the absence of Suv39h1 interaction with MyoD may be due to both the decrease of Suv39h1 protein and its phosphorylation by presently unknown kinase(s) induced during differentiation. Since phosphorylation of Suv39h1 regulates its function (Firestein et al, 2000), such modification may also alter its interaction with MyoD. The impact of Suv39h1 interaction with MyoD in muscle cells was further evident by the fact that muscle cells expressing exogenous Suv39h1 failed to express muscle and non-muscle genes, which are necessary and induced during differentiation. As a consequence, Suv39h1 overexpressing muscle cells failed to form multinucleated myotubes, a hallmark of differentiated muscle cells. The absence of myotubes may be due to the fact that C2C12 cells over-expressing Suv39h1 exhibit a delay of differentiation that causes the removal of these cells by Ara-C. Hence, I cannot fully rule out the possibility that muscle cells expressing ectopic Suv39h1 may exhibit a delay of differentiation instead of failure in the myotubes formation. This failure/delay may apparently originate due to Suv39h1 interaction with MyoD in these muscle cells before and after induced to differentiate. It suggests that ectopic Suv39h1 in muscle cells may impede MyoD ability to activate gene that are induced during differentiation. Consistent with this postulation was the finding that Suv39h1 blocks MyoD ability to activate myogenin and p21 expression in fibroblast cells. Ectopic expression of MyoD in fibroblasts leads to the activation of myogenin and other muscle genes that determine the muscle phenotype (Davis et al, 1987). Both myogenin and p21 are also expressed at the beginning of differentiation (Andres and Walsh, 1996), Together, this supports the notion that Suv39h1 association with MyoD in muscle cells determines myoblasts not to differentiate as a consequence of its effect on gene expression that are induced at the onset of differentiation.
Consistent to our previous report (Mal and Harter, 2003), the presence and absence of H3-K9 methylation on the chromatin of myogenin gene was observed when the gene is transcriptionally repressed or activated upon differentiation, respectively. Methylation of H3-K9 on the chromatin of myogenin promoter region was also observed when this and other differentiating genes remains repressed in muscle cells overexpressing Suv39h1. Suv39h1-associated MyoD binding to the chromatin of myogenin gene was detected in undifferentiated muscle cells, but Suv39h1-free MyoD when this gene was activated from its repressed state upon differentiation. In muscle cells overexpressing Suv39h1, I have found Suv39h1-associated MyoD binding to the chromatin of myogenin gene even when these cells were induced to differentiate but failed to express this and other differentiating genes. The MyoD binding with Suv39h1 to the chromatin of myogenin gene is consistent with their association in undifferentiated muscle cells and also muscle cells that failed to differentiate due to the expression of ectopic Suv39h1. Most significantly, the presence of Suv39h1 alongside with MyoD on the chromatin of myogenin gene is in harmony with the methylation of H3-K9 on the chromatin regulatory regions of this gene. Together, the results demonstrated that H3-K9 methylation by Suv39h1 associating with MyoD represses early critical muscle myogenin gene. Conversely, H3-K27 methylation by Ezh2HMT interacting with YY1 represses late muscle genes (Caretti et al, 2004). Therefore, it is clearly evident that H3-K9 methylase Suv39h1 and H3-K27 methylase Ezh2 play a pivotal role in the repression of early and late muscle gene expression in undifferentiated muscle cells, respectively.
Although I have found an absence of muscle gene expression in muscle cells overexpressing Suv39h1, a recent work described a situation in which Suv39h depletion appears to inhibit muscle differentiation (Ait-Si-Ali et al, 2004). This group concluded that the absence of muscle gene expression in Suv39h-depleted muscle cells to be due to an indirect effect of S-phase gene such as cyclin D1 and cyclin A2 deregulation in these cells. Paradoxically, I have found that muscle gene expression was unaffected in Suv39h-deplted muscle cells. The expression of muscle genes was also detected, although at low level, in Suv39h-depleted muscle cells in nonpermissible differentiation conditions where they assumed not to be expressed. The low level of muscle gene expression in nondifferentiation conditions suggests that the absence of Suv39h1 in conjunction with differentiation stimuli is needed for their full activation during differentiation. However, consistent with the Ait-Si-Ali group, cyclin A2 protein was induced in Suv39h-depelted muscle cells. The expression of p21 was also induced in muscle cells lack of Suv39h. As growth arrest is a prerequisite for muscle cells to differentiate, high levels of p21 most likely inhibited the cyclin A2 activity in Suv39h-depelted muscle cells. Thus, the results presented in this study clearly established that Suv39h prevents muscle gene expression in undifferentiated muscle cells.
