

Abstract
Emerin is a type II inner nuclear membrane (INM) protein of unknown function. Emerin function is likely to be important because, when it is mutated, emerin promotes both skeletal muscle and heart defects. Here we show that one function of Emerin is to regulate the flux of β-catenin, an important transcription coactivator, into the nucleus. Emerin interacts with β-catenin through a conserved adenomatous polyposis coli (APC)-like domain. When GFP-emerin is expressed in HEK293 cells, β-catenin is restricted to the cytoplasm and β-catenin activity is inhibited. In contrast, expression of an emerin mutant, lacking its APC-like domain (GFP-emerinΔ), dominantly stimulates β-catenin activity and increases nuclear accumulation of β-catenin. Human fibroblasts that are null for emerin have an autostimulatory growth phenotype. This unusual growth phenotype arises through enhanced nuclear accumulation and activity of β-catenin and can be replicated in wild-type fibroblasts by transfection with constitutively active β-catenin. Our results support recent findings that suggest that INM proteins can influence signalling pathways by restricting access of transcription coactivators to the nucleus.
Keywords: β-Catenin, emerin, laminopathies, nuclear envelope, nuclear lamina
Introduction
Emerin is a type II integral membrane protein of the inner nuclear membrane (INM). Emerin was identified as a 34 kDa protein encoded by the gene EMD located on the human X-chromosome, which when mutated gives rise to the X-linked form of Emery–Dreifuss muscular dystrophy (X-EDMD) (Bione et al, 1994; Manilal et al, 1996). The localisation of emerin at the INM is through its association with the type V intermediate filament proteins, lamins A/C (Sullivan et al, 1999; Vaughan et al, 2001), which form part of the nuclear lamina (Hutchison, 2002). Like lamins A/C, emerin expression is developmentally regulated and it normally appears at the time of organogenesis (Gareiss et al, 2005). Emerin on its own is dispensable for cell survival (Harborth et al, 2001) and normal development (Gruenbaum et al, 2002), but has overlapping functions in cell division and chromosome segregation with the INM protein MAN1 (Liu et al, 2003).
Because of its involvement in X-EDMD, there has been considerable interest in emerin function. A number of emerin binding partners have recently been identified, which hint at emerin function, but as yet no clear roles for emerin have been described. Emerin binds to the small dimeric chromatin-binding protein barrier to autointegration factor (BAF), through a conserved N-terminal sequence called the LEM domain (Lee et al, 2001; Segura-Totten and Wilson, 2004). BAF is essential for chromosome segregation (Margalit et al, 2005) and therefore emerin might assist BAF in this function. Emerin also binds to the transcription factor germ-cell-less (GCL) in a site overlapping the LEM domain (Holaska et al, 2003). The GCL/BAF-binding domain in emerin is also recognised by the death-promoting repressor Btf (Haraguchi et al, 2004). Despite reports that emerin binds to these transcription regulators, no function for emerin in regulating transcriptional activity has been definitively shown.
Emerin has also been implicated in cytoskeleton organisation. Emerin binds to both α- and β-actin (Lattanzi et al, 2003) and stabilises the pointed ends of F-actin (Holaska et al, 2004). Emerin also associates with the giant spectrin repeat proteins nesprin 1 (Mislow et al, 2002) and 2 (Libotte et al, 2005), and together with lamins A/C, nesprin 2 appears to be necessary for emerin localisation at the INM (Libotte et al, 2005; Zhang et al, 2005). Since nesprins have been implicated in actin bundling at the NE (Padmakumar et al, 2004), emerin might have a role in cortical actin organisation around the nucleus.
In cardiomyocytes, emerin colocalises with E-cadherin at intercalated disks (Cartegni et al, 1997). The authors noted that the C-terminal portion of the nucleoplasmic domain of emerin contained a sequence PVSASR-SSL-DLS that bore striking similarity to a consensus sequence in the adenomatous polyposis coli (APC) tumour suppressor protein. The APC consensus sequence referred to is involved in binding to the transcription coactivator β-catenin and is also found in E-cadherin (Pai et al, 1996). β-Catenin is the downstream target of the canonical Wnt signalling pathway and has been implicated in a range of developmental processes (e.g. Behrens et al, 1996; Molenaar et al, 1996). We therefore investigated the possibility that emerin is a β-catenin-binding protein in the nucleus. Here, we report that emerin is indeed a β-catenin-binding protein acts by influencing the nuclear accumulation of β-catenin through a CRM1-dependent export pathway. Furthermore, fibroblasts from patients with X-EDMD, which are null for emerin, display massive nuclear accumulation of β-catenin, which leads to an autostimulatory growth phenotype. We speculate that this growth phenotype might contribute to the accumulation of fibrotic tissue in both cardiac and skeletal muscle in X-EDMD patients.
Results
Emerin interacts with β-catenin through an APC homology domain
To investigate whether emerin is a β-catenin-binding protein, we first generated a number of emerin constructs. Three constructs were produced as either His-tagged or GFP-tagged proteins. The first construct (E1–220) encoded the entire nucleoplasmic domain of emerin, but lacked the transmembrane spanning and lumenal domains. The second construct (E1–176) encoded most of the nucleoplasmic domain, but contained a C-terminal deletion including 50% of the APC-like domain. The third construct (emerinΔ) contained an internal deletion of the APC-like domain together with seven additional amino acids (Supplementary Figure 1).
To investigate possible interactions between β-catenin and emerin, cell extracts from wild–type (wt) and emerin null fibroblasts were immunoprecipitated with antibodies against total β-catenin. β-Catenin was efficiently immunoprecipitated from wt and emerin null fibroblast extracts (Figure 1A). Emerin was efficiently co-immunoprecipitated with β-catenin in wt fibroblast extracts. The corresponding bands were absent in immunoprecipitates from emerin null fibroblast extracts, demonstrating that they indeed represent emerin (Figure 1A). The closely related LEM domain protein, LAP2β, was absent from β-catenin immunoprecipitates, demonstrating that the interaction between β-catenin and emerin was specific (Figure 1A). Emerin was not immunoprecipitated from wt fibroblast extracts using empty immunobeads, again suggesting that the interaction between emerin and β-catenin was specific.
Figure 1.
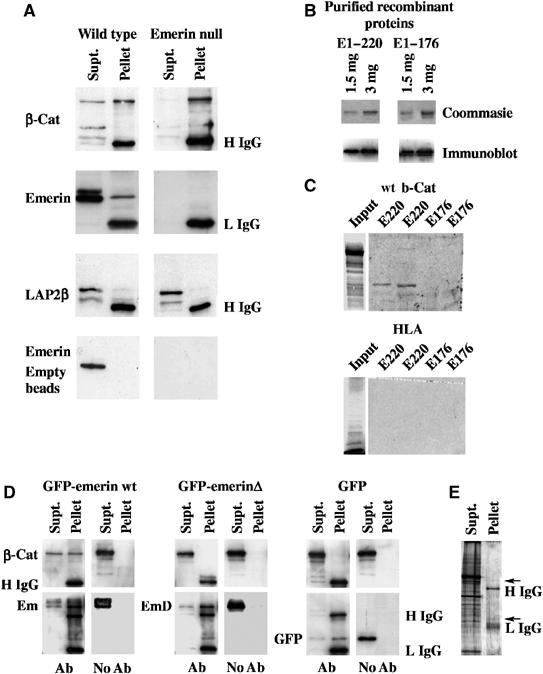
β-Catenin binds to emerin through a conserved APC homology domain. (A) β-Catenin was immunoprecipitated from cell extracts prepared from wt or emerin null fibroblasts. Immunoprecipitates were resolved on SDS–PAGE and immunoblotted with antibodies against total β-catenin (β-cat), emerin or LAP2β (top three panels). Alternatively, empty immunobeads were incubated with the same cell extracts and used for immunoblotting with anti-emerin antibody (bottom panel). The antibody used in each blot is indicated to the left-hand side of each panel, while the positions of IgG heavy or light chains (H IgG and L IgG, respectively) are indicated to the right-hand side of each blot. (B) Purified recombinant proteins E1–176 and E1–220 were resolved on SDS–PAGE, which were either stained with Coomassie Brilliant Blue (upper panels) or immunoblotted with anti-emerin antibody (lower panels). (C) 35S-met-labelled wt β-catenin or HLA were resolved on SDS–PAGE and subjected to autoradiography (input). Purified emerin E1–220 and E1–176 at 1.5 and 3 μg were resolved on SDS–PAGE and transferred to nitrocellulose. The filters were then overlayed with 35S-methionine-labelled proteins and subjected to autoradiography. In all panels, the first lane was loaded with 1.5 μg of recombinant protein, while the second lane was loaded with 3.0 μg. (D) β-Catenin and one of GFP-emerin, GFP-emerinΔ or GFP were coexpressed in HEK293 cells. Cell lysates were immunoprecipitated with anti-GFP antibody or empty immunobeads (indicated by Ab or no Ab, respectively, underneath each panel). Immunoprecipitates were immunoblotted with antibodies against β-catenin (upper panels) or GFP (lower panels). The positions of β-catenin (β-cat), GFP-emerin (em), GFP-emerinΔ (emΔ) or GFP are indicated at the left-hand side of each panel. The positions of IgG heavy chain (H IgG) and IgG light chain (L IgG) are indicated at the right-hand side of the final panel. (E) HEK293 cells that were co-transfected with GFP-emerin and β-catenin were immunoprecipitated with anti-GFP antibodies, resolved on SDS–PAGE and stained with ammoniacal silver. In all gels, Supt=material remaining in the supernatant. Pellet=material recovered in immunoprecipitates. Arrow indicates a band with expected mobility of GFP-emerin.
