

Abstract
Retroviral insertional mutagenesis preferentially identifies oncogenes rather than tumor suppressor (TS) genes, presumably because a single retroviral-induced mutation is sufficient to activate an oncogene and initiate a tumor, whereas two mutations are needed to inactivate a TS gene. Here we show that TS genes can be identified by insertional mutagenesis when the screens are performed in Blm-deficient backgrounds. Blm-deficient mice, like Bloom syndrome patients, have increased frequencies of mitotic recombination owing to a mutation in the RecQ protein-like-3 helicase gene. This increased mitotic recombination increases the likelihood that an insertional mutation in one allele of a TS gene will become homozygoused by non-sister chromatid exchange and the homozygosity of the insertion provides a marker for identifying the TS gene. We also show that known as well as novel TS genes can be identified by insertional mutagenesis in Blm-deficient mice and identify two JmjC family proteins that contribute to genome stability in species as evolutionarily diverse as mammals and Caenorhabditis elegans.
Keywords: Bloom syndrome, insertional mutagenesis, lymphoma, retrovirus, tumor suppressor genes
Introduction
To establish a genetic background in which homozygous mutations can be easily recovered, Luo et al (2000) generated ES cells and mice that were homozygous for a hypomorphic mutation (McDaniel et al, 2003) in the Bloom syndrome (BLM) RecQ protein-like-3 DNA helicase gene, Blmtm3Brd (subsequently referred to as Blmm3) (Ellis et al, 1995). This was important as two previous studies showed that Blm-null mutations are embryonic lethal (Chester et al, 1998; Goss et al, 2002). BLM is a recessive human genetic disorder associated with genomic instability (German, 1993). Helicases unwind double-stranded DNA, a process required for various aspects of DNA metabolism, including transcription, DNA repair, and replication. BLM predisposes patients to a wide variety of malignancies, which implies that the BLM helicase is required for maintaining genomic stability in many cell types. BLM genomic instability is unique in that it is characterized by increased mitotic recombination (German, 1993) and non-sister chromatid exchange (Luo et al, 2000). Mitotic recombination between non-sister chromatids that are heterozygous for a mutation of tumor suppressor (TS) gene can produce daughter cells that have lost the wild-type allele and are now homozygous for the mutant allele (Adams and Bradley, 2002). This increased ability to homozygous TS mutations is the reason that BLM patients and Blmm3-deficient mice are thought to develop a higher frequency of cancer.
The rate of loss of heterozygosity (LOH) in Blmm3-deficient ES cells is 4.2 × 10−4 events per locus per cell per generation (Luo et al, 2000), which is elevated 18-fold compared to wild-type ES cells. Guo et al (2004) have exploited this increased LOH to generate a genome-wide library of homozygous mutant ES cells from heterozygous mutant cells produced by infection of Blmm3-deficient ES cells with a gene trap retrovirus. They showed that they could use this library to identify bi-allelic gene trap mutations in novel mismatch repair genes. Likewise, Yusa et al (2004) treated Blmtm1Khor-deficient ES cells with N-ethyl-N-nitrosourea (ENU) and identified bi-allelic chemically induced mutations in genes required for glycosylphosphatidylinositol (GPI)-anchor biosynthesis. These studies showed that it is possible to perform phenotype-based recessive screens in Blm-deficient ES cells.
The Blmm3 allele has also been shown to increase the number of intestinal polyps in Apc heterozygous mutant mice (Luo et al, 2000; Goss et al, 2002). As Blmm3-deficient mice have elevated rates of mitotic recombination, this suggested that increased polyp numbers might result from increased non-sister chromatid exchange and subsequent loss of the wild-type Apc allele. Microsatellite mapping of polyp DNA confirmed this hypothesis and showed that Apc TS mutations are homozygoused at increased frequencies in Blmm3-deficient mice.
Here, we have asked whether retroviral insertional mutagenesis in Blmm3-deficient mice could be used as a tool for identifying TS genes. We reasoned that if a retrovirus integrated into the coding region of one allele of a TS gene, and interfered with its expression such as by splicing into viral sequences followed by transcription termination in the viral long terminal repeat (LTR), this integration might be homozygoused at an increased frequency in Blmm3-deficient mice owing to the increased frequency of non-sister chromatid exchange and we could use this homozygosity as a marker for identifying TS genes. Here we show that viral-induced TS mutations do become homozygoused in Blmm3-deficient mice and this homozygosity can be used as a marker for identifying TS genes.
Results
Reduced tumor latency in AKXD-Blmm3/m3 mice
To determine whether insertional mutagenesis in Blm-deficient mice can be used as a tool for identifying TS genes, we crossed the Blmm3 mutation (Luo et al, 2000) onto the AKXD-10, -14, and -27 inbred strain backgrounds by three successive rounds of backcross mating (Figure 1A). These three strains were derived from an F2 cross of AKR/J and DBA/2J mice (Gilbert et al, 1993). Each strain inherited ∼50% of its genome from AKR/J and ∼50% from DBA/2J mice, although the relative contribution to each strain differs due to random assortment during the inbreeding process. All three strains develop a high frequency of retrovirally induced B-cell lymphomas, which is caused by the expression of endogenous murine leukemia viruses that are transmitted in these strains (Gilbert et al, 1993). N3 backcross Blm+/m3 mice were then intercrossed and the Blm+/+ and Blmm3/m3 progeny aged for tumors. While these mice were aging, the Blmm3 mutation was backcrossed three more times to each of the three AKXD strains and the N6 Blm+/+ and Blmm3/m3 progeny also aged for tumors (Figure 1A). We then aged and monitored 181 N3 and 72 N6 mice for the development of tumors. Blmm3/m3 animals died significantly earlier than Blm+/+ animals when all of the data for the N3 and N6 generations were combined (Figure 1B). AKXD-Blmm3/m3 mice had an average lifespan of 400.9±10.7 days, whereas AKXD-Blm+/+ mice had an average lifespan of 448.7±10.9 days. Similar results were observed when the individual strains were analyzed separately, with the exception of the AKXD-27-Blm+/+ and AKXD-27- Blmm3/m3 mice, where there was less difference in survival (data not shown).
Figure 1.

Generation and aging of AKXD-Blmm3 mice. (A) Mating scheme for generating AKXD-Blm+/+ and AKXD-Blmm3/m3 mice. Mice carrying the Blmtm3Brd mutation were backcrossed three or six times to the AKXD-10, -14, or -27 inbred mouse strains. N3 or N6 backcrossed Blm+/m3 mice were then intercrossed and the Blm+/+ and Blmm3/m3 progeny aged for tumors. (B) AKXD-Blmm3/m3 (n=137) mice die earlier of retrovirus-induced lymphoma than AKXD-Blm+/+ (n=116) mice. AKXD-Blmm3/m3 mice and AKXD-Blm+/+ mice were monitored for lymphoma development. The age in days at the time of death is plotted for each Blm+/+ and Blmm3/m3 mouse. P<0.01 by Log-Rank and Wilcoxon test.
