

Abstract
The autoimmune disease pemphigus vulgaris (PV) manifests as loss of keratinocyte cohesion triggered by autoantibody binding to desmoglein (Dsg)3, an intercellular adhesion molecule of mucous membranes, epidermis, and epidermal stem cells. Here we describe a so far unknown signaling cascade activated by PV antibodies. It extends from a transient enhanced turn over of cell surface-exposed, nonkeratin-anchored Dsg3 and associated plakoglobin (PG), through to depletion of nuclear PG, and as one of the consequences, abrogation of PG-mediated c-Myc suppression. In PV patients (6/6), this results in pathogenic c-Myc overexpression in all targeted tissues, including the stem cell compartments. In summary, these results show that PV antibodies act via PG to abolish the c-Myc suppression required for both maintenance of epidermal stem cells in their niche and controlled differentiation along the epidermal lineage. Besides a completely novel insight into PV pathogenesis, these data identify PG as a potent modulator of epithelial homeostasis via its role as a key suppressor of c-Myc.
Keywords: β-catenin, desmosomes, epidermis, stem cells, plakoglobin
Introduction
Proliferation and terminal differentiation are part of the lifelong renewal of stratified squamous epithelia. During this process, the recruitment of stem cells into the proliferative compartment and subsequent commitment to terminal differentiation is finely regulated by the proto-oncogene c-Myc (Watt, 2001). In basal keratinocytes, c-Myc activation results in stem cell depletion and hyperproliferation as was shown in transgenic mouse models (Arnold and Watt, 2001; Waikel et al, 2001; Frye et al, 2003). When activated in suprabasal keratinocytes, c-Myc triggers proliferation and disrupts terminal differentiation in addition to premalignant transformations (Pelengaris et al, 1999; Waikel et al, 1999). Accordingly, in cultured mouse keratinocytes, c-Myc is induced at the onset of proliferation, then repressed as cells enter terminal differentiation (Kolly et al, 2005). In contrast to mouse, the consequences of c-Myc overexpression are unknown in human epidermis, as to date no skin disease with generalized c-Myc overexpression has been reported. However in human monolayer keratinocyte cultures, constitutive c-Myc expression is known to cause premature recruitment of epidermal stem cells into the proliferative compartment highlighting the importance of timely c-Myc suppression also in human skin (Gandarillas and Watt, 1997). The exact mechanisms of c-Myc suppression in both mouse and human keratinocytes, however, are not known.
Pemphigus vulgaris (PV) is a life-threatening autoimmune disease characterized by suprabasal acantholysis (i.e. loss of basal–basal and basal–suprabasal cell adhesion) in stratified squamous epithelia (Beutner and Jordon, 1964; Payne et al, 2004). The antigenic target is the desmosomal cadherin desmoglein (Dsg)3, which by virtue of its integration into desmosomes mediates cell–cell adhesion (Amagai et al, 1991). Dsg3 is mainly expressed in mucous membranes, the deep layers of epidermis and hair follicles, including epidermal stem cells (Koch et al, 1998; Shirakata et al, 1998; Mahoney et al, 1999; Wan et al, 2003). Most PV patient's antibodies target the adhesive interface between juxtaposed Dsg3 molecules of adjacent cells, and are thought to disrupt their trans-adhesion by steric hindrance (Tsunoda et al, 2003). Initially, this leads to depletion of nonkeratin–anchored, that is, Triton X-100-soluble Dsg3, and ultimately to loss of Dsg3 from fully assembled desmosomes (Aoyama and Kitajima, 1999; Sato et al, 2000). Although the molecular mechanism of Dsg3 depletion and subsequent acantholysis remains unknown, we have shown that the armadillo protein plakoglobin (PG), which associates with the cytoplasmic tail of Dsg3, is crucially involved in this process (Caldelari et al, 2001). Our previous work suggested that ‘secondary to antibody binding, modulation of available PG at the plasma membrane supports rapid degradation of Dsg3, and subsequently affects the ongoing differentiation process' (Caldelari et al, 2001). A number of other studies also indicated that besides adhesion, PG is involved in cell cycle regulation and the control of c-Myc. Findings include premature exit from the cell cycle triggered by moderately overexpressed PG in mouse epidermis (Charpentier et al, 2000), the ability of PG to limit excessive proliferation (Teuliere et al, 2004), the positive correlation between malignancy of epithelial tumors and loss of PG expression (Zhurinsky et al, 2000a) as well as the direct regulation of c-Myc by PG in hematopoietic cells (Muller-Tidow et al, 2004). Thus overall, a growing body of evidence now indicates that PG has a much broader biological role than just in adhesion.
In this study, we further characterize the role of PG in epidermal homeostasis suggested by our earlier study. Specifically, we investigate the control PG exerts over c-Myc, which in turn influences the delicate balance of stem cell recruitment, proliferation and terminal differentiation in keratinocytes.
Results
Pemphigus vulgaris antibodies (PVIgG) retain cultured keratinocytes in a proliferative state
The fate of Dsg3 and PG was addressed in PVIgG-exposed mouse keratinocytes at the transition between proliferation and terminal differentiation (i.e. during cell cycle exit/growth arrest). This stage largely reflects the status of basal keratinocytes with established Dsg3-based desmosomes, but not of suprabasal cells, which already are growth arrested (Caldelari et al, 2001) (Figure 1A). At 4 and 48 h postincubation with PVIgG or normal human control IgG (nhIgG), total steady-state PG appeared unchanged (Figure 1B). Given that most PG is associated with desmosomes, changes in nondesmosomal PG were masked. In subcellular fractions, the turn over of antibody targeted Triton X-100-soluble (nonkeratin-anchored) Dsg3 and associated PG at the plasma membrane transiently increased within the first hour after exposure to PVIgG and resulted in a significant depletion of these molecules from the cytoplasmic–vesicular pool (Supplementary Figure 1). Concomitantly, soluble PG levels failed to rise in the cytosol/low-detergent soluble membrane fraction and was followed by decreased nuclear import of PG as compared to normal terminally differentiating cells (Figure 1C; Supplementary Figure 2 shows relative levels of membrane, cytoplasmic and nuclear proteins). The reduction in PG protein was further paralleled by a decrease in PG steady-state mRNA (Figure 1D) suggesting the activation of a signaling cascade in response to the initial transient depletion of Dsg3 and PG at the plasma membrane. In contrast to PG, nuclear β-catenin and plakophilin1, two other armadillo-(like) proteins as well as β-catenin mRNA were unchanged (Figure 1C).
Figure 1.