To confirm that emerin and β-catenin interact and to map the site of interaction, His-tagged versions of the emerin constructs E1–220 and E1–176 were expressed in Escherichia coli and purified (Figure 1B). The purified proteins were resolved in SDS–PAGE, transferred to nitrocellulose and the filters were overlayed with either 35S-labelled β-catenin or as a control 35S-labelled human leukocyte antigen (HLA). β-Catenin interacted with E1–220 but not with E1–176. In contrast, HLA did not interact with any emerin peptide (Figure 1C). These findings suggested that emerin and β-catenin interact through the APC homology domain in emerin.
To confirm that emerin binds to β-catenin through its APC homology domain, HEK293 cells were co-transfected with β-catenin and one of GFP-emerin, GFP-emerinΔ or GFP. Cell extracts were used for immunoprecipitation (IP) with anti-GFP antibody. Each of the GFP-fusion proteins and GFP itself were efficiently immunoprecipitated with anti-GFP antibodies, but did not associate with empty immunobeads (Figure 1D). β-Catenin was efficiently co-immunoprecipitated with GFP-emerin but not with either GFP-emerinΔ or GFP. Neither did it associate with empty immunobeads (Figure 1D). To investigate the possibility that large numbers of proteins interact with emerin in IP assays, we resolved emerin IPs from HEK293 cell extracts on SDS–PAGE and silver-stained the gels. While IgG bands and a band with a molecular weight corresponding to GFP-emerin were clearly detected, major bands observed in cell extracts were not found in the IPs, indicating that the interaction between emerin and β-catenin were specific (Figure 1E). Based on these findings, we concluded that emerin was a β-catenin-binding protein and interacts with β-catenin through an APC-like domain.
Emerin inhibits β-catenin activity by preventing its accumulation in the nucleus
To investigate the influence of emerin on β-catenin activity, HEK293 cells were co-transfected with β-catenin constructs, TOPGLOW or FOPGLOW luciferase reporters, and one of GFP, GFP-emerin or GFP-emerinΔ. In the presence of TOPGLOW reporters, luciferase activity was depressed ∼2-fold in the presence of GFP-emerin, compared to GFP alone. In contrast, luciferase activity was activated ∼2-fold in the presence of GFP-emerinΔ compared to GFP alone (Figure 2A). Luciferase activity was negligible in the presence of FOP-GLOW reporters. These findings suggested that GFP-emerin antagonises β-catenin activity, while GFP-emerinΔ stimulates β-catenin activity. To further investigate the influence of emerin on β-catenin activity, we performed reporter assays on a colon carcinoma cell line carrying an activating mutation of β-catenin (van de Wetering et al, 2002). Overexpression of emerin in this cell line did not inhibit β-catenin activity. However, in common with other cancer cell lines (Vaughan et al, 2001), this cell line did not express lamins A/C at detectable levels and as a result emerin was located in the cytoplasm rather than at the INM (not shown). Therefore, instead, we coexpressed a mutant form of β-catenin (S37A) in HEK293 cells with either GFP or GFP-emerin. The activity of S37A was depressed by ∼40% in the presence of GFP-emerin compared to GFP alone, indicating that emerin is able to inhibit even activated forms of β-catenin (Figure 2B) if it is localised to the INM.
Figure 2.
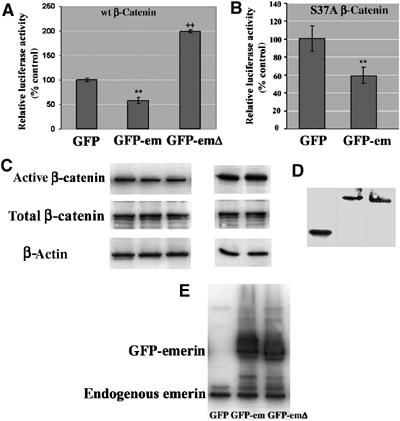
Emerin antagonises β-catenin activity. (A) HEK293 cells were transfected with combinations of wt β-catenin and GFP, GFP-emerin or GFP-emerinΔ together with TOPGLOW or FOPGLOW luciferase reporters and Renilla. (B) Alternatively, HEK293 cells were co-transfected with an activating mutation of β-catenin (S37A) and either GFP or GFP-emerin together with TOPGLOW or FOPGLOW luciferase reporters and Renilla. The levels of luciferase reporter assays were expressed relative to the level of luciferase activity in the presence of GFP. **Significantly reduced luciferase activity (P<0.001), ++Significantly increased luciferase activity (P<0.001). (C) HEK293 cells were transfected with β-catenin and GFP, GFP-emerin or GFP-emerinΔ or S37A and GFP or GFP-emerin, respectively. Cell extracts were prepared for immunoblotting with antibody against active β-catenin, total β-catenin or β-actin. Alternatively, GFP, GFP-emerin or GFP-emerinΔ were expressed in HEK293 cells. Cell lysates were prepared for immunoblotting with either anti-GFP (D) or anti-emerin antibodies (E).
To further investigate this phenomenon, the level of expression of total β-catenin was investigated by immunoblotting of transfected cells. There were negligible differences in the amounts of β-catenin in cells transfected with GFP, GFP-emerin or GFP-emerinΔ (Figure 2C). Next, the level of expression of each reporter was investigated and compared to endogenous emerin. The relative amounts of each reporter were also found to be equivalent in each transfected culture (Figure 2D). Moreover, by introducing GFP-emerin or GFP-emerinΔ into HEK293, the total amount of emerin was effectively doubled compared to GFP-transfected cells (Figure 2E).
The results reported above show that by doubling the amount of emerin in HEK293 cells, β-catenin activity is correspondingly reduced. In contrast, by introducing a mutant form of emerin into the INM, β-catenin activity is increased. These changes in β-catenin activity are not correlated with changes in expression levels of β-catenin. Several reports have indicated that β-catenin activity might be regulated by its rate of accumulation in the nucleus (Neufeld et al, 2000; Rosin-Arbesfeld et al, 2000). Therefore, we investigated the level of expression of nuclear β-catenin in cells transfected with the various GFP-reporters. Cells transfected with GFP alone showed moderate levels of β-catenin in the nucleus as well as staining at cell adhesion junctions (Figure 3A). In contrast, there was little nuclear β-catenin in cells transfected with GFP-emerin, but increased staining at cell adhesion junctions (Figure 3B). Cells transfected with GFP-emerinβ had very high levels of nuclear β-catenin (Figure 3C) compared GFP-transfected cells (Figure 3A) or untransfected cells. Taken together, these data suggest that overexpressing emerin restricts the amount of nuclear β-catenin, while expressing emerinΔ increases the amount of nuclear β-catenin. To investigate how emerin might influence the nuclear accumulation of β-catenin, transfected cells were treated with the drug leptomycin B, which inhibits CRM1-dependent nuclear export. Treatment with leptomycin B led to increases in the amount of nuclear β-catenin in GFP- and GFP-emerin-transfected cells, but did not give rise to further increases in the amount of nuclear β-catenin in GFP-emerinΔ cells (Figure 3A–C). Therefore, our data suggest that emerin participates in the CRM1-dependent nuclear export of β-catenin.