Common integration sites in AKXD lymphomas
Next, 1023 retroviral integration sites from AKXD-Blmm3/m3 lymphomas, and 515 retroviral integration sites from AKXD-Blm+/+ lymphomas, were cloned and sequenced following inverse polymerase chain reaction (PCR) (Li et al, 1999). All sequencing data are deposited in the Retroviral Tagged Cancer Gene Database (RTCGD) (http://rtcgd.ncifcrf.gov) and can be viewed on-line. Most integration sites could be positioned in the mouse genome by BLAST searches. Interestingly, nearly 44% of the integrations (674) were located at common integration sites (CISs) (Supplementary Table 1). CISs are regions in the genome that are targets of retroviral integration in two or more tumors and are thus more likely to contain a cancer gene (Suzuki et al, 2002). In total, 218 CISs were identified in the screen (Supplementary Table 1). A total of 68 CISs have been identified in previous studies, while 150 are novel. With the addition of these novel CISs, more than 430 CISs have been identified in mouse hematopoietic tumors using insertional mutagenesis (http://rtcgd.ncifcrf.gov). These are more candidate cancer genes than the 353 genes found mutated in human cancer (http://www.sanger.ac.uk/genetics/CGP/Census).
In all, 60 CIS genes are orthologs or homologs of genes mutated in human cancer, whereas the rest have not yet been examined for a role in cancer. These are the most interesting since these are the genes that represent potential novel human cancer genes. The majority of genes found in AKXD lymphomas fall into classes commonly associated with cancer (Supplementary Table 1). The largest class is transcription factors or proteins that regulate transcription. Other classes include chromatin remodeling proteins, kinases and phosphatases, cytokines and their receptors, Ras proteins and their regulators, signaling molecules, apoptosis-related proteins, actin-binding proteins, splicing factors and cell cycle regulatory proteins. Most of the integrations at CISs in Blm+/+ and Blmm3/m3 lymphomas are located upstream or downstream of the genes they are postulated to deregulate, suggesting that they function as oncogenes. Therefore, even in the Blmm3-deficient background, an appreciable number of oncogenes were still identified by insertional mutagenesis. This was not surprising as the virus used in the screen contains strong enhancers and promoters, which can drive the overexpression of oncogenes even in Blmm3-deficient tumors.
Candidate TS genes
To determine whether integrations in the coding region of TS genes in Blmm3-deficient tumors are homozygoused and can be used as a marker to identify TS genes, we first looked for CIS genes that were specific to Blmm3/m3 lymphomas and whose coding regions were consistently disrupted by viral integration. In total, 17 CISs genes met these criteria (Table I). For six of these genes, there is published evidence that they are TS genes. Rbl1 and Rbl2 are homologues of Rb1, a well-known TS gene. Mice lacking Rbl1 exhibit a myeloproliferative disorder characterized by ectopic myeloid hyperplasia in the spleen and liver (LeCouter et al, 1998). Likewise, Rbl2 is located in a region of homology with human 16q12.2 where deletions have been found in several human neoplasms, including breast, ovarian, hepatic and prostate cancers (Yeung et al, 1993). In addition, mice carrying loss-of-function mutations in Rb and Rbl1 or Rb and Rbl2 are highly cancer prone, indicating that in a variety of tissues, Rbl1 and Rbl2 suppress tumor development induced by loss of Rb and are functionally redundant with Rb (Dannenberg et al, 2004). Cdkn2c is a cyclin-dependent kinase inhibitor that controls cell cycle G1 progression. Cdkn2c knockout mice develop spontaneous pituitary tumors and lymphomas late in life and treatment of these mice with a chemical carcinogen induces tumors at an accelerated rate (Bai et al, 2003). Fanconi anemia, complementation group G (FANCG) mutations induce diverse clinical symptoms including developmental abnormalities, bone marrow failure, and early occurrence of malignancies. Primary splenocytes, bone marrow progenitor cells, and embryonic fibroblasts from Fancg knockout mice also exhibit increased chromosomal breakage and sensitivity to mitomycin C (Yang et al, 2001). Tnfrsf6 plays a central role in the regulation of programmed cell death. Tnfrsf6 mutant mice develop lymphadenopathy and a lupus-like disease. In addition, TNFRSF6 mutations have been observed in nasal natural killer/T-cell lymphomas and burn scar-related squamous cell carcinomas (Lee et al, 2000). E2f3 functions downstream of Rb and plays a crucial role in cell cycle control. Disruption of E2f3 promotes the development of metastatic medullary thyroid carcinomas in Rb1+/− mutant mice (Ziebold et al, 2003).
Table 1.
CISs encoding candidate TS genes
| Strain | Tumor | CIS gene | Protein classification | Chr | Location | LOH | Ex. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AKXD-14 | 4295 | Rbl1 | Cell cycle regulator | 2 | Intron 1 | Yes | Yes | ||||||||
| AKXD-10 | 11680 | Rbl1 | Cell cycle regulator | 2 | Intron 1 | Yes | Yes | ||||||||
| AKXD-27 | 8610 | Fancg | Fanconi anemia group G | 4 | Exon 4 | Yes | Yes | ||||||||
| AKXD-14 | 4301 | Fancg | Fanconi anemia group G | 4 | Intron 2 | Yes | Yes | ||||||||
| AKXD-27 | 8634 | Anp32b | Nuclear phosphoprotein | 4 | Intron 1 | No | ND | ||||||||
| AKXD-10 | 11976 | Anp32b | Nuclear phosphoprotein | 4 | Intron 1 | No | ND | ||||||||
| AKXD-10 | 13847 | Cdkn2c | Cdk4 inhibitor | 4 | Intron 1 | Yes | Yes | ||||||||
| AKXD-27 | 7221 | Cdkn2c | Cdk4 inhibitor | 4 | Intron 1 | Yes | Yes | ||||||||
| AKXD-27 | 8913 | Prdm16 | Transcription factor | 4 | Intron 2 | ND | ND | ||||||||
| AKXD-14 | 4313 | Prdm16 | Transcription factor | 4 | Intron 2 | No | ND | ||||||||
| AKXD-27 | 7107 | Prdm16 | Transcription factor | 4 | Intron 1 | ND | ND | ||||||||
| AKXD-27 | 7105 | Prdm16 | Transcription factor | 4 | Intron 1 | No | ND | ||||||||
| AKXD-27 | 6634 | Pou2f2 | Transcription factor | 7 | Intron 1 | ND | ND | ||||||||
| AKXD-27 | 6641 | Pou2f2 | Transcription factor | 7 | Intron 1 | Yes | ND | ||||||||
| AKXD-27 | 7105 | Pou2f2 | Transcription factor | 7 | Intron 1 | Yes | ND | ||||||||
| AKXD-10 | 11427 | Pou2f2 | Transcription factor | 7 | Exon 3 | Yes | ND | ||||||||
| AKXD-14 | 3046 | 1810054O13Rik | Undefined | 7 | Intron 1 | No | ND | ||||||||
| AKXD-10 | 14503 | 1810054O13Rik | Undefined | 7 | Intron 2 | No | ND | ||||||||
| AKXD-14 | 3197 | Il16 | Cytokine | 7 | Intron 15 | No | ND | ||||||||
| AKXD-10 | 11567 | Il16 | Cytokine | 7 | Intron 15 | No | ND | ||||||||
| AKXD-27 | 7338 | Rbl2 | Cell cycle regulation | 8 | Intron 4 | Yes | Yes | ||||||||
| AKXD-10 | 13739 | Rbl2 | Cell cycle regulation | 8 | Intron 5 | Yes | Yes | ||||||||
| AKXD-10 | 10092 | Coro2b | Actin-binding protein | 9 | Intron 3 | No | ND | ||||||||
| AKXD-10 | 12210 | Coro2b | Actin-binding protein | 9 | Intron 3 | No | ND | ||||||||