Collectively, the results in this study outline a model by which Suv39h1 interaction with MyoD impose transcriptional repression in myoblasts to initiate the differentiation program. Presumably, Suv39h1 exercises this activity by methylating H3-K9 at the promoter of the key muscle gene myogenin in proliferating myoblast cells. Finally, an intriguing issue is that modulating the Suv39h activity may make it possible to manipulate muscle cells from undifferentiated to differentiated state. This will be very useful for muscle regeneration following injury, in muscle degenerative diseases, and in myoblast gene therapy of Duchenne muscular dystrophy, where proliferation and differentiation are impaired and present a big challenge from a therapeutic viewpoint.
Materials and methods
Cell culture, transfection and retroviral infection
C3H10T1/2, 293T, HeLa and Pheonix-Ampho packaging cells and mEF cells derived from wt and Suv39h knockout (Suv39h−/−) mice were grown in D-MEM supplemented with 10% fetal bovine serum (FBS). C2C12 myoblast cells were cultured in D-MEM supplemented with 20% FBS (GM). I generated polyclonal population of C2C12 cells containing chromatin integrated 4RE-LacZ reporter gene through retroviral infection. Similarly, polyclonal population of C2C12 cells expressing ectopic Flag-Suv39h1, and C3H10T1/2 and mEFs expressing exogenous MyoD were generated. Infecting retrovirus generated with empty retroviral vector developed their respective control polyclone cells. Differentiation was induced by incubation in DM as previously described (Mal et al, 2000). Cells were transfected using Fugene 6 reagent (Roche). See the Supplementary Materials and methods for transfection, retrovirus production/infection and generation of polyclone cells.
Plasmids and antibodies
The MyoD-responsive reporter plasmids, expression vectors, retroviral vectors, construction of recombinant constructs and antibodies used are detailed in Supplementary Materials and methods.
Purification of proteins and reporter assay
Purification of GST fusion proteins was carried out as previously described (Mal et al, 2001) and purification of 35S-labelled Flag-Suv39h1 was described in Supplementary Materials and methods. Reporter assay was performed as described previously (Mal et al, 2001). Reporter Luciferase and β-galactosidase activities were determined in triplicate using the luciferase and β-galactosidase assay systems (Promega). The level of β-galactosidase and protein concentration was used for the normalization of reporter luciferase or β-galactosidase activity, respectively.
Cell extracts, immunoprecipitation and Western blotting
Preparation of total and nuclear extracts of cells, immunoprecipitation and immunoblotting were described previously (Mal et al, 2001). For immunoblot analysis, the signal was detected by ECL method (NEN).
HMT activity assays
The HMT activity assay was performed according to the method of Li et al (2002). In case of GST-MyoD, glutathione beads coated with equal quantities of GST or GST-MyoD fusion proteins were incubated with 100 μg nuclear extracts of HeLa cells. After washing, the beads were subjected to activity assay supplemented with 2.5 μg of H3 and 0.5 μCi of [3H]SAM. In case of C2C12 and mEF cells, MyoD immunoprecipitates retrieved from 250 μg nuclear extracts were washed and subjected to this assay. In the experiment shown in Figures 2B and 4B, full-length H3 was replaced by GST-H3(N) and unlabeled SAM (100 μM) was used as a methyl donor.
GST interaction assays
The GST interaction assays were carried out as described previously (Mal et al, 2001). Briefly, glutathione beads coated with equal quantities of GST, GST-MyoD proteins were incubated with 5 μl (∼5000 c.p.m.) of purified 35S-labeled Flag-Suv39H1 in binding buffer. Bound proteins associated with the beads were fractionated and analyzed by fluorography.
Suv39h knockdown in C2C12 cells by RNA interference
Inhibition of Suv39h expression in C2C12 cells was achieved by infection with recombinant lentiviral vector construct (pLV-bleo-Suv39h-siRNA) expressing hairpin siRNA against Suv39h controlled by H1 RNA gene promoter as previously described (Budanov et al, 2004). See the Supplementary Materials and methods for the production of high titer lentivirus stock, infection and the siRNA sequence used against Suv39h.
ChIP and Re-ChIP assays
ChIP and Re-ChIP assays were performed as described previously (Mal and Harter, 2003). Brief experimental procedure, antibodies and primers used and PCR amplification conditions can be found in Supplementary Materials and methods.
Supplementary Material
Supplementary Information
Acknowledgments
I thank Andrew Lassar, Eric N Olson, Pier Lorenzo Puri, Jin Q Cheng, Jiemin Wong, Judd C Rice, Peter M Chumakov, Andrei V Gudkov and Thomas Jenuwein for providing reagents. I wish to thank Dr Catherine Burkhart and Dr Andrei V Gudkov for critical reading of the manuscript. I am also thankful to Saumen Sarkar for graphics. This work is supported in part by funds provide by Lerner Research Institute and NIH NIAMS AR 051502 to AKM.