Figure 3.
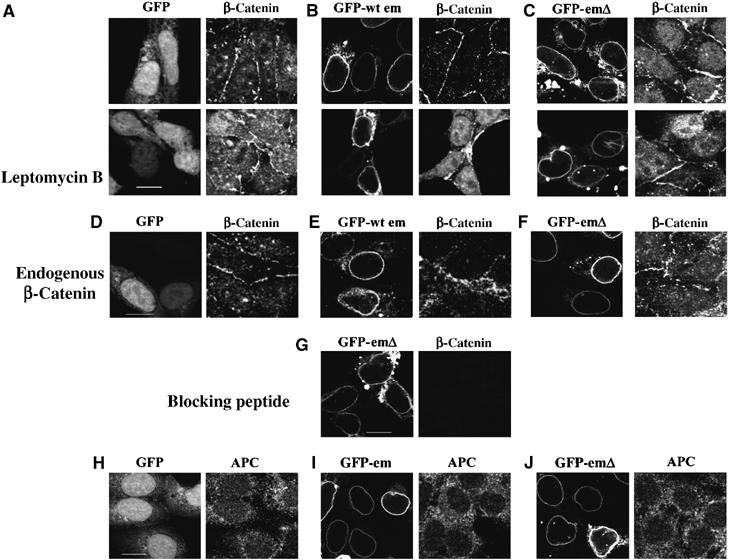
Emerin functions in a β-catenin export pathway. GFP (A), GFP-emerin (B) or GFP-emerinΔ (C) were co-transfected with β-catenin into HEK293 cells. Transfected cultures were grown in the presence or absence of leptomycin B and then fixed and stained with anti-β-catenin antibody followed by TRITC anti-mouse Ig. Alternatively, cultures were transfected with GFP, GFP-emerin and GFP-emerinΔ, then stained with anti-β-catenin antibody to investigate the influence of emerin on endogenous β-catenin (D–F). (G) Cells co-transfected with GFP-emerinΔ and β-catenin were stained with anti-β-catenin antibody that had been preabsorbed with a peptide corresponding to its epitope. Finally, cultures transfected with GFP, GFP emerin or GFP-emerinΔ were stained with anti-APC antibody followed by TRITC anti-mouse Ig (H–J). Micrographs show black and white images recorded from each fluorescence channel. Scale bars=10 μm.
To further investigate the influence of emerin on the localisation of β-catenin, we also looked at endogenous β-catenin. In cells expressing GFP alone (Figure 3D) or GFP-emerinΔ (Figure 3F), β-catenin was detected in the nucleus and at cell adhesion junctions. In contrast, in cells expressing GFP-emerin, β-catenin was absent from the nucleus and instead accumulated at cell adhesion junctions (Figure 3E).
In a recent report, nuclear immunostaining of some proteins in the Wnt signalling pathway were found to be nonspecific. To eliminate the possibility that the nuclear staining described here was nonspecific, we preabsorbed the β-catenin antibody used for immunostaining with a peptide corresponding to its epitope. Preabsorption with the peptide eliminated all staining (both within the nucleus and at cell adhesion junctions), indicating that both patterns of staining are specific (Figure 3G).
It has been reported that the nuclear export of β-catenin can be mediated by APC (Elftheriou et al, 2001). To investigate whether emerin might be involved in an APC pathway, we investigated the levels of nuclear APC in the presence of our GFP reporters. While the levels of nuclear APC were higher in the presence of GFP, than in the presence of GFP-emerin, we did not observe an increase in the level of nuclear APC in the presence of GFP-emerinΔ (Figure 3H–J). Based on these data, we could not conclude that emerin was acting within an APC pathway.
Abnormal growth phenotypes associated with the loss of emerin expression are caused by the activation and nuclear accumulation of β-catenin
To investigate whether emerin-dependent nuclear accumulation of β-catenin gives rise to predictable phenotypic changes, we used a collection of fibroblasts from patients with X-EDMD that are null for the expression of emerin (Markiewicz et al, 2002a). Previous reports have shown that β-catenin regulates cell proliferation in fibroblasts (Stockinger et al, 2001). Therefore, we initially compared the cell proliferation rates in three wt and three emerin null cell strains. All three emerin null strains proliferated more rapidly than wt strains. The density of emerin null fibroblast cultures was greater than control cultures 4 days after seeding (Figure 4A–D), while the rate of growth within the emerin null populations was significantly greater on each day of culture after seeding (Figure 4E).
Figure 4.
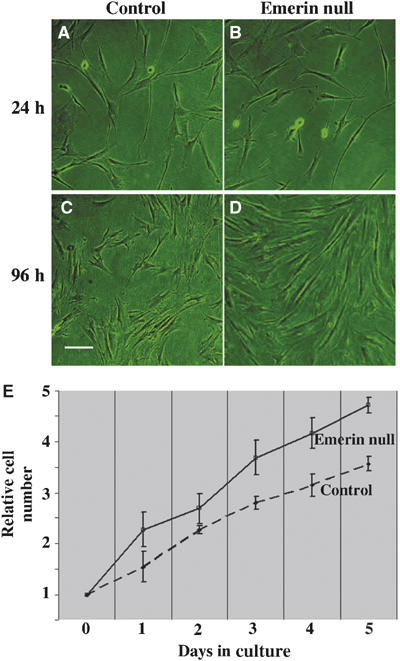
Emerin null fibroblasts display a rapid growth phenotype. wt (A, C) or emerin null fibroblasts (B, D) were grown in culture and photographed 24 h (A, B) and 96 h (C, D) after subculture. Alternatively, cells were harvested and counted each day. (E) Shows relative cell numbers after subculture for wt (control) and emerin null fibroblasts over 5 days. Each time point represents pooled data from three different wt and three different emerin null fibroblast strains. At each time point, the relative number for emerin null fibroblasts was always statistically greater than for wt fibroblasts. The P-values were as follows: day 1, 0.03; day 2, 0.05; day 3, 0.001; day 4, 0.005; and day 5, 0.002.
Next, we investigated whether the increased growth rate in emerin null fibroblasts was correlated with nuclear accumulation of β-catenin. Fibroblast cultures were costained with antibodies against emerin and β-catenin. In wt fibroblasts, emerin was concentrated at the NE, while β-catenin was located at cell adhesion plaques (Figure 5A). In contrast, in emerin null fibroblasts, emerin was not detected, but β-catenin was concentrated in the nucleus (Figure 5B). To determine whether the nuclear accumulation of β-catenin in emerin null fibroblasts resulted from increased levels of β-catenin, we performed semiquantitative immunoblotting. In all emerin null fibroblast strains, expression of active β-catenin was increased by two-fold or more compared to controls (Figure 5C and D). Thus, the absence of emerin in fibroblasts leads to a modest increase in the expression of active β-catenin but a massively increased nuclear accumulation of the protein.
Figure 5.