| AKXD-14 | 3162 | Gpx1 | Oxidoreductase | 9 | Intron 1 | No | ND | ||||||||
| AKXD-27 | 7244 | Gpx1 | Oxidoreductase | 9 | Exon 2 | No | ND | ||||||||
| AKXD-14 | 4149 | Prdm1 | Zinc-finger protein | 10 | Intron 4 | No | ND | ||||||||
| AKXD-10 | 11978 | Prdm1 | Zinc-finger protein | 10 | Intron 4 | No | ND | ||||||||
| AKXD-27 | 7088 | Prdm1 | Zinc finger protein | 10 | Intron 4 | No | ND | ||||||||
| AKXD-14 | 3134 | Prdm1 | Zinc-finger protein | 10 | Intron 3 | No | ND | ||||||||
| AKXD-10 | 14100 | C330016O10Rik | Protein targeting | 11 | Exon 4 | Yes | Yes | ||||||||
| AKXD-10 | 11978 | C330016O10Rik | Protein targeting | 11 | Intron 3 | Yes | Yes | ||||||||
| AKXD-14 | 3296 | E2f3 | Transcription factor | 13 | Intron 1 | Yes | Yes | ||||||||
| AKXD-10 | 14328 | E2f3 | Transcription factor | 13 | Exon 1 | Yes | Yes | ||||||||
| AKXD-27 | 8309 | 3110004L20Rik | Undefined | 13 | Intron 1 | No | ND | ||||||||
| AKXD-27 | 6648 | 3110004L20Rik | Undefined | 13 | Intron 1 | No | ND | ||||||||
| AKXD-10 | 14043 | Cldn10 | Tight junction protein | 14 | Intron 1 | No | ND | ||||||||
| AKXD-14 | 3280 | Cldn10 | Tight junction protein | 14 | Intron 1 | No | ND | ||||||||
| AKXD-10 | 12169 | Tnfrsf6 | Tnf receptor superfamily | 19 | Intron 1 | Yes | Yes | ||||||||
| AKXD-27 | 6634 | Tnfrsf6 | Tnf receptor superfamily | 19 | Intron 1 | Yes | Yes | ||||||||
| CIS loci listed in bold are new candidate cancer causing genes identified in the present study, whereas CIS listed in nonbold have been identified in previous insertional mutagenesis screens. Location column shows where each viral integration was located within the gene. LOH column shows whether LOH was detected by Southern analysis, and Ex column shows whether expression was analyzed by Northern blot. ND means not determined. 3110004L20Rik and Cldn10 Southerns did not show mutant rearranged bands, suggesting that these genes probably function in tumor progression rather than tumor initiation and it was thus not possible to determine whether these integrations underwent LOH. | CISs, common integration sites; LOH, loss of heterozygosity. | ||||||||||||||
As viral integration into the coding region of a CIS can induce the expression of truncated proteins that function as oncogenes, we expected that at least some of the genes listed in Table I function as oncogenes rather than TS genes. This was in fact the case as at least two of the genes listed in Table I are oncogenes. Prdm16 is a myeloid leukemia oncogene that is activated in t(1;3)(p36;q21)-positive leukemia cells (Mochizuki et al, 2000). This translocation induces the expression of a short form of PRDM16 that lacks the PR domain. The PR domain is common to a subset of zinc-finger proteins that function as negative regulators of tumorigenesis. It is also related to the SET domain found in genes that regulate chromatin structure, such as MLL. The lack of a PR domain could therefore inactive the chromatin-associated functions of a protein without affecting its other functions. Viral integrations in Prdm16 also promote the expression of a truncated protein that lacks the PR domain (Du et al, 2005). Prdm1 is related to Prdm16 and also contains a PR domain. Viral integrations in the coding regions of Prdm1 are therefore also likely to be selected because they too induce the expression of a protein that lacks the PR domain.
Viral integrations into TS genes undergo LOH
To determine whether any of the integrations listed in Table I have become homozygoused in tumors, control brain and tumor DNAs were analyzed by Southern analysis using probes specific for each locus. The wild-type and virally rearranged mutant alleles were then quantitated using a luminescent image analyzer. If duplication of the viral allele did not occur following viral integration, the wild-type allele should be equal or more intense than the mutant allele as the lymphomas are always contaminated with normal tissue. If, however, duplication of the viral allele occurred, the wild-type allele, representing only contaminating normal tissue, will be less intense than the mutant allele in tumors where there is not much contaminating normal tissue. Representative Southern results for the six genes listed in Table I predicted to encode TS genes are shown in Supplementary Figure 1A and the quantitated results in Figure 2A. In all cases, the mutant allele was more intense than the wild-type allele consistent with duplication of the viral allele. In addition, integrations at two other genes listed in Table I, C330016O10Rik and Pou2f2, also showed evidence of LOH (Supplementary Figure 1A, Figure 2A). As expected, integrations at Prdm1 and Prdm16 did not become duplicated following viral integration (Supplementary Figure 1B, Figure 2B) nor did the integrations in any of the other genes listed in Table I (data not shown). C330016O10Rik is the mouse ortholog of human NEDD4-binding protein 3 (N4BP3). This FEZ1 domain-containing gene is related to the FEZ1/LZTS1 leucine zipper, putative tumor suppressor-1. Pou2f2 is a POU domain transcription factor that controls the expansion and/or maintenance of mature B cells (Schubart et al, 2001).
Figure 2.
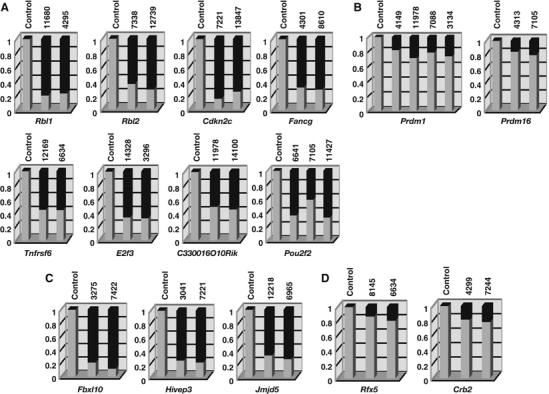
Quantitative analysis of retroviral integrations in tumor DNAs of AKXD-Blmm3 mice. Southern blots of genomic tumor DNAs were performed using flanking probes for each CIS gene. Tumor ID numbers are shown at the top of each column. A tumor that does not have a viral integration at that locus was used for the control. Gel images were acquired using a luminescent image analyzer and the band intensities quantified using the Image Gauge ver.4 software. The ratio of the intensity of the wild-type band (light gray) and mutant band (dark gray) is shown for each tumor sample. Among the CIS genes whose coding regions were consistently disrupted by viral integration, some of the genes (A) showed evidence of bi-allelic integration, but others (B) did not. (C) Bi-allelic integrations were also detected for three other CIS genes whose expressions were downregulated by viral integration, (D) but was not detected in CIS genes whose expressions may be upregulated by viral integration in AKXD-Blm+/+ lymphomas.
Another CIS gene, Fbxl10, also showed evidence of LOH (Supplementary Figure 2A, Figure 2C). Viral integrations in the coding sequence of Fbxl10 were identified in two Blm-deficient lymphomas, but in a third tumor, the viral integration was located in the first coding exon and did not disrupt the coding region of Fbxl10. Thus, Fbxl10 was not included in Table I. Fbxl10 is a member of the jumonji family of proteins (Balciunas and Ronne, 2000). A Caenorhabditis elegans homolog of Fbxl10, T26A5.5, has been identified in an RNA interference (RNAi) screen designed to detect mutator genes that contribute to genome stability in C. elegans somatic cells (Pothof et al, 2003). These genes protect the C. elegans genome against insertions and deletions and are therefore excellent candidates for TS genes in higher eukaryotes.