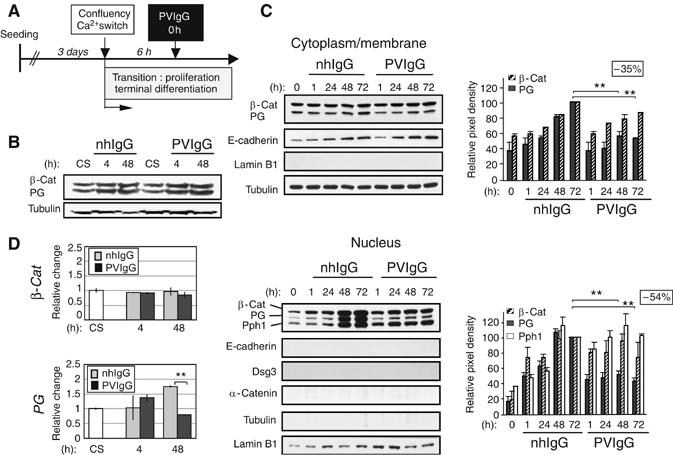
Nuclear PG is reduced in PVIgG-treated mouse keratinocytes. (A) Experimental setup used in this and our previous study (Caldelari et al, 2001). (B) Western blot analysis of total lysates from keratinocytes treated as indicated. CS designates the sample collected at calcium switch. Tyrosine tubulin was used as a loading control. (C) Western blot analyses of cytoplasmic/membrane and nuclear fractions. Proportionally 10 times more protein (in terms of cell equivalents) was loaded for the nuclear than the cytoplasmic/membrane fractions (see Supplementary Figure 2A for respective amounts of adhesion components in each fraction). Tubulin, E-cadherin, Dsg3 and α-catenin and the nuclear envelope protein lamin B1 served as a loading or purity control, respectively. Graphs depict the relative change of indicated proteins with respect to control cells at 72 h (set as 100). Insets indicate percentage PG reduction in PVIgG-treated cells at 72 h as compared to control keratinocytes. Note that nuclear β-catenin was similar in control and PVIgG-treated cells. Nuclear accumulation of armadillo proteins coincides with the phosphorylation of GSK3β on Ser9 (Calautti et al, 2005). Pph1=plakophilin1. **P<0.009, n=5. Error bars are ±s.d. (D) Graphs show steady-state mRNA levels determined by Q-PCR presented as relative change compared to CS. **P<0.01, n=6. Error bars are ±s.d.
The consequences of decreased nuclear PG on transcription of proliferation and terminal differentiation markers were addressed. Consistent with unchanged levels of nuclear β-catenin, the mRNA level of its target gene cyclin D1 (Tetsu and McCormick, 1999) was not reduced in PVIgG-treated cells (Figure 2A). In opposite, cyclin D1 mRNA was moderately but significantly increased. Accordingly, p21WAF1 mRNA, which encodes a crucial determinant of the exit from the cell cycle in keratinocytes and suppressor of cyclin D1 (Missero et al, 1996), failed to increase. The steady-state level of the corresponding proteins followed a similar pattern with cyclin D1 and p21WAF1 persisting at higher levels up to day 15 in PVIgG-exposed cells (Figure 2B). Apparent sustained proliferation in these cells was paralleled by hampered accumulation of Dsg3 mRNA and protein during the first 48 h (Supplementary Figure 3). Indicative for a role of PG in the process, PG−/− keratinocytes showed similar mRNA and protein levels as PVIgG-treated cells (Supplementary Figure 3), and noticeable were unresponsive to PVIgG as shown in our previous study (Caldelari et al, 2001).
Figure 2.

PVIgG retain keratinocytes in a proliferative state. (A) Relative change of indicated mRNA levels in wild-type keratinocytes assessed by Q-PCR. *P<0.05; **P<0.01; 4 h, 2 days, n=8; 6 days, 8 days, n=2. Error bars represent the ±s.d. or range (6 days, 8 days). (B) Western blot analysis from parallel cultures to those in (A). Tubulin was assessed on the same blot than cyclin D1 and p21WAF1. (C) Proliferation curves are shown for indicated cell types. Counts started at confluency when the calcium switch was introduced. Note that untreated wild-type keratinocytes undergo growth arrest between days 4 and 6 after calcium switch as seen by a plateau. *=detachment of intact cellular sheets; **=detachment of single cells. Error bars are ±s.d. A representative experiment of two carried out in triplicates is shown. WT=wild-type keratinocytes.
Consistently, growth curves with PVIgG-treated keratinocytes showed sustained proliferation as compared to control cells (Figure 2C). A similar fate was also observed for PG−/− keratinocytes in 3D cultures (Supplementary Figure 4) as well as conventional submerged cultures (Figure 2C). Remarkably, the growth rate of PG−/− cells was strictly independent of PVIgG. In line with their unresponsiveness to PVIgG but despite weaker adhesion than wild-type cells (Caldelari et al, 2001), the PG−/− keratinocytes detached from the culture dish in large intact cell sheets around day 10, as opposed to single cells in PVIgG-exposed wild-type cultures. All cultures entered at least a transient plateau around growth arrest (days 4–6 after calcium switch). This could be owing to cell–cell contact-induced signals like Notch-1 (Kolly et al, 2005) that may precede the major changes (as defined in the following), leading to sustained proliferation in PG−/− as well as PVIgG-treated keratinocytes. Together, these results suggest that proliferation is sustained in cultured keratinocytes with reduced or lack of nuclear PG.
The status in PV patients reflects the findings in cultured keratinocytes
The proliferation status was addressed in PV patients using immunofluorescence analysis performed on perilesional and nonlesional biopsies from six patients with variable duration of disease. For comparison, biopsies from healthy donors, patients with other autoimmune bullous diseases such as pemphigus foliaceous (PF) and bullous pemphigoid (BP) as well as chronic eczematous dermatitis (as an example of an inflammatory skin disorder with epidermal hyperproliferation (Altekrueger and Ackerman, 1994)) were also investigated.
Direct immunofluorescence confirmed the typical clustered staining pattern of IgG deposits and PG in the epidermis and oral mucosa of PV patients (Carlotti et al, 1993; A de Bruin, unpublished) (Figure 3A and Supplementary Figure 5). Staining for PG appeared weaker while that of β-catenin was unchanged. Consistent with the results obtained in culture, the number of cycling keratinocytes (Ki67-positive cells) was diffusely upregulated (up to 15 times) in perilesional and nonlesional epidermis as well as in mucosa of all PV patients analyzed (Figure 3B). Moreover, the proliferating Ki67-positive cells localized to basal and suprabasal layers, a typical feature of hyperproliferative epidermis (Andreadis et al, 2001). Despite this, differentiation markers were still expressed, but their expression pattern was disrupted in particular in patients with longstanding disease (Supplementary Figure 5). None of these features were observed in PF, an autoimmune bullous disease so far considered closely related to PV. BP and eczematous dermatitis showed some increased Ki67-positive cells, probably depending on the relevant inflammatory exocytosis involved.
Figure 3.