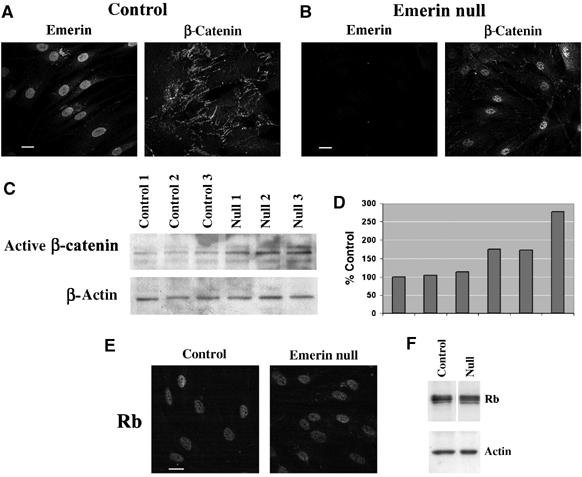
Nuclear accumulation and increased expression of β-catenin in emerin null fibroblasts. Wt (A—control) or emerin null (B) human fibroblasts were costained with antibodies against emerin or active β-catenin to determine the relative distributions of each protein. Micrographs show black and white images taken from each fluorescence channel. Alternatively, extracts were prepared from three different wt and three different emerin null human fibroblast strains and used for immunoblotting with either anti-active β-catenin or anti-β-actin antibodies (C). Densitometry was performed to quantify the level of expression of active β-catenin in each culture relative to the first control culture (D). Wt (control) or emerin null fibroblasts were stained with an antibody against Rb (E) Alternatively, wt (control) or emerin null fibroblasts were prepared for Western blotting using antibodies against total Rb or β-actin (F). Scale bars=10 μm.
A rapid growth phenotype is also characteristic of fibroblasts from an Lmna −/− mouse. However, this phenotype arises because the retinoblastoma (Rb) protein is proteolysed (Johnson et al, 2004). We therefore also investigated Rb expression and localisation in the emerin null fibroblasts. The distribution of Rb in emerin null fibroblasts was identical to controls (Figure 5E). In addition, the levels of expression of different Rb isoforms, judged by Western blotting, were also identical in emerin null and wt fibroblasts (Figure 5F). Therefore, we concluded that the rapid growth phenotype did not arise from defects in the Rb pathway.
We wondered whether the increase in nuclear β-catenin would be correlated with responsiveness to withdrawal of growth factors. To investigate this possibility, we transferred wt and emerin null fibroblasts to low serum medium and stained each culture with the proliferation marker Ki67 immediately afterwards or 96 h later. Immediately after transfer to low serum medium, >60% of cells in each culture were proliferating as judged by the expression of Ki67 (Figure 6A, B and H). In contrast, <10% of cells in wt fibroblast cultures were proliferating 96 h later, whereas ∼60% of emerin null fibroblasts were proliferating at the same time (Figure 6C, D and H). These findings suggested that emerin null fibroblasts did not respond to withdrawal of growth factors by entry into a quiescent state. To investigate whether this growth phenotype resulted from the absence of expression of emerin, we transfected emerin null fibroblasts with GFP-emerin and grew them under selection to establish a stable cell line. In emerin null fibroblasts expressing GFP-emerin, β-catenin was not detected in the nucleus, but instead was detected in the cytoplasm (Figure 6E and F). When the same cultures were transferred to low serum medium and stained with Ki67 antibodies, while almost 60% of cells were proliferating immediately after transfer to low serum medium, only 12% proliferating 96 h later (Figure 6G and H). Thus, autostimulatory growth in emerin null fibroblasts is a direct result of the absence of emerin expression and is correlated with nuclear accumulation of β-catenin.
Figure 6.
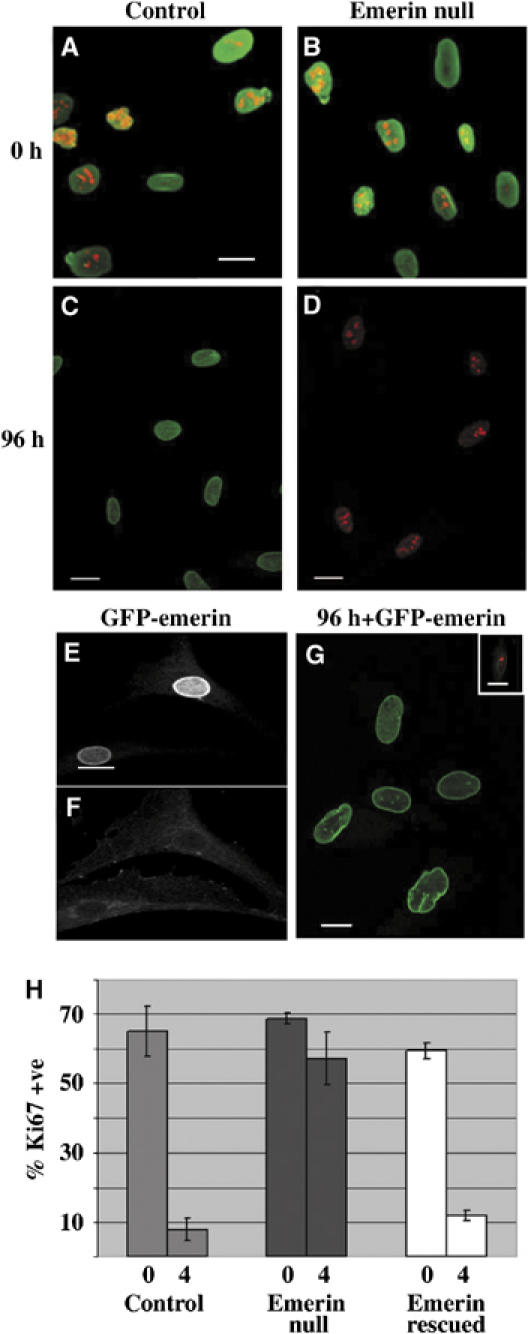
Emerin null fibroblasts display autostimulatory growth in culture. Wt or emerin null fibroblasts were transferred to low serum medium. Proliferative capacity was investigated by costaining cultures with either anti-lamin A/C and anti-Ki67 antibodies (A, B) or anti-emerin and anti-Ki67 antibodies (C, D) immediately after transfer to low serum medium (0 h) or 96 h later. Each panel shows two colour merged images in which the green signal represents either lamin A/C or emerin and the red signal represents Ki67. A stable cell line expressing GFP-emerin was established from one emerin null fibroblast strain (E–G). The cell line was stained with anti-β-catenin antibody followed by TRITC anti-mouse Ig. The panels show representative black and white images of GFP-emerin (E) and TRITC (F) fluorescence. Alternatively, cultures were transferred to low serum medium for 4 days and then fixed and stained with anti-Ki67 antibody followed by TRITC anti-rabbit Ig (G). The panel shows representative two colour merged images in which GFP-emerin is in green and Ki67 is in red. Inset in (G) shows Ki67 expression in the same cell line before transfer to low serum medium. Scale bars in all micrographs are 10 μm. To quantify the level of expression of Ki67 in each culture used, 200 cells were scored on duplicate slides and in triplicate experiments immediately after transfer to low serum medium (0) and 96 h later (H). The data are expressed as the mean percentage of Ki67 +ve cells±s.e.
To obtain direct evidence that loss of emerin function leads to activation of β-catenin signalling, we performed reporter assays on emerin null and wt fibroblasts. β-Catenin activity was almost five-fold higher in emerin null fibroblasts compared to control fibroblasts (Figure 7A). To determine whether the increased activity of β-catenin leads to transcription of downstream target genes, we investigated the expression of c-myc, which is the immediate downstream target of β-catenin/TCF-4 (van de Wetering et al, 2002). We found that c-myc was massively upregulated in emerin null fibroblasts compared to controls (Figure 7B), again suggesting activation of β-catenin. Previous studies have shown that ectopic expression of the Wnt agonist axin inhibits Wnt-dependent cell proliferation in haematopoietic stem cells (Reya et al, 2003). To determine whether activation of β-catenin causes cell proliferation in emerin null fibroblasts, we transfected wt and emerin null fibroblasts with axin. We found that cell proliferation was depressed by ∼25% in axin-transfected wt fibroblasts. Importantly, cell proliferation was almost completely inhibited in axin-transfected emerin null fibroblasts, indicating that proliferation in these cells was stimulated by β-catenin/TCF signalling (Figure 7C).
Figure 7.