We also examined two CIS genes that were mutated exclusively in Blmm3-deficient lymphomas, Hivep3 and Jmjd5, in which the viral integrations were located in intronic sequences but upstream of the first coding exon (Supplementary Table 1). In both cases, the viral integrations appeared to be duplicated in tumors (Supplementary Figure 2A, Figure 2C). These viral integrations are therefore predicted to block rather than upregulate gene expression, which was confirmed by Northern analysis (see below). Hivep3 is a member of the human immunodeficiency virus type 1 enhancer-binding protein (HIVEP) family. HIVEPs are large zinc-finger proteins that regulate transcription through the kappa-B enhancer motif. Antisense or dominant-negative HIVEP3 enhances NF-kappa-B-dependent transactivation and JNK phosphorylation and inhibits apoptosis and cytokine gene expression (Oukka et al, 2002). Interestingly, Jmjd5 is also a JmjC family protein, and a C. elegans homolog of Jmjd5, C06H2.3, was identified in the same RNAi-based mutator screen used to identify T26A5.5 (Pothof et al, 2003). Therefore, Jmjd5 is also an excellent candidate for a TS gene.
As a control, we analyzed two CIS genes, Rfx5 and Crb2, that were identified in Blmm3-deficient tumors but whose coding regions were not disrupted by viral integration. In both cases, the wild-type allele was more intense than the virally induced mutant allele (Supplementary Figure 2B, Figure 2D), consistent with the expectation that these are oncogenes.
In another control, we checked to see whether any CIS genes were consistently disrupted by viral integration in AKXD-Blm+/+ tumors. Only one gene, Psma5, was identified (Supplementary Table 1). Psma5 encodes the alpha-type 5 subunit of the proteasome and is an unlikely candidate for a TS gene. Consistent with this, viral integrations at Psma5 did not show evidence of LOH (data not shown). In contrast, 17 CIS genes were disrupted by viral integration in AKXD-Blmm3/m3 tumors and eight of them showed evidence of LOH (Table I). The Blm mutation therefore appears to increase the frequency at which TS genes can be identified by insertional mutagenesis and can be used as a marker to identify TS genes.
Reduced TS gene expression in tumors
A total of 11 CIS genes including Fbxl10, Hivep3, Jmjd5 and the eight CIS genes listed in Table I that showed LOH met our criteria for a TS gene. To confirm their expression was reduced in lymphomas as expected for a TS gene, we compared their expression in tumors with viral integrations at the CIS to similar tumors without viral integrations at the CIS by Northern analysis. Representative Northern results for Rbl1, Rbl2 and Cdkn2c are shown in Figure 3A and quantitated in Figure 3B. Expression results for the other TS genes (with the exception of Pou2f2 which could not be detected by Northern analysis) are shown in Supplementary Figure 3A and quantitated in Supplementary Figure 3B. In all cases examined, expression was reduced in tumors by 80% or more compared to tumors without integrations at the CIS. These results further confirm that these are TS genes.
Figure 3.
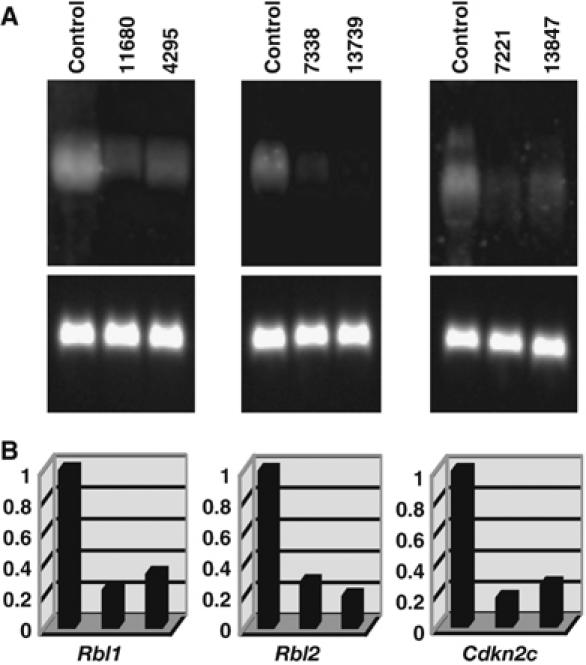
Expression analysis of candidate TS genes in AKXD-Blmm3/m3 lymphomas. (A) Northern blots. Total RNAs from the indicated tumors were fractionated on 1.0% agarose–formaldehyde gels and transferred onto nylon membranes. Tumor ID numbers are shown at the top of each gel. A similar type of tumor that does not have a viral integration at that locus was used as a control. Probes for each candidate TS gene were labeled with digoxigenin and used for hybridization. The blot was treated with anti-DIG antibody conjugated with alkaline phosphatase and then developed with CDP-Star reagents. The same blot was stripped and rehybridized with a glyceraldehyde phosphate dehydrogenase (GAPDH) probe (lower panel) to control for RNA loading. (B) Quantitative analysis of the Northern blots. Gel images were acquired using a luminescent image analyzer and the intensities of the bands quantified. Arbitrary units were shown on vertical axis regarding the expression level of control cells as 1.
Fbxl10 and Jmjd5 are mutator genes
Based on the C. elegans studies, we decided to determine whether Fbxl10 and Jmjd5 are mutator genes. To do this, we established an RNAi-based system for suppressing their expression in eukaryotic cells and then measured the effect of this suppression on genome stability. Six retroviral vectors expressing small hairpin RNAs (shRNAs) were constructed for each gene and introduced into NIH 3T3 cells. Infected cells were then selected for their puromycin resistance and the expression levels of each gene quantitated by Northern analysis (Supplementary Figure 4). Two shRNA-expressing constructs for each gene were used for suppression studies so that off-target effects of the shRNAs could be avoided.
Next, we developed an assay that could be used to determine whether cells that expressed these shRNAs exhibited defects in genome stability. For this a slippage vector was constructed (Figure 4A). This vector contains 17 CA-dinucleotide repeats cloned just downstream from the initiation codon of the enhanced green fluorescent protein (eGFP) gene, which puts the eGFP-coding sequence out of frame. Instability resulting in gains of one or loss of two CA repeats, for example, will restore the reading frame and activate eGFP (Figure 4A). The slippage vector was then introduced into NIH3T3 cells and stable cell lines were established. The reporter cells were then infected with shRNA-expressing retroviruses and stably infected cells selected with puromycin. Single cell clones were then expanded for 2 weeks and eGFP-positive cells counted using a fluorescence-activated cell sorter (FACS). Luria–Delbruck fluctuation analyses (Luria and Delbruck, 1943) were then used to calculate the mutation rate. The mutation rate for control cells expressing an shRNA for the luciferase gene was 7 × 10−7 events per cell per generation (Figure 4B, column 1). Empty vector-infected cells also showed a similar mutation rate (data not shown) as did cells infected with retroviruses that expressed shRNAs that did not inhibit Jmjd5 and Fbxl10 expression (Figure 4B, columns 2 and 3, respectively). In contrast, infection with two retroviruses that reduced Jmjd5 expression 14 and 26% produced mutation rates that were five and three times higher than control cells, respectively (Figure 4B, columns 4 and 5). Similarly, infection with two retroviruses that reduced Fbxl10 expression 19 and 40% produced mutation rates that were about three times higher than control cells (Figure 4B, columns 6 and 7). As a positive control, we examined the mutation rate of cells expressing shRNAs for Msh6 and Msh2, two well-known mismatch repair genes. Theses cells showed seven and 17 times higher mutation rates than control cells, respectively (data not shown). Thus, we conclude that suppression of Jmjd5 and Fbxl10 confers a mutator phenotype to the cells.