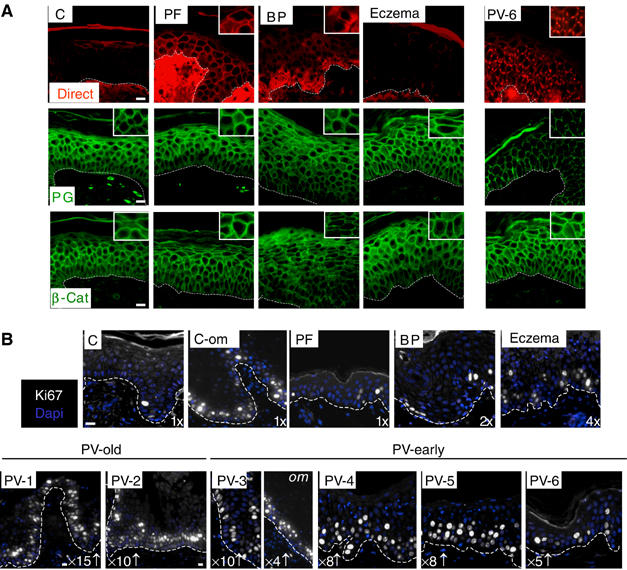
The status in PV patients reflects results obtained in cultured keratinocytes. (A) Paraffin-embedded tissue from nonlesional skin of a PV patient (PV6) and patients with PF, BP and chronic eczematous dermatitis (Eczema) were investigated by immunofluorescence microscopy and compared to control biopsies (epidermis (C)). Direct labeling demonstrates IgG deposits. Insets represents a three times higher magnification of representative areas to depict clustered desmosomal, but not adherens junction (β-catenin) proteins in PV patient six (nonlesional epidermis; PV patients 1–5, see Supplementary Figure 4). Note that biopsies were processed simultaneously and photographic procedures held constant to obtain semiquantitative results. Scale bars, 200 μm. (B) Consecutive cuts of the biopsies in (A) and in Supplementary Figure 4 were stained with Ki67 and counterstained with Hoechst. Numbers indicate the increase of Ki67-positive cells (in nonacantholytic areas) as compared to the normal situation (set as 1 for C and C-om (oral mucosa)). Scale bar, 200 μm (scale was reduced for PV-1 and PV-2 to give a broader overview).
PVIgG induce a generalized c-Myc overexpression in keratinocytes
The PV phenotype with hyperproliferation despite continuing expression of differentiation markers in suprabasal cells was reminiscent of that described for transgenic mice with activated c-Myc in basal keratinocytes (Arnold and Watt, 2001; Waikel et al, 2001). Consistent with this, the relative c-Myc mRNA levels were generally up to 1.5 times higher (maximally up to two-fold (data not shown)) in PVIgG-treated keratinocyte cultures than in control cells (Figure 4A). During the 8 days investigated, c-Myc levels in PVIgG-treated cells always exceeded those of confluent control cells at calcium switch and, importantly, reached to the level reported in proliferating keratinocytes (Kolly et al, 2005). On the protein level, the change in mRNA was paralleled by a substantial increase in the 64 and 46 kDa c-Myc isoforms (Figure 4B). The 46 kDa isoform of c-Myc likely represents the unphosphorylated c-Myc, which is characteristic for rapidly growing cells and is constitutively overexpressed in colon tumor cells (Tao et al, 2002). In human (Figure 4B) and mouse (Figure 4C, WT) keratinocytes, the increase in both c-Myc isoforms was already substantial after 1 day of PVIgG exposure, and was observed in both the cytoplasm and the nucleus (Figure 4C, WT). C-Myc accumulation correlated with progressive weakening of intercellular adhesive strength, as examined by an adhesion assay that applies mechanical stress to monolayer cultures (Figure 4D).
Figure 4.
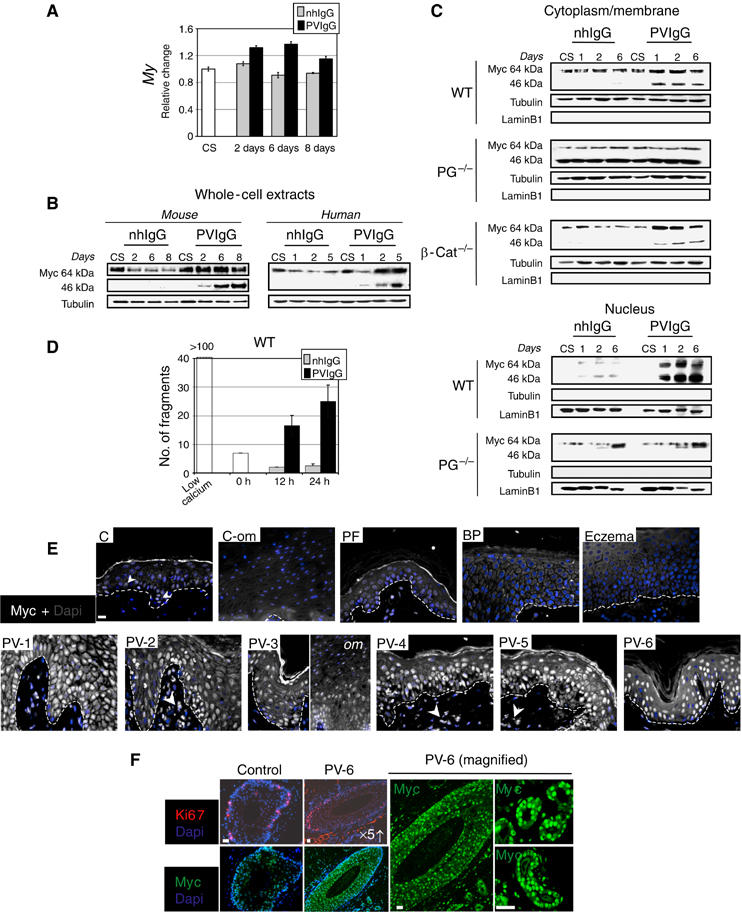
PVIgG upregulate c-Myc. (A) Graph indicates the relative change in c-Myc mRNA levels as compared to CS. One representative result carried out in duplicates of three independent experiments is shown. Error bars represent the range. (B) Western blot analyses for c-Myc was performed on total cell lysates obtained from parallel cultures to those in (A) (mouse) or from human keratinocytes. (C) Western blot analyses of cytoplasmic/membrane and nuclear fractions. (D) Graph indicates number of fragments generated after the application of mechanical stress to wild-type mouse monolayer cultures. Zero hours indicate beginning of PVIgG or nhIgG treatment (6 h after calcium switch). One experiment of two carried out in duplicates is shown. Scale bars represent the range. (E) Consecutive sections of paraffin-embedded PV and control biopsies, as in Figure 3 (B), were stained for c-Myc, counterstained with Hoechst. c-Myc-positive cells in the dermis (arrow-heads) likely are leukocytes as judged from H&E stains (data not shown), which is consistent with their absence from nonlesional skin (PV-6). Arrows point to faint c-Myc staining in control skin. (F) Hair follicles stained with Ki67 or c-Myc and Hoechst (left panel). The right panel (PV-6 magnified) is a two-fold magnification of the hair follicle in the left panel and a six-fold magnification of sebaceous glands. All biopsies were processed simultaneously and photographic procedures held constant to obtain semiquantitative results. Scale bars, 200 μm.