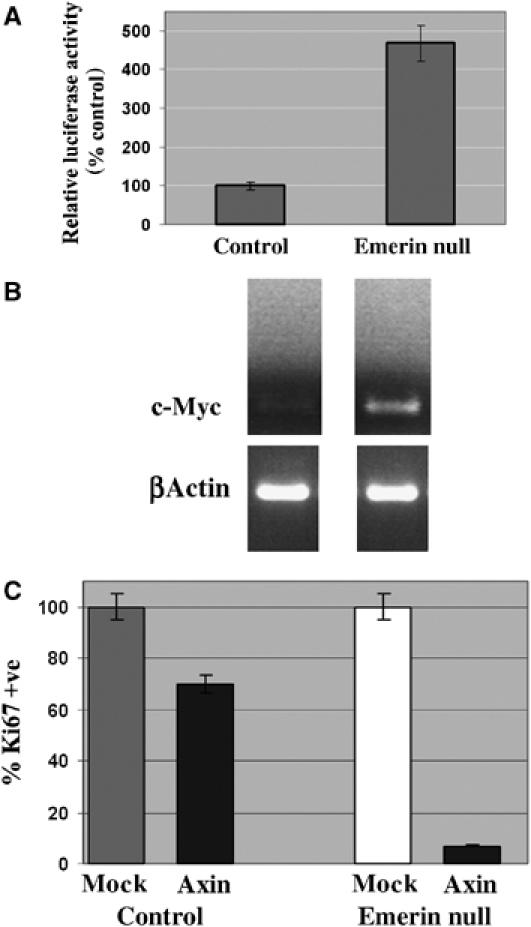
β-Catenin activation causes proliferation in emerin null fibroblasts. Wt and emerin null fibroblasts were transfected with β-catenin and either TOPGLOW or FOPGLOW reporters together with Renilla. The levels of luciferase in emerin null fibroblasts are expressed relative to the levels of luciferase in wt fibroblasts (A). RT–PCR was performed on RNA extracted from wt (control) or emerin null fibroblasts using primers specific for c-myc or as a loading control β-actin (B). Wt (control) or emerin null fibroblasts were transfected with FLAG-tagged axin or were mock transfected. After 48 h, cultures were stained with antibodies against Ki67 (or anti-FLAG antibodies to determine transfection efficiency). Two hundred cells were counted on each of triplicate slides to determine the proliferation index. For both control or emerin null cells, the proliferation index in axin-transfected cultures were expressed as a relative percentage of the proliferation index in mock-transfected cultures (C).
To investigate whether autostimulatory growth could be attributed directly to the activation of β-catenin, normal fibroblasts cell lines that permanently expressed exogenous wt or constitutively active β-catenin (S37A) were created. In the cell line expressing S37A, β-catenin was localised in the nucleus of all cells, even though the cells expressed endogenous emerin (Supplementary Figure 2A). In the cell line expressing wt β-catenin, it was located in the nucleus in only a minority of cells (Supplementary Figure 2D). Each cell line was transferred to low serum medium to investigate responsiveness to growth factors. At 12 h after transfer to low serum medium, >80% of cells expressing S37A were proliferating and had nuclear β-catenin (Supplementary Figure 2B and G). At the same time, 70% of cells expressing wt β-catenin were Ki67 positive, but none displayed nuclear β-catenin (Supplementary Figure 2E and G). At 96 h after transfer to low serum medium, >50% of cells expressing S37A were still proliferating and displaying high levels of nuclear β-catenin (Supplementary Figure 2C and G). In contrast, only 10% of cells expressing wt β-catenin proliferate at the same time (Supplementary Figure 2F and G). Thus, activation of β-catenin did give rise to an autostimulatory growth phenotype.
Discussion
Emerin function in nuclear export of β-catenin
In this study, we show that emerin binds to β-catenin through a C-terminal APC-like domain. Overexpression of emerin inhibits β-catenin signalling by preventing the nuclear accumulation of β-catenin. In contrast, expression of a mutant form of emerin that lacks the β-catenin-binding domain dominantly stimulates β-catenin nuclear accumulation and signalling. Emerin appears to act by stimulating the export of β-catenin, since the nuclear export inhibitor, leptomycin B, abrogates the effect of overexpression of emerin.
The shuttling of β-catenin into and out of the nucleus is seen as central to its function as a transcription coactivator. β-Catenin appears to be exported from the nucleus by at least two independent pathways, one of which is dependent on CRM1, while the other is independent of CRM1 (Wiechens and Fagotto, 2000; Elftheriou et al, 2001). Several reports have indicated that export of β-catenin from the nucleus is mediated by other proteins. A number of studies have shown that APC shuttles between the nucleus and cytoplasm (Henderson, 2000; Neufeld et al, 2000; Rosin-Arbesfeld et al, 2000) through centrally located nuclear export signal sequences (Rosin-Arbesfeld et al, 2003) and mediates the CRM1-dependent nuclear export of β-catenin. Emerin-mediated export of β-catenin does not appear to occur through an APC-dependent pathway, since neither overexpression of emerin nor expression of emerinΔ dramatically affects the levels of nuclear APC. More recent investigations have implicated the negative regulator of the Wnt pathway, axin, as a mediator of CRM1-dependent nuclear export of β-catenin (Cong and Varmus, 2004; Wiechens et al, 2004). It remains to be seen whether emerin is acting through this pathway.
It is clear that many proteins that control cell proliferation and survival are regulated by the rate at which they accumulate in the nucleus (Fabbro and Henderson, 2003). The nuclear accumulations of two proteins that control cell proliferation have recently been shown to be mediated by proteins of the INM. The tumour suppressor Rb protein is anchored in the nucleus by lamins A/C (Markiewicz et al, 2002b). In cells that are null for lamins A/C, Rb is not retained in the nucleus and as a consequence is targeted for destruction by the proteasome (Johnson et al, 2004). More recently, it has been shown that the INM protein MAN1 regulates the nuclear translocation of the TGF-β effector, rSMAD, and therefore antagonises BMP signalling pathways (Osada et al, 2003; Pan et al, 2005). Emerin is now the third example of an INM protein that influences transcription by restricting the rate of nuclear accumulation of a transcription regulator. Therefore, influencing the rate of accumulation of transcription regulators in the nucleus is an important emerging function of INM proteins. Based on the examples that are available currently, this function appears to occur in two distinct ways, in that emerin and MAN1 both restrict the nuclear accumulation of transcription coactivators and therefore antagonise signalling pathways, while lamins A/C promote the nuclear accumulation of Rb and therefore re-inforce gene silencing.
EmerinΔ dominantly stimulated β-catenin signalling when present at the INM. Since emerinΔ cannot bind to β-catenin, it was not surprising that it did not inhibit β-catenin signalling. However, it was surprising that β-catenin signalling was dominantly upregulated by emerinΔ. Previous reports have suggested that emerin can bind to itself in vitro (Clements et al, 2000). In addition, emerin also forms hetero-oligomeric complexes with other nuclear envelope proteins, such as the lamins (Vaughan et al, 2001). Therefore, if emerin function is determined by its existence within dimeric or trimeric complexes in vivo, then the presence of emerinΔ in such complexes might render wt emerin nonfunctional for the export of β-catenin and thus replicate the phenotype of an emerin null cell.
While our data clearly show that emerin influences the nuclear accumulation of β-catenin, the mechanism of action is less clear. Overexpression of emerin in cells lacking lamins A/C does not inhibit β-catenin activity, but neither does emerin localise to the INM in these cells. Therefore, the correct localisation of emerin at the INM is necessary for its influence on nuclear accumulation of β-catenin. The nucleoplasmic domain of emerin exists in a very flexible state with little defined secondary structure in the absence of binding partners (Wolff et al, 2001; Laguri et al, 2001). Therefore, in the absence of overlapping binding partners, residues 168–186 would be available for binding to β-catenin in vivo. This might have the dual effects of retaining β-catenin close to the nuclear envelope and altering emerin conformation once β-catenin is bound, thereby aiding export of β-catenin through nuclear pore complexes. It is also notable that the major sites of emerin phosphorylation (ser163, tyr167 and ser175) are all either immediately upstream of or within the β-catenin-binding domain (Brill et al, 2004; Hirano et al, 2005). Therefore, β-catenin–emerin interactions in vivo could be regulated by phosphorylation and therefore might be very transient, which again could be an aid to nuclear export. Other proteins that bind to emerin in vitro also have a very transient interaction with emerin in vivo. For example, the small DNA-binding protein BAF interacts very transiently but very frequently with emerin during interphase; however, while this interaction can be detected by techniques such as FRET, BAF localisation in immunofluorescence assays is nucleoplasmic rather than at the NE (Shimi et al, 2004). We propose that emerin might interact with β-catenin during interphase in a similar manner and that indeed a “touch and go” mode of association would be an aid to efficient nuclear export.