Figure 4.
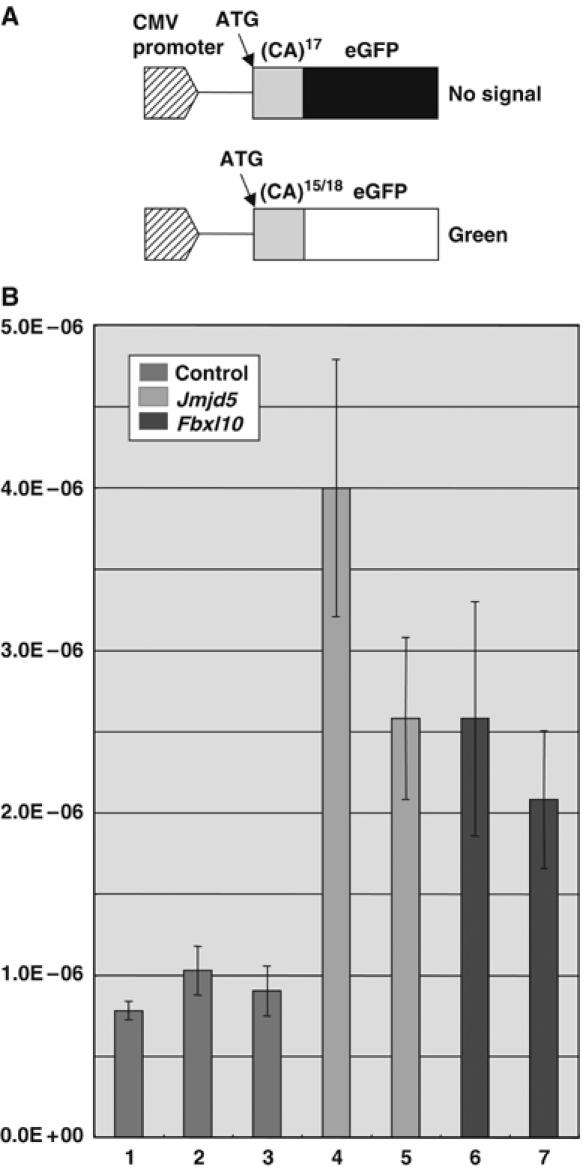
Jmjd5 and Fbxl10 genes contribute to genome stability. (A) A slippage vector used to quantitate the effect of Jmjd5 and Fbxl10 shRNA-based downregulation on genome stability. The eGFP slippage vector contains 17 CA-dinucleotide repeats cloned just downstream from the initiation codon of the eGFP gene, which puts the eGFP-coding sequence out of frame. Instability resulting in gains of one or loss of two CA repeats, for example, will restore the reading frame and activate eGFP expression. (B) Mutation rates of cells infected with shRNA-expressing retroviruses that downregulate Jmjd5 (column 4 by shRNA-A3 and column 5 by shRNA-A6, see Supplementary Figure 4) and Fbxl10 (column 6 by shRNA-B2 and column 7 by shRNA-B4) expression were determined by Luria–Delbruck fluctuation analyses. As controls, cells were infected with retroviruses expressing luciferase shRNA (column 1) or shRNAs that did not inhibit Jmjd5 and Fbxl10 expression (column 2 by shRNA-A4 and column 3 by shRNA-B1, respectively).
To determine whether Jmjd5 and Fbxl10 might function in DNA mismatch repair similar to Msh2, we examined the survival of cells with downregulated Jmjd5 and Fbxl10 expression after exposure to N-methyl-N′-nitro-N-niotrosoguanidine (MNNG). MNNG cytotoxicity is caused by its ability to introduce O6-methylguanine (O6-meG) into DNA (Karran and Bignami, 1996). It can direct misincorporation of thymine during replication, generating O6-meG:T mismatches. Recognition of these mismatches by the mismatch repair system leads to cell cycle arrest and apoptosis. Cells defective in mismatch repair are resistant to killing by MNNG. This effect has been called tolerance (Karran and Bignami, 1994). NIH3T3 cells infected with shRNA-expressing retroviruses were treated with a range of MNNG concentrations. After 10–14 days of culture, surviving colonies were counted. In control experiments, cells were infected with shRNA retroviruses that failed to downregulate Jmjd5 and Fbxl10 and as expected they had the same sensitivities as control cells. In contrast, cells expressing Msh2 shRNA that downregulated Msh2 expression 17% were more resistant to MNNG than control cells (Figure 5). Similarly, among cells expressing Jmjd5 shRNA that downregulated Jmjd5 expression, 14% were more resistant to MNNG than control cells, although the increase of tolerance was not as large as with Msh2 shRNA. These results suggest that Jmjd5, like Msh2, might also function in DNA mismatch repair. The same effect was not seen, however with Fbxl10 shRNA. Cells expressing Fbxl10 shRNA that downregulated Fbxl10 expression 19% did not increase MNNG tolerance. Although we cannot exclude the possibility that the level of suppression we were able to achieve was too low to have an effect in the MNNG assay, it is possible that Fbxl10 might function in DNA repair processes other than mismatch repair.
Figure 5.
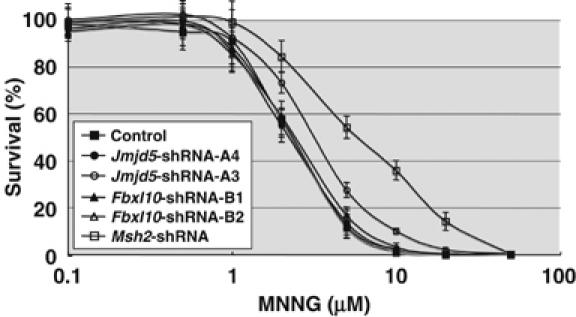
Cell tolerance to the DNA-methylating agent MNNG. NIH3T3 cells were infected with shRNA-expressing retroviruses that suppress Jmjd5 (open circle by shRNA-A3, see Supplementary Figure 4), Fbxl10 (open triangle by shRNA-B2), and Msh2 (open square). As controls, cells were infected with retroviruses expressing luciferase shRNA (closed square) or shRNAs that did not inhibit Jmjd5 and Fbxl10 expression (closed circle by shRNA-A4 and closed triangle by shRNA-B1, respectively). These cells were exposed to increasing amounts of MNNG for 1 h in the presence of O6-benzylguanine. After 10–14 days, surviving colonies were counted. The experiments were repeated 10 times and the average survival rates were shown.
Discussion
Here, we show that TS genes can be identified by insertional mutagenesis when the screens are performed in Blmm3-deficient mice. TS genes were identified as CIS genes whose expression is disrupted by viral integration and the viral integrations are homozygoused in Blmm3-deficient tumors. Homozygosity was determined by Southern analysis due to the fact that these tumors were produced on partially inbred strain backgrounds. While this did not allow us to prove unambiguously that homozygosity resulted from non-sister chromatid exchange, this is by far the most likely mechanism. Homozygosity was never detected in control lymphomas from wild-type (Blm+/+) animals and heterozygous Apc mutations have previously been shown to be homozygoused by non-sister chromatid exchange in intestinal tumors in Apc+/− Blmm3/m3 mice (Luo et al, 2000).