PG−/− keratinocytes had 1.5 times higher c-Myc mRNA levels than normal differentiating wild-type cells (data not shown). This correlated with a high protein level predominantly of the cytoplasmic 46 kDa c-Myc isoform (Figure 4C, PG−/−). Furthermore, cytoplasmic c-Myc was not regulated after calcium switch or in response to PVIgG. Consistent with a 2-day delay of enhanced growth as compared to PVIgG-treated cells (Figure 2C), nuclear accumulation of the 64 kDa isoform was only increased in the PG−/− keratinocytes at day 6 after calcium switch and in both nhIgG- and PVIgG-treated cells. This suggests that the PVIgG-induced enhanced turn over of PG in wild-type cells (Supplementary Figure 1), which does not occur in PG−/− cells, amplifies c-Myc activity by increasing its nuclear accumulation. In contrast to PG−/− cells, c-Myc levels in β-catenin−/− keratinocytes corresponded to those of wild-type cells and were upregulated in response to PVIgG (Figure 4C, β-cat−/−). This is consistent with the finding that proliferation and onset of terminal differentiation proceed normally in these cells (Posthaus et al, 2002), that epidermal deletion of β-catenin did not upregulate c-Myc (Teuliere et al, 2004) and 3D cultures of β-catenin−/− keratinocytes grew to the thickness of wild-type cultures in contrast to PG−/− keratinocytes, which form hyperplastic sheets (Supplementary Figure 4). Accordingly, β-catenin−/− cells responded to PVIgG with loss of intercellular adhesion like wild-type cells (data not shown) while PG−/− cells did not (Caldelari et al, 2001).
Consistent with results obtained in cell culture, the six PV patients revealed a generalized (diffuse) strong c-Myc staining in all layers of the epidermis and oral mucous membrane, with highest intensity in the nucleus as demonstrated by semiquantitative immunofluorescence analysis (Figure 4E). Importantly, c-Myc was as significantly upregulated in nonlesional PV skin, hair follicles and sebaceous glands (patient 6; Figure 4E and F). In each case, the c-Myc levels exceeded those in basal keratinocytes of healthy donors, and in particular in biopsies of patients with other autoimmune bullous diseases or eczematous dermatitis.
Like in the two transgenic mouse models with c-Myc activation in basal keratinocytes (Arnold and Watt, 2001; Waikel et al, 2001), staining for β1-integrin was substantially lower and keratins 6 and 14 were increased in the epidermis of all six PV patients (Supplementary Figure 5). Impaired wound healing and in particular enhanced exit of epidermal stem cells from their niche were attributed to these changes (Arnold and Watt, 2001; Waikel et al, 2001; Frye et al, 2003). Together, these results revealed for the first time that PVIgG trigger c-Myc accumulation and hyperproliferation, which is accompanied by reciprocal regulation of β1-integrin, and keratins 6 and 14.
PG is a suppressor of c-Myc in epidermal keratinocytes
In mouse embryonic stem cells and keratinocytes, a dual mode of c-Myc regulation has been proposed that consists of transcriptional regulation in conjunction with post-translational stabilization (Cartwright et al, 2005; Kolly et al, 2005). Transcription of c-Myc can be regulated by Tcf/Lef transcription factors together with PG (Kolligs et al, 2000; Muller-Tidow et al, 2004). These factors are expressed in mouse keratinocytes (Posthaus et al, 2002), of which Lef-1 was related to hyperproliferation in mouse epidermis if functionally inactivated (Niemann et al, 2002).
We initially addressed whether PG can inhibit Lef-1 or Tcf4-mediated transcription from the human c-Myc promoter described previously (Kolligs et al, 2000). In a reporter gene assay, PG inhibited transcription together with Lef-1 in a dose-dependent manner, while it activated transcription in the presence of Tcf4 or, as reported, on its own (suggesting the use of endogenous Tcf/Lef family members) (Kolligs et al, 2000) (Figure 5A). PG-mediated suppression was Lef-1 dependent (as demonstrated by truncated Lef-1, which fails to bind PG (Huber et al, 1996)) and was exerted by the C-terminus of PG known to harbor the transactivation site (Hecht et al, 1999) (as shown by deletion mutants of PG (Zhurinsky et al, 2000b)). PG/Lef-1-mediated suppression was comparable in transfected wild-type and β-catenin−/− keratinocytes, further demonstrating that this effect was directly mediated by PG and not indirectly via endogenous β-catenin displaced by PG from its cellular binding partners (Figure 5A shows selected transfections). In addition, suppression by Lef-1/PG further was characteristic for the c-Myc promoter as it was not seen when using an artificial Tcf-responsive promoter (Molenaar et al, 1996) (Figure 5B). In line with a role of Lef-1 in basal keratinocytes, Lef-1 protein was mainly confined to the nuclei in the deep mouse and human epidermis (Figure 5C).
Figure 5.
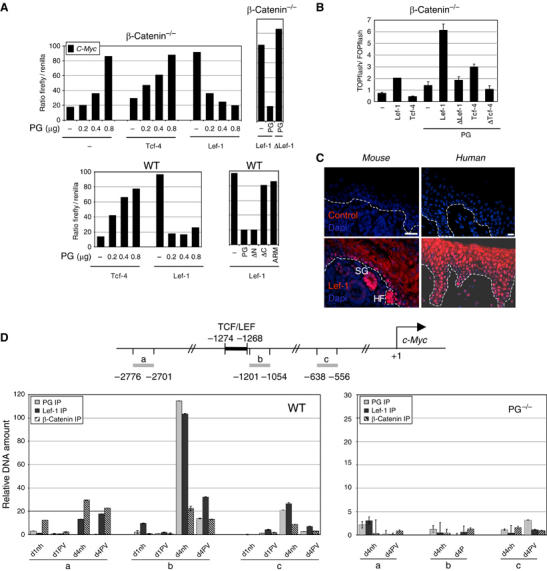
PG is a suppressor of c-Myc. (A) Graphs of reporter gene assays using the human c-Myc promoter on indicated cell types. The ratio of firefly over renilla luciferase activity is indicated. One representative experiment of at least four independent experiments carried out in single measures per cell type is shown. ΔLef-1 lacks the armadillo-binding domain; PG ΔC, ΔN and ARM lack the C-terminal, N-terminal domain or both, respectively. Individual mutation of three LEF/TCF binding sites in the c-Myc promoter (two reported, one unreported) reduces Lef-1-mediated activation by over 50% (C Kolly et al, unpublished). (B) TOPflash/FOPflash promoter activity in the presence of indicated exogenous factors. One representative experiment of three is shown. Error bars represent the range. (C) Paraffin-embedded tissue from normal newborn mouse epidermis and human skin stained with Lef-1 antibodies. Controls were incubated with secondary antibody and processed for enhancement. Lef-1 staining in mouse epidermis is highest in remnants of hair follicles (HF) and sebaceous glands (SG), and fainter staining is confined to the nuclei of mainly basal keratinocytes. Human epidermis shows strong Lef-1 nuclear staining in basal and suprabasal keratinocytes. Scale bars, 200 μm. (D) Top panel: Scheme of the mouse c-Myc promoter and TCF/LEF binding site. Gray bars indicate positions of primer sets relative to the transcription initiation site which were used to amplify precipitated DNA by Q-PCR. Bottom panel: ChIP. Graphs show relative amounts of amplification products obtained by Q-PCR with indicated primer sets. One of three (WT) and two (PG−/−) independent experiments measured in duplicates is shown. Error bars indicate the range.