Possible function of emerin in controlling cell proliferation and consequences for disease
Emerin is anchored in the nucleus through interactions with lamins A/C (Sullivan et al, 1999; Vaughan et al, 2001) and the genes encoding emerin (EMD) and lamins A/C (LMNA) can both give rise to different forms of EDMD when mutated. Mutations in EMD give rise to X-EDMD (Bione et al, 1994), while mutations in LMNA give rise to autosomal-dominant and autosomal-recessive EDMD (Bonne et al, 1999; Raffaele Di Barletta et al, 2000). The functional link between the two proteins and the fact that both are implicated in very similar diseases suggested that both proteins operate through convergent pathways. At face value this appears to be true. A recent report has shown that mouse embryo fibroblasts (MEFs) from an Lmna −/− mouse, which is a model of EDMD, have a rapid growth phenotype, similar to the one described here for emerin null fibroblasts. Moreover, in the fibroblasts emerin is absent from the INM (Sullivan et al, 1999). However, it has been reported that the rapid growth phenotype in Lmna −/− MEFs arises because of the loss of anchorage function in Rb, leading to its proteasomal degradation and therefore effectively an Rb null phenotype (Johnson et al, 2004). In emerin null human fibroblasts, the expression and phosphorylation of Rb is unaffected and autostimulatory growth arises through the activation of β-catenin. This divergence in molecular pathways might of course reflect fundamental differences between human and mouse, since fibroblasts from a unique patient with a homozygous nonsense mutation (Y259X) in LMNA displayed slow growth in culture and premature senescence (Muchir et al, 2003). Alternatively, since we have shown that loss of lamin A/C leads to mis-localisation of emerin to the cytoplasm and loss of function, up-regulated β-catenin signalling might also contribute to the rapid growth phenotype observed in Lmna −/− MEFs.
In an emerin knockout mouse, skeletal muscle regeneration is impaired both in vivo and in vitro, and this appears to occur through upregulated phosphorylation of Rb and therefore impairment of Rb inhibition of E2F (Melcon et al, 2006). The mouse itself does not have an EDMD-like phenotype and Wnt signalling is not implicated in skeletal muscle regeneration, while Rb function is. However, in an earlier investigation of upregulated and downregulated genes in fibroblasts from X-EDMD patients, changes in expression of genes within the Wnt signalling pathway were reported (Tsukahara et al, 2002). Therefore, the apparent involvement of Rb pathways in the Melcon study versus the lack of Rb involvement in our study might be owing to the different cell lineages used or differences between mouse and human, which is reflected by the fact that the emerin knockout mouse is not a model for X-EDMD.
Despite the fact that mutations in LMNA appear to affect Rb pathways, while mutations in EMD affect β-catenin signalling in fibroblasts, both lead to abnormal growth phenotypes, which could contribute to the aetiology of EDMD. A striking feature of cardiac and skeletal muscle associated with patients with mutations in EMD and LMNA is the accumulation of fatty fibrotic tissue, which replaces cardiomyocytes and muscle fibres (e.g. van der Kooi et al, 1996; Fatkin et al, 1999). It has been proposed recently that mutations in LMNA might contribute to this phenotype by enhanced cell turnover leading to accelerated ageing and increased fibrosis in the heart and in muscle blocks (van Berlo et al, 2005). Wnt signalling is implicated in the generation and function of heart muscle (Eisenberg and Eisenberg, 2006), thus aberrant Wnt signalling might promote cardiomyopathy. Therefore, based on our current findings, we propose that mutations in EMD might contribute to skeletal muscle and heart pathology, through accelerated cell turnover, which might lead to the expansion of fibroblast populations and as a result increased fibrosis.
Materials and methods
Fibroblast cultures
Fibroblast cultures from three Polish patients with X-linked EDMD and three age-matched normal individuals and their growth conditions have been described previously (Markiewicz et al, 2002a). For cell counting, cells were harvested at 24 h intervals and cell numbers estimated by counting duplicate samples using a Neubauer haemocytometer. Alternatively, 2 days after subculture cells were induced to enter a quiescent state by transferring them to DMEM supplemented with 0.5% FBS plus antibiotics. Cultures were used between passages 6 and 12.
Transfection of fibroblasts
Cells were seeded onto six-well plates at an initial density of 1.5 × 105 cells/well. After 36 h, cultures were transfected with GFP-emerin by liposome-mediated transfection using GeneJammer (Stratagene). At 24 h after transfection, cultures were treated with Genaticin (GibcoBRL) to kill untransfected cells. At 5 days after transfection, cells were subcultured onto glass coverslips and then transferred to low serum medium. Alternatively, cells were transfected with wt or mutant β-catenin (Morin et al, 1997) as described above and grown under selection. Finally, fibroblasts were transiently transfected with FLAG-Axin in pcDNA4 (5 μg DNA) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfection efficiency was >75% and transfected (FLAG-positive) cells were scored for Ki67 proliferation marker in duplicates slides in triplicate experiments.
Transient expression of emerin and β-catenin constructs in HEK293 cells
Two micrograms of plasmid DNA (GFP, GFP-emerin wt, GFP-emerinΔ, wt β-catenin or S37A β-catenin) in pcDNA3, 0.12 M CaCl2 and HBS (70 mM NaCl, 0.5 mM Na2HPO4, HEPES, pH 7.0) were added to 2 ml of culture medium and added to cultures in six-well plates. The medium was replaced after 24 h, and the proteins were transiently expressed in cells for an additional 24 h.
Indirect immunofluorescence microscopy
Cells growing on 12 mm diameter glass coverslips were processed for immunofluorescence according to published protocols (Vaughan et al, 2001). Cells were stained with rabbit anti-Ki67 (DAKO), monoclonal anti-lamin A/C (JoL2, Serotec), monoclonal anti-emerin antibodies (NCL, Novocastra), monoclonal anti-active β-catenin (clone 8E7, Upstate Biochemicals) or monoclonal anti-APC (Ab-7, Calbiochem). Samples were observed with a Zeiss Axioskop fitted with a × 40 1.4 N/A PlanNeofluor lens and equipped for epifluorescence. Alternatively, images were collected using a BioRad MRC 600 laser scanning confocal microscope. For blocking of the antibody against active β-catenin, a specific peptide was created with the sequence H-His-Ser-Gly-Ala-Thr-Thr-Thr-Ala-Pro-NH2, identical to the epitope recognized by the antibody (residues 34–44 of β-catenin clone 8E7). The peptide (10 μg) was mixed with the antibody (1 μg) prior to indirect immunofluorescence.
Western blotting experiments
Fibroblast cultures were established in 25 cm2 flasks were scraped into ice-cold RIPA buffer (Markiewicz et al, 2002a) containing a cocktail of protease inhibitors (Sigma) and boiled for 3 min. The equivalent of 3 × 105 cells were loaded per lane of a 10% SDS–PAGE and resolved. Proteins were transferred to nitrocellulose and immunoblotted with anti-active β-catenin or anti-β-actin (1:1000, Sigma) using chemiluminescence. To estimate the relative levels of expression of β-catenin, densitometry was performed using UVI Bandmap software (UVItec Ltd).