Of the 11 genes we identified that met our criteria for a TS gene, there is published evidence that six are TS genes. These include the two Rb1 homologues, Rbl1 (p107) and Rbl2 (p130); the cyclin-dependent kinase inhibitor Cdkn2c; the Fanconi anemia complementation group Fancg gene; the FAS antigen Tnfrsf6 gene and E2f3. Not surprisingly, none of these genes has been previously identified in insertional mutagenesis screens performed in wild-type mice. In addition, we identified five novel candidate TS genes. These include the mouse ortholog of human NEDD4-binding protein 3 C330016O10Rik; the POU domain transcription factor Pou2f2; the human immunodeficiency virus type 1 enhancer-binding protein Hivep3 and two JmjC domain-containing genes Fbxl10 and Jmjd5. Interestingly, these two JmjC family genes have C. elegans homologs that were recently identified in an RNAi-based mutator screen designed to identify genes that protect the C. elegans genome against insertions and deletions (Pothof et al, 2003). Using a similar RNAi-based mutator screen in mammalian cells, we confirmed that these genes also protect the mammalian genome against insertions and deletions. Both genes are therefore excellent candidates for TS genes in mammalian cells.
Surprisingly, two of the novel candidate TS genes we identified are mutated by viral integration in wild-type lymphomas. Two viral integrations in Pou2f2 have been identified in AKXD lymphomas wild type at Blm (Suzuki et al, 2002). Likewise, two viral integrations have been identified in lymphomas isolated from Cd3-deficient mice (Bijl et al, 2005). The Pou2f2 integrations identified in Blmm3-deficient lymphomas are located in intron 1 (three integrations) or in exon 1 (one integration) (Table I). In all eight cases, the integrations are predicted to disrupt the Pou2f2-coding region. Similarly, one integration in Fbxl10 intron 12 has been identified in a wild-type AKXD lymphoma and another integration in intron 12 was identified in a BXH-2 leukemia (Li et al, 1999; Suzuki et al, 2002). An integration in Fbxl10 intron 2 was similarly identified in a p27Kip1+/− lymphoma (Hwang et al, 2002). This is in addition to the three integrations we identified in Blmm3-deficient lymphomas. All these six integrations disrupt the Fbxl10-coding region (five integrations) or result in decreased Fbxl10 expression (one integration). While the reason for this is unclear, one possibility is that these genes function as haploinsufficient TS genes in wild-type lymphomas but in Blmm3-deficient tumors there is selective pressure to inactive the wild-type allele.
Our studies show that TS genes can be identified by insertional mutagenesis in Blmm3-deficient mice. However, even in Blmm3-deficient mice, there are still more oncogenes than TS genes identified by insertional mutagenesis (Supplementary Table 1). This was expected as the retrovirus that induces disease in AKXD mice has very strong enhancers and promoters, which can efficiently activate the expression of an oncogene when it lands nearby even in Blmm3-deficient lymphomas. In order to inactivate a TS gene, the virus must first integrate into the coding region and then be homozygoused by LOH or the other allele inactivated by another mechanism. While the rate of loss of heterozygosity in Blmm3-deficient ES cells is elevated 18-fold compared to wild-ype ES cells, the overall rate is still only 4.2 × 10−4 events per locus per cell generation. Given that the Blmm3 mutation is hypomorphic and chromosome instability and tumor predisposition is inversely correlated with BLM protein levels (McDaniel et al, 2003), it might be possible to increase the number of TS genes identified by insertional mutagenesis by performing these screens in Blm null mice. However, as the Blm null mutation is lethal (Chester et al, 1998; Goss et al, 2002), this would require the generation of a Blm conditional allele or the use of compound heterozygotes that carry a null allele on one chromosome and a Blmm3 allele on the other (McDaniel et al, 2003). One could of course also weaken or delete the promoters or enhancers carried by the virus so it would be less likely to activate an oncogene. However, this would also cripple the ability of the virus to replicate and induce tumors. Alternatively, one could use another insertional mutagen, such as the transposon Sleeping Beauty, which can be built to lack promoter/enhancer activity and function solely as a gene trap (Dupuy et al, 2005).
Several laboratories have recently shown that the JmjC domain we identified in two of our novel candidate TS genes is the signature motif for the long sought after histone demethylase (Tsukada et al, 2006). There are over 100 JmjC domain-containing proteins across the evolutionary phyla and essentially all of them are predicted to be enzymes (Clissold and Ponting, 2001). The JmjC domain-containing proteins FBXL10 and FBXL11 (renamed JHDM1B and JHDM1A, respectively) have been shown to demethylate di- and monomethylated H3-K36 to mono- or unmethylated products (Tsukada et al, 2006), the JMJD2 subfamily has been shown to demethylate trimethylated H3-K9 to di- and monomethylated products (Whetstine et al, 2006), whereas JHDM2A has been shown to demethylate mono- and dimethyl-H3-K9 to mono- and unmethylated products (Yamane et al, 2006). JHDM2A also exhibits hormone recruitment to androgen-receptor target genes resulting in H3-K9 demethylation and transcriptional activation. This links histone demethylation to hormone-dependent transcriptional activation (Yamane et al, 2006). Whetstine et al (2006) have shown that RNAi depletion of the C. elegans homologue of JMJD2A also increases general H3-K9 trimethylation and localizes H3-K36 trimethylation levels on meiotic chromosomes, triggering p53-dependent germline apoptosis. In the C. elegans germ line, DNA damage-induced apoptosis, but not physiological apoptosis, requires the functional p53 homolog, CEP-1. JMJD2A-induced apoptosis was shown to be abrogated in a cep-1 deletion mutant, suggesting a link between JMJD2A inactivation and DNA damage-induced apoptosis. As seen previously in cells undergoing DNA damage-induced apoptosis, increased numbers of RAD-51 foci were also observed in the nucleus at mid-pachytene. RAD-51 is a member of the highly conserved RecA protein family involved in strand invasion/exchange during double strain break (DSB) repair. Increased RAD-51 foci levels may therefore reflect an increase in the levels of DSBs or a delay in the progression of meiotic DSB repair.
While it remains to be determined exactly how loss-of-function mutations in Fbxl10 and Jmjd5 cause cancer, the findings of Whetstine suggest that imbalances in histone demethylation brought about by loss of Fbxl10 might affect DNA repair either through increased DSBs or delayed repair of DSBs. Our studies suggest that loss of Jmjd5 might also produce defects in mismatch repair. In addition, histone methylation has been implicated in multiple other biological processes including heterochromatin formation, X-inactivation, genomic imprinting, and silencing of homeotic genes (reviewed in Martin and Zhang, 2005). Loss of Fbxl10 and Jmjd5 are therefore likely to produce a myriad of effects in the cell and might not be limited to the repair of DNA mismatches and DSBs.
Among histone modifications, acetylation has been unambiguously associated with cancer (Santos-Rosa and Caldas, 2005). Histone acetyltransferases such as PCAF, P300, CBP, and MOZ are rearranged or mutated in several human cancers, acting as TS genes. Histone deacetylases (HDACs) are also frequently overexpressed in human cancer, suggesting that they function as oncogenes. However, a clear correlation between histone methylation and cancer has not been conclusively demonstrated, although some histone methyltransferase genes including MLL1, MLL2, SMYD3, and EZH2 have been shown to be amplified and/or overexpressed in human cancer. In this study, we have provided evidence that two JmjC domain-containing proteins, Fbxl10 and Jmjd5, are TS genes in hematopoietic cancer. Our findings suggest that both histone methylases and demethylases may play important roles in cancer. Identification of JmjC proteins as protein demethylases provides a unique opportunity to explore the relationship between this rapidly growing family of histone-modifying enzymes and cancer.