Using chromatin immunoprecipitation (ChIP), we further examined whether PG/Lef-1 is recruited to the c-Myc promoter at cell cycle exit, and if β-catenin is involved in this process. Before growth arrest (1 day after calcium switch (Figure 2C and Kolly et al (2005)) neither PG nor β-catenin was detectably bound to the TCF/LEF binding site in the mouse c-Myc promoter, as seen by lack of DNA amplification above back ground with a primer set immediately adjacent to the TCF/LEF binding site (chosen according to quantitative real-time PCR (Q-PCR) requirements) (Figure 5D, left panel; compare primer set a to b). Lef-1 antibodies precipitated a small amount of DNA from the TCF/LEF domain. Results were identical for untreated cells as well as for cells 1 day before calcium switch (data not shown). In contrast, in cells undergoing growth arrest (4 days after calcium switch (Figure 2C and Kolly et al (2005)), TCF/LEF-specific amplification increased over 100-fold in Lef-1 and PG antibody precipitates, while significantly less TCF/LEF-specific fragments were precipitated from PVIgG-treated keratinocytes. Compatible with our findings so far, TCF/LEF-specific β-catenin precipitates were in any case below the background defined with the upstream primers in the same sample. No specific amplification of c-Myc promoter fragments was obtained from PG−/− cells (Figure 5D, right panel).
This set of experiments shows that at exit from the cell cycle and onset of terminal epidermal differentiation, PG but apparently not β-catenin represses the c-Myc promoter in an Lef-1-dependent manner and that PVIgG compromise this repression by reducing PG/Lef-1 binding.
PVIgG-triggered c-Myc overexpression weakens intercellular adhesion
To establish the link between PV antibody binding to Dsg3, c-Myc activity and loss of intercellular adhesion, lesion formation was examined in a passive transfer study (Anhalt et al, 1982) in the presence or absence of c-Myc inhibitors that prevent the Myc–Max interaction required for c-Myc activity (Yin et al, 2003). The importance of Dsg3 as trigger of the signaling cascade leading to c-Myc activation was addressed by the mouse monoclonal Dsg3-specific antibody AK23 (Tsunoda et al, 2003). AK23 is a pathogenic PV antibody, which targets the adhesive interface between juxtaposed Dsg3 molecules. The mouse monoclonal antibody NAK3 was used as a negative control (M Amagai et al, unpublished). NAK3 also targets Dsg3, is unable to induce blisters on its own, but has weak pathogenic activity in combination with other blister-inducing antibodies.
NhIgG and PVIgG, AK23 and NAK3 were subcutaneously injected in the lumbar area of neonatal mice that had been preinjected at the same site with c-Myc inhibitors or vehicle. After 24 h, blisters were observed in PVIgG/vehicle- and AK23/vehicle-injected mice but not in their siblings pretreated with c-Myc inhibitors (Figure 6A). Despite binding of NAK3 to the surface of keratinocytes, no blisters developed under any condition, as seen in nhIgG-injected mice (Figure 6A shows NAK3). However, consistent with its weak pathogenic activity (M Amagai et al, unpublished), c-Myc levels seen by semiquantitative immunofluorescence microscopy were higher in NAK3-injected than in control mice, but significantly lower than in PVIgG- and AK23-injected mice with or without c-Myc inhibitors (Figure 6B). At that early time point, c-Myc was mainly localized in the cytoplasm and only to some extent in the nucleus.
Figure 6.
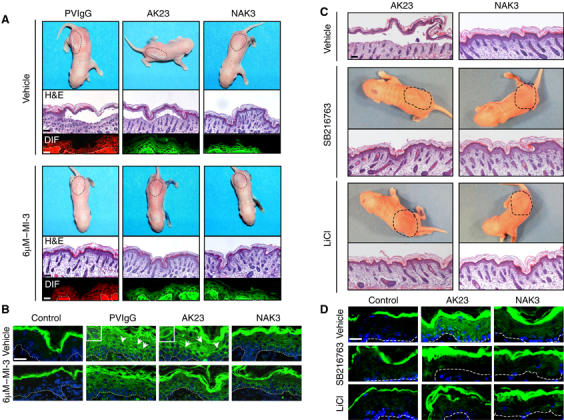
c-Myc accumulation accounts for the loss of intercellular adhesion. (A) Top pictures show neonatal mice subcutaneously injected with vehicle (top panel) or 6 μM c-Myc inhibitor 3 (bottom panel) followed by PVIgG, AK23 or NAK3 together with half the minimal dose of a human PF serum. Discontinued lines indicate the area of injection. Micrographs underneath show lesions (H&E) and IgG binding in the perilesional area assessed by direct immunofluorescence (DIF) (red=anti-human antibodies; green=anti-mouse antibodies). (B) Consecutive sections of the perilesional areas shown in (A) were stained for c-Myc. c-Myc was mainly present in the cytoplasm of PVIgG- and AK23-injected mice, and to some extent also of NAK3-injected mice. Arrows point to the nuclei (insets) with slightly elevated c-Myc. (C) As in (A), except that mice were preinjected with inhibitors to GSK3. (D) Immunofluorescence analysis of c-Myc in consecutive sections of perilesional areas shown in (C). Scale bars, 1.6 mm for H&E, otherwise 200 μm.
To further confirm that abrogation of PG's nuclear import is key for c-Myc accumulation and consequently PV pathogenesis, the passive transfer study was repeated in the presence or absence of the GSK3 inhibitors SB216763 (Coghlan et al, 2000) and LiCl (Beaulieu et al, 2004). GSK3β is a serine/threonine kinase that targets PG to the proteasome degradation machinery (Aberle et al, 1997). Its inhibition in keratinocytes results in PG stabilization and nuclear accumulation (Kodama et al, 1999). Furthermore, GSK3β was found to be inactivated at cell cycle exit in cultured mouse keratinocytes (Calautti et al, 2005), suggesting that this event might be responsible for the nuclear trafficking of PG at growth arrest (Figure 1C). In presence of GSK3β inhibitors, AK23-induced blister formation was abrogated and c-Myc levels were significantly decreased (Figure 6C and D).
Taken together, these results demonstrate that constitutive c-Myc activation initiated by PVIgG-bound Dsg3 directly affects intercellular adhesive strength in mouse epidermis and underscore that this event is critically dependent on de-regulated nuclear trafficking of PG.
Discussion
Here we show that PG is a crucial suppressor of c-Myc in keratinocytes and that PV antibodies disrupt this activity by selective depletion of PG from soluble Dsg3 pools and reduced nuclear PG resulting in abolished PG/Lef-1-mediated c-Myc suppression. Given that the c-Myc upregulation in PV at the protein level was larger than would be expected from the observed increase in mRNA, it appears that post-transcriptional control mechanisms also contribute to c-Myc regulation in these cells. Such mechanisms were already discussed for normal keratinocyte proliferation and differentiation (Kolly et al, 2005) and fit well with both our current results and emerging reports of regulatory pathways in keratinocytes that link intercellular adhesion to c-Myc regulation via PG (as discussed below).
Pathogenic PV antibodies (like the monoclonal antibody AK23) are thought to disrupt trans-adhesion between Dsg3 molecules (Tsunoda et al, 2003), which results, as shown here, in sustained proliferation. Under normal culture conditions, proliferation is stopped in keratinocytes by confluency, that is, cell–cell contact that results in the stabilization of cadherins through trans-adhesion (Kolly et al, 2005). Hence, the current study suggests that the disruption of trans-adhesion between surface-exposed soluble Dsg3 and associated PG is sufficient to generate a signal of ‘no cell contact' in confluent monolayers.