References
- Ait-Si-Ali S, Guasconi V, Fritsch L, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A (2004) A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J 23: 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres V, Walsh K (1996) Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol 132: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes CA, Bergstrom DA, Penn BH, Seaver KJ, Knoepfler PS, Tapscott SJ (2004) Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell 14: 465–477 [DOI] [PubMed] [Google Scholar]
- Berkes CA, Tapscott SJ (2005) MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 16: 585–595 [DOI] [PubMed] [Google Scholar]
- Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD (2005) An initial blueprint for myogenic differentiation. Genes Dev 19: 553–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM (2004) Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 304: 596–600 [DOI] [PubMed] [Google Scholar]
- Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V (2004) The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18: 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987–1000 [DOI] [PubMed] [Google Scholar]
- de la Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS, Tapscott SJ, Imbalzano AN (2005) MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol 25: 3997–4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodou E, Xu SM, Black BL (2003) mef2c is activated directly by myogenic basic helix–loop–helix proteins during skeletal muscle development in vivo. Mech Dev 120: 1021–1032 [DOI] [PubMed] [Google Scholar]
- Firestein R, Cui X, Huie P, Cleary ML (2000) Set domain-dependent regulation of transcriptional silencing and growth control by SUV39H1, a mammalian ortholog of Drosophila Su(var)3–9. Mol Cell Biol 20: 4900–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcales SV, Puri PL (2005) Signaling to the chromatin during skeletal myogenesis: novel targets for pharmacological modulation of gene expression. Semin Cell Dev Biol 16: 596–611 [DOI] [PubMed] [Google Scholar]
- Gerber AN, Klesert TR, Bergstrom DA, Tapscott SJ (1997) Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev 11: 436–450 [DOI] [PubMed] [Google Scholar]
- Kouzarides T (2002) Histone methylation in transcriptional control. Curr Opin Genet Dev 12: 198–209 [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Sullivan RJ, Jenuwein T (2003) An epigenetic road map for histone lysine methylation. J Cell Sci 116: 2117–2124 [DOI] [PubMed] [Google Scholar]
- Li J, Lin Q, Yoon HG, Huang ZQ, Strahl BD, Allis CD, Wong J (2002) Involvement of histone methylation and phosphorylation in regulation of transcription by thyroid hormone receptor. Mol Cell Biol 22: 5688–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, Olson EN (2000) Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6: 233–244 [DOI] [PubMed] [Google Scholar]
- Mal A, Chattopadhyay D, Ghosh MK, Poon RY, Hunter T, Harter ML (2000) p21 and retinoblastoma protein control the absence of DNA replication in terminally differentiated muscle cells. J Cell Biol 149: 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal A, Harter ML (2003) MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc Natl Acad Sci USA 100: 1735–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal A, Sturniolo M, Schiltz RL, Ghosh MK, Harter ML (2001) A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J 20: 1739–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437: 436–439 [DOI] [PubMed] [Google Scholar]
- Naya FS, Olson E (1999) MEF2: a transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr Opin Cell Biol 11: 683–688 [DOI] [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Naguibneva I, Fritsch L, Duquet A, Ait-Si-Ali A, Robin P, Vervisch A, Pritchard LL, Cole P, Harel-Bellan A (2001) CBP/p300 and muscle differentiation: no HAT, no muscle. EMBO J 20: 6816–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Sabourin LA, Rudnicki MA (2000) The molecular regulation of myogenesis. Clin Genet 57: 16–25 [DOI] [PubMed] [Google Scholar]
- Sartorelli V, Caretti G (2005) Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr Opin Genet Dev 15: 528–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Puri PL, Hamamori Y, Ogryzko V, Chung G, Nakatani Y, Wang JY, Kedes L (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol Cell 4: 725–734 [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N (2003) Establishment of histone h3 methylation on the inactive x chromosome requires transient recruitment of eed–enx1 polycomb group complexes. Dev Cell 4: 481–495 [DOI] [PubMed] [Google Scholar]
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL (2004) p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 36: 738–743 [DOI] [PubMed] [Google Scholar]
- Sims RJ III, Nishioka K, Reinberg D (2003) Histone lysine methylation: a signature for chromatin function. Trends Genet 19: 629–639 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Fukushima T, Shinkai Y (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem 276: 25309–25317 [DOI] [PubMed] [Google Scholar]
- Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ (1991) Muscle-specific transcriptional activation by MyoD. Genes Dev 5: 1377–1386 [DOI] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN (2002a) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110: 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Olson EN (2002b) Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol 22: 7302–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information