Materials and methods
Mice and tumor classification
AKXD-10, -14, and -27 mice develop a high incidence of B-cell lymphomas (Gilbert et al, 1993). These mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and aged in our colony at the National Cancer Institute, Frederick. Mice carrying the Blmtm3Brd mutation (Luo et al, 2000) were kindly provided by Alan Bradley (The Wellcome Trust Sanger Institute, Hinxton, Cambridge, UK). The Blmtm3Brd mutation was crossed onto the three AKXD strain backgrounds as described in the Results. The mice were then aged and observed for the onset of lymphomas. Moribund mice were examined by histopathological analysis and the lymphomas immunostained with CD45R and CD3 antibodies, which are specific for B and T cells, respectively.
Inverse PCR cloning and DNA sequencing
Inverse PCR was performed as described with slight modifications (Suzuki et al, 2002). Briefly, 5 μg tumor DNA was digested to completion with SacII, BamHI, SacI, and XbaI (Takara, Japan). Digested-DNA was then self-circularized by dilution and ligation using T4 DNA ligase (2450 U; Takara, Japan) in a total volume of 600 μl at 16°C for 16 h. Circular DNA was then precipitated with ethanol and dissolved in 40 μl TE buffer. In total, 2 μl was used in the primary PCR in a 50 μl PCR reaction volume containing 20 nmol each dNTP, 10 pmol each forward and reverse primer, 1 × buffer 2 and 2.5 U enzyme mix using the Expand Long Template PCR System (Roche, USA). We used a GeneAmp PCR System 9700 (Applied Biosystems, USA) programmed at 94°C for 2 min, followed by 30 cycles of 94°C for 20 s, 60°C for 40 s, 68°C for 12 min and a final extension step at 68°C for 7 min. The primary PCR product was then quantitated following electrophoresis on 1% agarose genes and 0.1–1.0 μl of the primary PCR product used as the template in the secondary PCR reaction. The secondary PCR was carried out using the same conditions as the primary PCR, except that secondary amplification primers were used. We separated the secondary PCR product on a 1% agarose gel, purified it using the QIAquick Gel Extraction Kit (Qiagen, USA) and directly cloned it using a Perfectly Blunt Cloning Kit (Novagen, Germany). The primer sequences for inverse PCR have been described (Li et al, 1999). DNA sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) on an ABI PRISM 3100-Avant Genetic Analyzer (Applied Biosystems, USA) using SP6 and T7 sequencing primers.
Sequence comparisons
We compared the retroviral integration site (RIS) sequences against the public mouse genome database, August 2005 draft assembly (http://genome.ucsc.edu/) and identified annotated candidate genes located near each RIS. We compared these RISs with previously identified RISs in the web-accessible Mouse Retroviral Tagged Cancer Gene Database (http://genome2.ncifcrf.gov/RTCGD). To define common integrations sites, we used the conditions described previously (Suzuki et al, 2002).
Southern and Northern blotting
High-molecular weight genomic DNAs were prepared from frozen tissues (tumors and control brains) as described previously (Jenkins et al, 1982). Restriction enzyme-digested DNAs (5 μg) were electrophoresed in 0.8% agarose gels and transferred to positively charged nylon membranes (Roche, USA) followed by UV crosslinking. The probes were labeled with Gene Images Alkphos Direct Labeling (Amersham, USA) and the bands detected using CDP-Star reagents (Roche). For Northern blotting, total RNAs from the tumors were extracted with Trizol solution (Invitrogen, USA). A 10 μg aliquot of total RNA was separated on a 1.0% agarose gel containing 2% formaldehyde and transferred onto a nylon membrane. The probes were labeled with DIG (digoxigenin) DNA Labeling Mix (Roche) by the PCR and used for hybridization. The blot was treated with anti-DIG antibody conjugated with alkaline phosphatase (Roche) and then developed with CDP-Star reagents. The gel images were acquired with a luminescent image analyzer LAS-3000 (Fujifilm, Japan). The intensities of the bands were quantified using Image Gauge ver.4 software (Fujifilm, Japan).
Production of the retroviruses expressing shRNAs
To downregulate expression of candidate TS genes, we cloned complementary oligonucleotides for each gene into the RNAi-ready pSIREN-RetroQ vector (BD Clontech, USA). Two complimentary oligonucleotides for each siRNA target site were synthesized according to the manufacturer's protocol and cloned into the pSIREN-RetroQ vector. The expression vector was then transfected into the virus-packaging cell line Plat E (a kind gift from Dr Toshio Kitamura, University of Tokyo), using the Genejuice transfection reagent (Novagen, Germany). Virus-containing supernatants were collected 48 h after transfection. For infection, 1 × 105 NIH 3T3 cells were plated in 60 mm dishes and cultured for 6 h with virus-containing supernatants (5 × 105 colony-forming unit (CFU)/ml as assessed by puromycin resistance in NIH 3T3 cells) in the presence of 6 μg/ml polybrene (Sigma). Cells were then washed with PBS, incubated for 24 h with Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, and then exposed to selection in medium containing 1 μg/ml puromycin (Sigma). shRNA constructs that suppressed best were identified by Northern analysis of infected cells following drug selection or 2 weeks.
Quantitative analysis for mutator (mismatch repair) activity
For the construction of the slippage-eGFP vector, the eGFP-coding sequence with a mutated initiation codon (ATG to CTG) was generated by PCR. A new initiation codon that leads to out of frame eGFP expression was supplied with the oligonucleotides containing CA repeats shown below and cloned into the BamHI and NotI site of pEGFP-N1 (BD Clontech, USA), replacing the wild-type eGFP-coding sequence. The oligonucleotides containing CA repeats were synthesized as follows: EcoRI-initiation-(CA)17, (5′-AATTCCGCCATGGAA(CA)17G) and BamHI-(CA)17, (5′-GATCC(TG)17TTCCATGGCGG). The annealed oligonucleotides were then ligated into the EcoRI and BamHI site of the modified pEGFP-N1 vector to create the slippage-eGFP vector. The vector was then linearized by ApaL1 digestion and transfected into NIH3T3 cells using the Genejuice transfection reagent (Novagen, Germany). Transfected cells were subsequently exposed to selection in medium containing 500 μg/ml G418 (Nakarai, Japan), and the drug-resistant colonies picked after 10 days of selection. Cell lines stably expressing the slip-eGFP transcripts were selected by Northern blot. The slip-eGFP NIH3T3 cells were then infected with each shRNA-expressing retrovirus as described above, and selected with puromycin. The puromycin-resistance cell clones were then expanded and the eGFP-positive cells detected by a FACS (Calibur, BD, USA). Similar experiments were done at least six times and the data subjected to Luria–Delbruck fluctuation analyses (Luria and Delbruck, 1943).
MNNG sensitivity
Survival after MNNG treatment was determined by clonogenic assay. NIH3T3 cells were infected with shRNA-expressing retroviruses and selected with puromycin. The resistant cells at clonal density (100 cells/35 mm dish) were treated 18 h after seeding with MNNG (WAKO chemicals, Japan). The treatments were performed by removing media from cells and by adding serum-free media. MNNG was then added to the indicated final concentration. After 1 h of drug exposure, the cells were rinsed extensively with PBS, and fed on complete growth media. After 10–14 days, surviving colonies were fixed with PBS containing 2% paraformaldehyde, stained with Giemsa solution and counted. During the whole procedure from 2 h before drug exposure, O6-benzylguanine (20 μM; Sigma, USA) was included in the media to inhibit endogenous methyltransferase activity that might otherwise remove the methyl groups added by MNNG (Dolan et al, 1990).