Under normal conditions, the stabilization of cadherins through trans-adhesion is accompanied by recruitment of activated phosphoinosite 3-kinase (PI3K) to the cadherin–catenin complexes, which is followed by GSK3β inhibition (Calautti et al, 2005). PG thereby serves as the main docking partner for PI3K in the cadherin–catenin complex, while GSK3β inhibition stabilizes cytoplasmic PG and β-catenin (Kodama et al, 1999), prompting their nuclear translocation and the PG-mediated c-Myc suppression shown here. Recruitment of PI3K to trans-adhering cadherins also activates, among others, the small GTPase Rac-1 (Fukuyama et al, 2006). Deletion of Rac-1, as we show for PG, results in c-Myc overexpression (Benitah et al, 2005), suggesting that PG and Rac-1 are in the same pathway. Accordingly, as demonstrated in Rac−/− mice (Benitah et al, 2005), abolished negative regulation by p21-activated kinase 2 might contribute to c-Myc post-translational accumulation in PV. Collectively, our findings along with these other reports support the notion that cadherin-bound PG links the status of intercellular adhesion to c-Myc transcriptional and possibly post-translational regulation via a singling cascade involving PI3K and GSK3β, which in turn regulates nuclear trafficking of PG. Although this cascade of events must be investigated more deeply, our findings in PV support this possibility. Downstream of cadherin-bound PG, PVIgG negatively interfere with a signaling pathway that relates to terminal differentiation and involves GSK3β: the transiently enhanced turn over of Dsg3 and PG in response to PVIgG was followed by delayed terminal differentiation and consequently sustained proliferation; GSK3β inhibition was sufficient to abrogate blister formation in PVIgG-injected neonatal mice, supporting the possibility that the PVIgG-targeted signaling pathway involves the PI3K/GSK3β axis that in turn is in control of PG nuclear trafficking and consequently c-Myc.
The cascade of events from cadherin-bound PG at the plasma membrane through to PG-mediated transcriptional control mechanisms underscores the notion that PG acts as an integrator of environmental cues (Calautti et al, 2005; Yin et al, 2005a, 2005b). Our results also demonstrate that PG can integrate these cues when bound to trans-adhering soluble Dsg3, but conversely is restricted from doing so in the absence of the Dsg3 stabilization seen in confluent monolayers in the presence of PV antibodies. Further support of PG-mediated crosstalk between desmosomal adhesion and c-Myc is evident in that observation that PG−/− keratinocytes fail to respond to environmental cues in the same manner as wild-type cells, that is, they are unresponsive to PVIgG for all parameters analyzed, fail to integrate growth factor-induced signals (Yin et al, 2005a), have elevated Src activity (Yin et al, 2005b), a tyrosine kinase that is activated upstream of PI3K (Calautti et al, 2005), and consequently constitutively express c-Myc and hyperproliferate.
The fact that PV antibodies trigger c-Myc overexpression in basal and suprabasal epidermis suggests that PG acts as a suppressor of c-Myc both at cell cycle exit and in terminally differentiated cells. Moreover, the basal compartment also harbors stem cells, which crucially depend on c-Myc suppression (Owens and Watt, 2003). Stem cells express Dsg3 and therefore are likely targets of PV antibodies (as also judged from the continuous pattern of IgG deposits). Hence, c-Myc suppression by PG in the stem cell compartment cannot currently be ruled out. Further indications that PG regulates c-Myc also in the stem cell compartment are evident when considering that tumor development in epithelial cells likely is initiated by stem cells (Owens and Watt, 2003). Lack of PG was found to be involved in a range of tumors, including loss of heterozygosity in breast and ovarian carcinomas (Aberle et al, 1995), and reduced PG levels predict an adverse outcome in patients with epithelial tumors (Zhurinsky et al, 2000b). The opposite is generally reported for β-catenin, which is considered a tumor promoter in many tissues (Conacci-Sorrell et al, 2002), and this is in line with the lack of tumor formation in β-catenin-deleted skin (Watt, 2001). Thus, the idea that PG expression is crucial both to preserve epithelial homeostasis and prevent carcinogenesis is not new. However, our finding that PG is a key suppressor of the proto-oncogene c-Myc, both at cell cycle exit in keratinocytes and probably also in the stem cell compartment, provides a molecular basis for these observations for the first time. Thus, the molecular mechanism of PV pathogenesis described here provides insights into an unexpected role of PG in epidermal homeostasis that will have consequences for epidermal stem cell and tumor biology.
Materials and methods
Keratinocyte cultures
Isolation and characterization of mouse wild-type, PG−/− and β-catenin−/− keratinocytes (all C57BL/6 genetic background) were described previously (Caldelari et al, 2001; Posthaus et al, 2002). Human keratinocytes from foreskin were a kind gift form CELLnTEC Advanced Cell Systems AG, Switzerland. Conventional submerged cultures and stimulation conditions were according to our previously established protocol for studies on PV (Figure 1) (Caldelari et al, 2001), except that the medium was changed to CnT-02 (CELLnTEC, Switzerland) or Cnt-02 supplemented with 1.2 mM calcium (referred to as ‘high calcium medium'). Human cells were cultivated in CnT-07 (CELLnTEC, Switzerland). For 3D cultures, cells were grown at the air–liquid interface for 14 days as described earlier (Suter et al, 1991).
Passive transfer studies using neonatal mice
Around 16 h after birth, alebino DDY mice were subcutaneously injected in the lumbar area with 6 μM c-Myc inhibitors (5521700 (MI-1) or 5404711 (MI-3); Chembridge) or vehicle (DMSO/PBS). After 2 h, 15 mg PVIgG or nhIgG, or alternatively 75 μg mouse monoclonal AK23 (Tsunoda et al, 2003) or NAK3 antibody (M Amagai, unpublished) were injected in the same area in combination with half the minimal dose of a human PF serum inducing blister formation (Mahoney et al, 1999). Vehicle or c-Myc inhibitors alone were used as negative controls. After 24 h, the animals were evaluated for macroscopic blistering. Biopsies were taken from lesional areas, fixed in 4% buffered formaldehyde overnight at 4°C and paraffin embedded. Biopsies were deparaffinized and stained with H&E or processed for direct immunofluorescence as described under immunofluorescence analyses. Four animals per treatment (PVIgG, AK23, NAK3, nhIgG, no treatment), each with vehicle or c-Myc inhibitors, were analyzed in two independent experiments, which showed consistent results. The time-course and injection protocols using chemical GSK3 inhibitors, SB216763 (100 μM; 2 μg/g body weight; Calbiochem) and LiCl (200 μg/g body weight) (Beaulieu et al, 2004), were identical to the c-Myc inhibitor experiment. In total, 18 animals were preinjected with the inhibitors and subsequently with AK23 or NAK3 and two with PBS. Six animals were preinjected with vehicle followed by AK23, NAK3 or vehicle alone. Results obtained were identical per treatment group.