β-Catenin/TCF transactivation assay
Cells growing in six-well plates (Greiner, UK) at a cell density 2.5 × 105/well were transfected with mixtures of 1 μg of TOPGLOW or FOPGLOW plasmid DNA, 0.1 μg TK Renilla and different combinations of GFP, GFP-emerin wt, GFP-emerinΔ and wt β-catenin in pcDNA3 (1 μg each). HEK293 cells were transfected using 0.12 M CaCl2 and HBS (70 mM NaCl, 0.5 mM Na2HPO4, HEPES, pH 7.0). Human fibroblasts were transfected with Lipofectamine 2000 (Invitrogen). The medium was replaced after 24 h, and the proteins were transiently expressed in cells for an additional 24 h. Luciferase and Renilla activities were assayed using a luciferase assay system (Promega) and luminescence was measured using a Lumat LB 9507 luminometer (Berthold Technologies). Control luciferase activity (with TOPGLOW and Renilla in the presence of GFP vector) was assigned the value of 100% and represents β-catenin activity in the absence of exogenous emerin. A standard t-test was performed for data analysis.
Immunoprecipitation
Mouse IgG Dynabeads (Dynal Biotech) were coupled to β-catenin or anti-GFP antibody by incubation for 12 h at 4°C in the presence of 1% BSA. Cell cultures were preincubated with hypotonic solution containing 10 mM KCl, 10 mM Tris–HCl, pH 7.4, 1.5 mM MgCl2, 0.1% Triton X-100 and protease inhibitors cocktail (Sigma) before homogenisation with buffer containing 0.5 M NaCl. Soluble fractions were dialysed to PBS/0.1% Triton X-100 for 12 h at 4°C and processed for IP according to published protocols (Markiewicz et al, 2002b).
Leptomycin B treatment of HEK293 cells
HEK293 cells were seeded onto glass coverslips and co-transfected with GFP, GFP-emerin wt, GFP-emerinΔ and β-catenin. At 48 h post-transfection, leptomycin B (50 ng/ml medium) was added to the cultures and cells left for additional 2.5 h. Cells were fixed in 4% paraformaldehyde in PBS and processed for indirect immunofluorescence microscopy.
Solid-phase overlay assay
cDNAs for β-catenin and HLA cDNA in pCDNA3 were transcribed in vitro by using T7 polymerase (Promega) and RNAs were translated in vitro by using rabbit reticulocyte lysate (Promega) and [35S]methionine (Amersham), according to the manufacturer's instructions. His-tagged purified recombinant emerin fragments, E1–220 and E1–176 were separated by SDS–PAGE. Gels were stained with Coomassie Brilliant Blue or transblotted onto nitrocellulose (Bio-Rad). Nitrocellulose membranes were blocked in overlay buffer (10 mM Tris, pH 7,4, 150 mM NaCl, 1 mM EDTA, 0.1% Tween 20) containing 5% dried milk for 1 h and probed with in vitro translated 35S-labeled proteins, for 14 h at 4°C. After extensive washing, the nitrocellulose filter was air-dried and bound proteins were detected by autoradiography.
Construction of GFP-emerinΔ
Site-specific deletion of nucleotides 504–558 in GFP-emerin cDNA was created using QuickChange Site-Directed Mutagenesis (Stratagene) according to the manufacturer's protocol. The oligonucleotide primers used were 5′-CAG AGC ATC ACG CAC TAC TCC ACC TCT TTT ATG TCC-3′ (forward) and 5′-GGA CAT AAA AGA GGT GGA GTA GTG CGT GAT GCT CTG-3′ (reverse). Obtained clones containing GFP-emerin with deleted amino-acid sequence RPVSASRSSLDLSYYPTSS were verified by DNA sequencing.
RT–PCR
mRNA was extracted from control and emerin null fibroblasts using TRIZOL reagent (Invitrogen) and reverse transcribed using Reverse Transcription System (Promega). Resulting cDNA products were used as templates for specific PCR amplifications using PCR Master Mix (Promega). Primers used were as follows:
5′-TCCACCTCCAGCTTGTACCTG-3′ and
5′-CGCCTCTTGACATTCTCCTCG-3′ for c-myc and
5′-GGCACCACACCTTCTACAATGAGC-3′ and
5′-CGTCATACTCCTGCTTGCTGATCCAC-3′ for β-actin.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Legends to Supplementary Figures
Acknowledgments
We thank our colleagues Dr Colin Jahoda (Durham, UK) and Dr Yigal Pinto (Maastricht, The Netherlands) for helpful comments and Dr Stephen Byers for the gift of β-catenin constructs. We also wish to thank Dr Naomi Willis (Durham) for assistance with RT–PCR. This work was supported by a grant from the Muscular Dystrophy Campaign to CJH and JAE, a Wellcome Trust Study abroad fellowship award to CJH, a Royal Society Travelling Fellowship to CJH and a grant from the Polish State Committee of Research (No. 4P05B 061 18) to IHP.
References
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W (1996) Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382: 638–642 [DOI] [PubMed] [Google Scholar]
- Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. (1994) Identification of a novel X-linked gene responsible for Emery–Dreifuss muscular dystrophy. Nat Genet 8: 323–327 [DOI] [PubMed] [Google Scholar]
- Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery–Dreifuss muscular dystrophy. Nat Genet 21: 285–288 [DOI] [PubMed] [Google Scholar]
- Brill LM, Salomon AR, Ficarro SB, Mukherjic M, Stettler-Gill M, Peters EC (2004) Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilised metal affinity chromatography and tandem mass spectrometry. Anal Chem 76: 2763–2772 [DOI] [PubMed] [Google Scholar]
- Cartegni L, di Barletta MR, Barresi R, Squarzoni S, Sabatelli P, Maraldi N, Mora M, Di Blasi C, Cornelio F, Merlini L, Villa A, Cobianchi F, Toniolo D (1997) Heart-specific localization of emerin: new insights into Emery–Dreifuss muscular dystrophy. Hum Mol Genet 13: 2257–2264 [DOI] [PubMed] [Google Scholar]
- Clements L, Manilal S, Love DR, Morris GE (2000) Direct interaction between emerin and lamin A. Biochem Biophys Res Commun 267: 709–714 [DOI] [PubMed] [Google Scholar]
- Cong F, Varmus H (2004) Nuclear-cytoplasmic shuttling of axin regulates subcellular localization of beta-catenin. Proc Natl Acad Sci USA 101: 2882–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg LM, Eisenberg CA (2006) Wnt signal transduction and the formation of the myocardium. Dev Biol 293: 305–315 [DOI] [PubMed] [Google Scholar]
- Elftheriou A, Yoshida M, Henderson BR (2001) Nuclear export of human β-catenin can occur independent of CRM1 and the adenamatous polyposis coli tumour suppressor. J Biol Chem 276: 25883–25888 [DOI] [PubMed] [Google Scholar]
- Fabbro M, Henderson BR (2003) Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp Cell Res 282: 59–69 [DOI] [PubMed] [Google Scholar]
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 341: 1715–1724 [DOI] [PubMed] [Google Scholar]
- Gareiss M, Eberhardt K, Kruger E, Kandert S, Bohm C, Zentgraf H, Muller CR, Dabauvalle MC (2005) Emerin expression in early development of Xenopus laevis. Eur J Cell Biol 84: 295–309 [DOI] [PubMed] [Google Scholar]
- Gruenbaum Y, Lee KK, Liu J, Cohen M, Wilson KL (2002) The expression, lamin-dependent localization and RNAi depletion phenotype for emerin in C. elegans. J Cell Sci 115: 923–929 [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Holaska JM, Yamane M, Koujin T, Hashiguchi N, Mori C, Wilson KL, Hiraoka Y (2004) Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery–Dreifuss muscular dystrophy. Eur J Biochem 271: 1035–1045 [DOI] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114: 4557–4565 [DOI] [PubMed] [Google Scholar]
- Henderson BR (2000) Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol 2: 653–660 [DOI] [PubMed] [Google Scholar]
- Hirano Y, Segawa M, Ouchi FS, Yamakawa Y, Furukawa K, Takeyasu K, Horigome T (2005) Dissociation of emerin from barrier-to-autointegration factor is regulated through mitotic phosphorylation of emerin in a Xenopus egg cell free system. J Biol Chem 280: 39925–39933 [DOI] [PubMed] [Google Scholar]
- Holaska JM, Kowalski AK, Wilson KL (2004) Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol 2: E231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska JM, Lee KK, Kowalski AK, Wilson KL (2003) Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J Biol Chem 278: 6969–6975 [DOI] [PubMed] [Google Scholar]