Supplementary Material
Supplementary Figures and Table
Acknowledgments
We thank D Gilbert, N O'Sullivan, R Morimoto, Y Shioyama, and A Okada for excellent technical help. This research was supported by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research (NAJ and NGC), and by Grants-in-Aid for Scientific Research, and Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology of Japan (TS).
References
- Adams DJ, Bradley A (2002) Induced mitotic recombination: a switch in time. Nat Genet 30: 6–7 [DOI] [PubMed] [Google Scholar]
- Bai F, Pei XH, Godfrey VL, Xiong Y (2003) Haploinsufficiency of p18(INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol Cell Biol 23: 1269–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciunas D, Ronne H (2000) Evidence of domain swapping within the jumonji family of transcription factors. Trends Biochem Sci 25: 274–276 [DOI] [PubMed] [Google Scholar]
- Bijl J, Sauvageau M, Thompson A, Sauvageau G (2005) High incidence of proviral integrations in the Hoxa locus in a new model of E2a-PBX1-induced B-cell leukemia. Genes Dev 19: 224–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester N, Kuo F, Kozak C, O'Hara CD, Leder P (1998) Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom's syndrome gene. Genes Dev 12: 3382–3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold PM, Ponting CP (2001) JmjC: cupin metalloenzyme-like domains in jumonji, hairless and phospholipase A2beta. Trends Biochem Sci 26: 7–9 [DOI] [PubMed] [Google Scholar]
- Dannenberg JH, Schuijff L, Dekker M, van der Valk M, te Riele H (2004) Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev 18: 2952–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan ME, Moschel RC, Pegg AE (1990) Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci USA 87: 5368–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Jenkins NA, Copeland NG (2005) Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood 106: 3932–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA (2005) Mammalian mutagenesis using a highly mobile somatic sleeping beauty transposon system. Nature 436: 221–226 [DOI] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83: 655–666 [DOI] [PubMed] [Google Scholar]
- German J (1993) Bloom syndrome: a Mendelian prototype of somatic mutational disease. Medicine (Baltimore) 72: 393–406 [PubMed] [Google Scholar]
- Gilbert DJ, Neumann PE, Taylor BA, Jenkins NA, Copeland NG (1993) Susceptibility of AKXD recombinant inbred mouse strains to lymphomas. J Virol 67: 2083–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J (2002) Enhanced tumor formation in mice heterozygous for Blm mutation. Science 297: 2051–2053 [DOI] [PubMed] [Google Scholar]
- Guo G, Wang W, Bradley A (2004) Mismatch repair genes identified using genetic screens in Blm-deficient embryonic stem cells. Nature 429: 891–895 [DOI] [PubMed] [Google Scholar]
- Hwang HC, Martins CP, Bronkhorst Y, Randel E, Berns A, Fero M, Clurman BE (2002) Identification of oncogenes collaborating with p27Kip1 loss by insertional mutagenesis and high-throughput insertion site analysis. Proc Natl Acad Sci USA 99: 11293–11298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins NA, Copeland NG, Taylor BA, Bedigian HG, Lee BK (1982) Ecotropic murine leukemia virus DNA content of normal and lymphomatous tissues of BXH-2 recombinant inbred mice. J Virol 42: 379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P, Bignami M (1994) DNA damage tolerance, mismatch repair and genome instability. BioEssays 16: 833–839 [DOI] [PubMed] [Google Scholar]
- Karran P, Bignami M (1996) Drug-related killings: a case of mistaken identity. Chem Biol 3: 875–879 [DOI] [PubMed] [Google Scholar]
- LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL, Rudnicki MA (1998) Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol Cell Biol 18: 7455–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Shin MS, Kim HS, Park WS, Kim SY, Jang JJ, Rhim KJ, Jang J, Lee HK, Park JY, Oh RR, Han SY, Lee JH, Lee JY, Yoo NJ (2000) Somatic mutations of Fas (Apo-1/CD95) gene in cutaneous squamous cell carcinoma arising from a burn scar. J Invest Dermatol 114: 122–126 [DOI] [PubMed] [Google Scholar]
- Li J, Shen H, Himmel KL, Dupuy AJ, Largaespada DA, Nakamura T, Shaughnessy JD Jr, Jenkins NA, Copeland NG (1999) Leukaemia disease genes: large-scale cloning and pathway predictions. Nat Genet 23: 348–353 [DOI] [PubMed] [Google Scholar]
- Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A (2000) Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet 26: 424–429 [DOI] [PubMed] [Google Scholar]
- Luria SE, Delbruck M (1943) Mutation of bacteria from virus sensitivity to virus resistance. Genetics 28: 491–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Zhang Y (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6: 838–849 [DOI] [PubMed] [Google Scholar]
- McDaniel LD, Chester N, Watson M, Borowsky AD, Leder P, Schultz RA (2003) Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair (Amst) 2: 1387–1404 [DOI] [PubMed] [Google Scholar]
- Mochizuki N, Shimizu S, Nagasawa T, Tanaka H, Taniwaki M, Yokota J, Morishita K (2000) A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood 96: 3209–3214 [PubMed] [Google Scholar]
- Oukka M, Kim ST, Lugo G, Sun J, Wu LC, Glimcher LH (2002) A mammalian homolog of Drosophila schnurri, KRC, regulates TNF receptor-driven responses and interacts with TRAF2. Mol Cell 9: 121–131 [DOI] [PubMed] [Google Scholar]
- Pothof J, van Haaften G, Thijssen K, Kamath RS, Fraser AG, Ahringer J, Plasterk RH, Tijsterman M (2003) Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev 17: 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Caldas C (2005) Chromatin modifier enzymes, the histone code and cancer. Eur J Cancer 41: 2381–2402 [DOI] [PubMed] [Google Scholar]
- Schubart K, Massa S, Schubart D, Corcoran LM, Rolink AG, Matthias P (2001) B cell development and immunoglobulin gene transcription in the absence of Oct-2 and OBF-1. Nat Immunol 2: 69–74 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, Jenkins NA, Copeland NG (2002) New genes involved in cancer identified by retroviral tagging. Nat Genet 32: 166–174 [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439: 811–816 [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125: 467–481 [DOI] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y (2006) JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125: 483–495 [DOI] [PubMed] [Google Scholar]
- Yang Y, Kuang Y, De Oca RM, Hays T, Moreau L, Lu N, Seed B, D'Andrea AD (2001) Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood 98: 3435–3440 [DOI] [PubMed] [Google Scholar]
- Yeung RS, Bell DW, Testa JR, Mayol X, Baldi A, Grana X, Klinga-Levan K, Knudson AG, Giordano A (1993) The retinoblastoma-related gene, RB2, maps to human chromosome 16q12 and rat chromosome 19. Oncogene 8: 3465–3468 [PubMed] [Google Scholar]
- Yusa K, Horie K, Kondoh G, Kouno M, Maeda Y, Kinoshita T, Takeda J (2004) Genome-wide phenotype analysis in ES cells by regulated disruption of Bloom's syndrome gene. Nature 429: 896–899 [DOI] [PubMed] [Google Scholar]
- Ziebold U, Lee EY, Bronson RT, Lees JA (2003) E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol 23: 6542–6552 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Table