Human biopsies
Lesional and perilesional biopsies of skin, scalp and oral mucosa performed for clinical purposes were used according to the rules of the Medical Faculty. The diagnosis of PV (six patients), PF (two patients) and BP (five patients) relied on clinical, light microscopical, immunohistological (DIF) and serological (IIF) criteria. At the time of biopsy, all patients with autoimmune bullous disease showed positive antibody titers in IIF. Chronic eczematous dermatitis (two patients) was confirmed by light microscopy. Control biopsies were obtained from two healthy donors. All biopsies were fixed with 4% formaldehyde, paraffin embedded and consecutive sections of the same biopsy were used for immunofluorescence analyses. The PV biopsies were classified according to light microscopy in ‘old' lesions (patients 1 and 2), presenting with extensive suprabasal acantholysis and mixed inflammatory infiltrates, and ‘early' lesions (patients 3–6), exhibiting only focal acantholysis and/or eosinophilic spongiosis. For patient 6, a biopsy far from any blister was analyzed (nonlesional).
Sera and antibodies
Protein A-Sepharose-purified PVIgG contained antibodies against Dsg3 but not Dsg1 (Caldelari et al, 2001) or any other major protein (Supplementary Figure 4). nhIgG were derived from >1000 healthy donors (Sandoglobulin) and purified by Protein A-Sepharose. The affinity-purified mouse monoclonal anti-Dsg3 antibodies AK23 (Tsunoda et al, 2003) and NAK3 (M Amagai, unpublished) were kind gifts of M Amagai (Keio University School of Medicine, Tokyo, Japan). Primary antibodies were against the extracellular domain EC5 of Dsg3 (J Stanley, University of Philadelphia, Philadelphia, PA; Koch et al, 1998), Dsg1/2 (DG3.10 Progen), E-cadherin (gp-48, R Kemler, Max-Planck Institute, Freiburg, Germany), β-catenin and PG (Transduction Laboratories), Ki67 and lamin B1 (Zymed), p21WAF1 and cyclin D1 (BD PharMingen), tubulin-tyrosine (Sigma), c-Myc (Upstate Biotechnology, sc-764 Santa Cruz and BD PharMingen; the first was used for immunofluorescence analysis of human biopsies, the second for mouse biopsies and the first and third in combination for immunoblots) and Lef-1 (R Grosschedl, Max-Planck Institute, Freiburg, Germany; Travis et al, 1991).
Immunofluorescence analyses
Biopsies were deparaffinized and antigens retrieved for DIF by trypsinization (1 × trypsin (S2012 DAKO) in 0.1% CaCl (pH 7.8), 1.5 h at 37°C) and otherwise by microwaving three times 5 min at 720 W in 0.01 M sodium citrate buffer (pH 6.0). Nuclei were counterstained with Hoechst 33258 (H-1398, Molecular Probes). Lef-1 was revealed with the TSA-Plus Cyanine 3 enhancer kit (Perkin-Elmer Life Sciences) according to the manufacturer's protocol. Experimental procedures and microscopical processing as described (Caldelari et al, 2001) were held constant for the same antibody to obtain semiquantitative results.
Extraction of cellular proteins
Total cell lysates were obtained by scraping keratinocytes into SDS loading buffer. Cytoplasmic/low-detergent soluble membrane and nuclear extracts were prepared as described for keratinocytes by Corsini et al (1996) with some modifications. Briefly, cells were lysed with hypotonic lysis buffer containing 0.58% NP-40, scraped and centrifuged. The supernatant (referred to as cytoplasm/membrane fraction) was removed. Pelleted nuclei were resuspended with the aid of a 23 G needle and additionally centrifuged through a 0.85 M sucrose cushion for 15 min at 11 700 g to remove nucleus-associated endoplasmic reticulum and cellular debris. Western blot analysis was performed and the purity of nuclear fractions was confirmed by the absence of E-cadherin, Dsg3, α-catenin, keratin 14 and tyrosine tubulin from the extracts; lamin B1 was used as loading control.
Q-PCR
Total RNA was extracted and analyzed using TaqMan™ Q-PCR technology and primers designed (Supplementary Table 1) and validated as described previously (Kolly et al, 2005). All samples were normalized against cyclophilin S1 mRNA and results are reported as n-fold change relative to the sample at calcium switch. Analyses were carried out in duplicates of at least three independent experiments.
Proliferation assay
A total of 3 × 104 cells/cm2 were seeded into 8.8 cm2 culture dishes and incubated with PVIgG, nhIgG or high calcium medium alone as described (Caldelari et al, 2001). Attached cells were trypsinized and counted in a hemacytometer. Two independent experiments were carried out in triplicates.
Reporter gene assay
The assay was performed using the DualLuciferase™ Reporter Assay System (Promega, Wallisellen, Switzerland). One day after seeding, wild-type or β-catenin−/− cells were transfected with polyethylenimine (PEI, linear, MW-25 000; Polysciences Inc., Warrington, PA). Briefly, 50 μl medium was mixed with 2 μg of DNA per 15 μl of PEI (1 mg/ml), incubated for 8 min, mixed with 450 μl medium/10% FCS and added onto cells in six-well plates 24 h postseeding. After 2 h, cells were washed with PBS, and incubated with fresh medium for 24–28 h prior to lysis. In each case, 1.25 μg for reporter genes (kind gifts of E Fearon, Ann Arbor, MI (Kolligs et al, 2000) and H Clevers, Netherlands Institute for Developmental Biology, Utrecht, Netherlands (Molenaar et al, 1996)) were transfected and, unless stated otherwise, 0.4 μg of plasmids encoding Lef-1, Tcf-4, ΔLef-1 and PG (all kind gifts of R Kemler, Freiburg, Germany (Huber et al, 1996)), respectively, and deletion mutants of PG (kind gift A Ben-Ze'ev, Rehovot, Israel (Zhurinsky et al, 2000a)). In each case, vector was added to obtain a total of 0.8 μg plasmid encoding exogenous factors. Seven nanograms of renilla luciferase-encoding plasmid were co-transfected as normalizer for transfection efficiency.
ChIP
The ChIP assay was carried out according to Frank et al (2001). Briefly, cells were fixed with formaldehyde to crosslink DNA and associated proteins. Samples were sonicated to obtain DNA fragments between 500 and 1000 bp in length as confirmed by gel electrophoresis (data not shown). Prior to immunoprecipitation, samples were adjusted for DNA input as measured by photospectrometry and confirmed by Q-PCR. Each sample was split and immunoprecipitated with Lef-1, PG or β-catenin antibodies, or rabbit IgG as negative control. Thirty percent of the precipitated material was controlled by Western blotting, the remaining reverse crosslinked and the DNA subjected to Q-PCR using three sets of primers to the mouse c-Myc promoter (Figure 5A, Supplementary Table 1). The MatInspector program was used to define the TCF/LEF binding site in the mouse c-Myc promoter and primers to areas of interest were designed as described (Kolly et al, 2005). The primer express program (Applied Biosystems) did only allow one to design Q-PCR primers adjacent to the TCF/LEF binding site. Results are presented as relative change compared to the IgG control. The experiment was carried out three times with PVIgG- and nhIgG-treated wild-type mouse keratinocytes and two times with PG−/− cells.