- Hutchison CJ (2002) Lamins: building blocks or regulators of gene expression? Nat Rev Mol Cell Biol 3: 848–858 [DOI] [PubMed] [Google Scholar]
- Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK (2004) A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci USA 101: 9677–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguri C, Gilquin B, Wolff N, Romi-Lebrun R, Courchay K, Callebaut I, Worman HJ, Zinn-Justin S (2001) Structural characterisation of the LEM motif common to three human inner nuclear membrane proteins. Structure 9: 503–511 [DOI] [PubMed] [Google Scholar]
- Lattanzi G, Cenni V, Marmiroli S, Capanni C, Mattioli E, Merlini L, Squarzoni S, Maraldi NM (2003) Association of emerin with nuclear and cytoplasmic actin is regulated in differentiating myoblasts. Biochem Biophys Res Commun 303: 764–770 [DOI] [PubMed] [Google Scholar]
- Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL (2001) Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci 114: 4567–4573 [DOI] [PubMed] [Google Scholar]
- Libotte T, Zaim H, Abraham S, Padmakumar VC, Schneider M, Lu W, Munck M, Hutchison C, Wehnert M, Fahrenkrog B, Sauder U, Aebi U, Noegel AA, Karakesisoglou I (2005) Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 16: 3411–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee KK, Segura-Totten M, Neufeld E, Wilson KL, Gruenbaum Y (2003) MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. Proc Natl Acad Sci USA 100: 4598–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manilal S, Nguyen TM, Sewry CA, Morris GE (1996) The Emery–Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet 5: 801–808 [DOI] [PubMed] [Google Scholar]
- Margalit A, Segura-Totten M, Gruenbaum Y, Wilson KL (2005) Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc Natl Acad Sci USA 102: 3290–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewicz E, Venables R, Mauricio Alvarez R, Quinlan R, Dorobek M, Hausmanowa-Petrucewicz I, Hutchison C (2002a) Increased solubility of lamins and redistribution of lamin C in X-linked Emery–Dreifuss muscular dystrophy fibroblasts. J Struct Biol 140: 241–253 [DOI] [PubMed] [Google Scholar]
- Markiewicz E, Dechat T, Foisner R, Quinlan RA, Hutchison CJ (2002b) Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell 13: 4401–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, Mitchell S, Nader G, Bakay M, Rottman JN, Hoffman EP, Stewart CL (2006) Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet 15: 637–651 [DOI] [PubMed] [Google Scholar]
- Mislow J, Holaska J, Kim M, Lee K, Segura-Totten M, Wilson K, McNally E. (2002) Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett 525: 135. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van der Wetering M, Oosterwegel M, Petersib-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H (1996) Xtcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86: 391–399 [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of β-catenin-Tcf signalling in colon cancer by mutations in β-catenin or APC. Science: 275: 1787–1790 [DOI] [PubMed] [Google Scholar]
- Muchir A, van Engelen BG, Lammens M, Mislow JM, McNally E, Schwartz K, Bonne G (2003) Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp Cell Res 291: 352–362 [DOI] [PubMed] [Google Scholar]
- Neufeld KL, Zhang F, Cullen BR, White RL (2000) APC-mediated down-regulation of beta catenin activity involves nuclear sequestration and nuclear export. EMBO Rep 1: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada S, Ohmori SY, Taira M (2003) XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development 130: 1783–1794 [DOI] [PubMed] [Google Scholar]
- Pai LM, Kirkpatrick C, Blanton J, Oda H, Takeichi M, Peifer M (1996) Drosophila a-catenin and E-cadherin bind to distinct regions of Drosophila armidillo. J Biol Chem 271: 32411–32420 [DOI] [PubMed] [Google Scholar]
- Padmakumar VC, Abraham S, Braune S, Noegel AA, Tunggal B, Karakesisoglou I, Korenbaum E (2004) Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp Cell Res 295: 330–339 [DOI] [PubMed] [Google Scholar]
- Pan D, Estevez-Salmeron LD, Stroschein SL, Zhu X, He J, Zhou S, Luo K (2005) The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repres. J Biol Chem 280: 15992–16001 [DOI] [PubMed] [Google Scholar]
- Raffaele Di Barletta M, Ricci E, Galluzzi G, Tonali P, Mora M, Morandi L, Romorini A, Voit T, Orstavik KH, Merlini L, Trevisan C, Biancalana V, Hausmanowa-Petrusewicz I, Bione S, Ricotti R, Schwartz K, Bonne G, Toniolo D (2000) Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery–Dreifuss muscular dystrophy. Am J Hum Genet 66: 1407–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423: 409–414 [DOI] [PubMed] [Google Scholar]
- Rosin-Arbesfeld R, Cliffe A, Brabletz T, Bienz M. (2003) Nuclear export of the APC tumour suppressor controls beta-catenin function in transcription. EMBO J 22: 1101–11013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin-Arbesfeld R, Townsley F, Bienz M (2000) The APC tumour suppressor has a nuclear export function. Nature 406: 10009–10012 [DOI] [PubMed] [Google Scholar]
- Segura-Totten M, Wilson KL (2004) BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol 14: 261–266 [DOI] [PubMed] [Google Scholar]
- Shimi T, Koujin T, Segura-Totten M, Wilson KL, Haraguchi T, Hiraoka Y. (2004) Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J Struct Biol 147: 31–41 [DOI] [PubMed] [Google Scholar]
- Stockinger A, Eger A, Wolf J, Beug H, Foisner R (2001) E-cadherin regulates cell growth by modulating proliferation- dependent beta-catenin transcriptional activity. J Cell Biol 154: 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147: 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara T, Tsujino S, Arahata K (2002) CDNA microarray analysis of gene expression in fibroblasts of patients with X-linked Emery–Dreifuss muscular dystrophy. Muscle Nerve 25: 898–901 [DOI] [PubMed] [Google Scholar]
- Van Berlo JH, Voncken JW, Kubben N, Broers JLV, Duisters R, van Leeuwen RE, Crijns HJ, Ramaekers FCS, Hutchison CJ, Pinto YM (2005) A-type lamins are essential for TGF-{beta}1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet 14: 2839–2849 [DOI] [PubMed] [Google Scholar]
- van der Kooi AJ, Ledderhof TM, de Voogt WG, Res CJ, Bouwsma G, Troost D, Busch HF, Becker AE, de Visser M (1996) A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann Neurol 39: 636–642 [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H (2002) The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111: 241–250 [DOI] [PubMed] [Google Scholar]
- Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JLV, Ramaekers FCS, Wehnert M, Morris GE, Whitfield WGF, Hutchison CJ (2001) Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci 114: 2577–2590 [DOI] [PubMed] [Google Scholar]
- Wiechens N, Fagotto F (2000) CRM-1 and Ran-independent nuclear export of β-catenin. Curr Biol 11: 18–27 [DOI] [PubMed] [Google Scholar]
- Wiechens N, Heinle K, Englmeier L, Schohl A, Fagotto F (2004) Nucleo-cytoplasmic shuttling of Axin, a negative regulator of the Wnt-beta-catenin pathway. J Biol Chem 279: 5263–5267 [DOI] [PubMed] [Google Scholar]
- Wolff N, Gilquin B, Courchay K, Callebaut I, Worman HJ, Zinn-Justin S (2001) Structural analysis of emerin, an inner nuclear membrane protein mutated in X-linked Emery–Dreifuss muscular dystrophy. FEBS Lett 501: 171–176 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ragnauth CD, Skepper JN, Worth NF, Warren DT, Roberts RG, Weissberg PL, Ellis JA, Shanahan CM (2005) Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci 118: 673–687 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Legends to Supplementary Figures