Adhesion assay
The adhesion assay for PVIgG- and nhIgG-exposed keratinocytes was carried out as described (Yin et al, 2005b). Experiments were carried out in duplicates and repeated two times.
Statistical analysis
Using the Lilliefors test, normal distribution of data was excluded. Statistical significance (P<0.05) was then assessed by the Kruskal–Wallis test for multiple group comparisons, followed by the Mann–Whitney test for comparisons of two groups.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Table 1
Acknowledgments
Our special thanks go to Dr M Amagai (Keio University School of Medicine, Tokyo, Japan) who generously provided the mouse monoclonal antibodies AK23 and NAK3 and to all the researcher who generously gave us the tools used in this study. We are indebted to Dr P Lerch (ZLB-Behring, Bern) for support in PVIgG isolation, Drs K Hunziger (University of Kansas, Lawrence) and P Fernandez (Institute Animal Pathology, Vetsuisse Faculty Bern) for technical support in setting up the ChIP assay, M Wyder for excellent technical assistance and P Girling for editorial help. We also are indebted to Professors A Ben-Ze'ev (Weizmann Institute of Science, Department for Molecular Cell Biology, Rehovot, Israel) and R Kemler (Max-Planck Institute, Freiburg, Germany) for critical and helpful comments on the manuscript. This work was supported by the Martha Stiftung, Zürich, Switzerland (Reto Caldelari) and by the Swiss National Science Foundation Grant Nos. 31-59456.99 and 3100A0-107243.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) Beta-catenin is a target for the ubiquitin–proteasome pathway. EMBO J 16: 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberle H, Bierkamp C, Torchard D, Serova O, Wagner T, Natt E, Wirsching J, Heidkamper C, Montagna M, Lynch HTea (1995) The human plakoglobin gene localizes on chromosome 17q21 and is subjected to loss of heterozygosity in breast and ovarian cancers. Proc Natl Acad Sci USA 92: 6384–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altekrueger I, Ackerman AB (1994) ‘Eczema' revisited. A status report based upon current textbooks of dermatology. Am J Dermatopathol 16: 517–522; discussion 523–531 [PubMed] [Google Scholar]
- Amagai M, Klaus-Kovtun V, Stanley JR (1991) Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67: 869–877 [DOI] [PubMed] [Google Scholar]
- Andreadis ST, Hamoen KE, Yarmush ML, Morgan JR (2001) Keratinocyte growth factor induces hyperproliferation and delays differentiation in a skin equivalent model system. FASEB J 15: 898–906 [DOI] [PubMed] [Google Scholar]
- Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA (1982) Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 306: 1189–1196 [DOI] [PubMed] [Google Scholar]
- Aoyama Y, Kitajima Y (1999) Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol 112: 67–71 [DOI] [PubMed] [Google Scholar]
- Arnold I, Watt FM (2001) c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr Biol 11: 558–568 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG (2004) Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA 101: 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah SA, Frye M, Glogauer M, Watt FM (2005) Stem cell depletion through epidermal deletion of Rac1. Science 309: 933–935 [DOI] [PubMed] [Google Scholar]
- Beutner EH, Jordon RE (1964) Demonstration of skin autoantibodies in sera of pemphigus vulgaris patients by indirect immunofluorescence staining. Proc Soc Exp Biol Med 117: 505–510 [DOI] [PubMed] [Google Scholar]
- Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PF (2005) Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J Biol Chem 280: 32856–32865 [DOI] [PubMed] [Google Scholar]
- Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Muller E (2001) A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol 153: 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlotti A, Balaton AJ, de Muret A, Vaillant L, Wechsler J, Villela M (1993) Autoimmune pemphigus. A distinct staining pattern with an anti-desmoglein antibody. Arch Dermatol 129: 596–599 [DOI] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S (2005) LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132: 885–896 [DOI] [PubMed] [Google Scholar]
- Charpentier E, Lavker RM, Acquista E, Cowin P (2000) Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol 149: 503–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7: 793–803 [DOI] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Zhurinsky J, Ben-Ze'ev A (2002) The cadherin–catenin adhesion system in signaling and cancer. J Clin Invest 109: 987–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini E, Schubert C, Marinovich M, Galli CL (1996) Role of mitochondria in tributyltin-induced interleukin-1alpha production in murine keratinocytes. J Invest Dermatol 107: 720–725 [DOI] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B (2001) Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Gardner C, Li ER, Arnold I, Watt FM (2003) Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development 130: 2793–2808 [DOI] [PubMed] [Google Scholar]
- Fukuyama T, Ogita H, Kawakatsu T, Inagaki M, Takai Y (2006) Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene 25: 8–19 [DOI] [PubMed] [Google Scholar]
- Gandarillas A, Watt FM (1997) c-Myc promotes differentiation of human epidermal stem cells. Genes Dev 11: 2869–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A, Litterst CM, Huber O, Kemler R (1999) Functional characterization of multiple transactivating elements in beta-catenin, some of which interact with the TATA-binding protein in vitro. J Biol Chem 274: 18017–18025 [DOI] [PubMed] [Google Scholar]
- Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R (1996) Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev 59: 3–10 [DOI] [PubMed] [Google Scholar]
- Koch PJ, Mahoney MG, Cotsarelis G, Rothenberger K, Lavker RM, Stanley JR (1998) Desmoglein 3 anchors telogen hair in the follicle. J Cell Sci 111: 2529–2537 [DOI] [PubMed] [Google Scholar]
- Kodama S, Ikeda S, Asahara T, Kishida M, Kikuchi A (1999) Axin directly interacts with plakoglobin and regulates its stability. J Biol Chem 274: 27682–27688 [DOI] [PubMed] [Google Scholar]
- Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR, Fearon ER (2000) Gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev 14: 1319–1331 [PMC free article] [PubMed] [Google Scholar]
- Kolly C, Suter MM, Muller EJ (2005) Proliferation, cell cycle exit, and onset of terminal differentiation in cultured keratinocytes: pre-programmed pathways in control of C-Myc and Notch1 prevail over extracellular calcium signals. J Invest Dermatol 124: 1014–1025 [DOI] [PubMed] [Google Scholar]
- Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR (1999) Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceous and vulgaris. J Clin Invest 103: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missero C, Di Cunto F, Kiyokawa H, Koff A, Dotto GP (1996) The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev 10: 3065–3075 [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H (1996) XTcf-3 transcription factor mediates b-catenin-induced axis formation in Xenopus embryos. Cell 86: 391–399 [DOI] [PubMed] [Google Scholar]
- Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, Sargin B, Kohler G, Stelljes M, Puccetti E, Ruthardt M, deVos S, Hiebert SW, Koeffler HP, Berdel WE, Serve H (2004) Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol 24: 2890–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann C, Owens DM, Hulsken J, Birchmeier W, Watt FM (2002) Expression of DeltaNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development 129: 95–109 [DOI] [PubMed] [Google Scholar]
- Owens DM, Watt FM (2003) Contribution of stem cells and differentiated cells to epidermal tumours. Nat Rev Cancer 3: 444–451 [DOI] [PubMed] [Google Scholar]
- Payne AS, Hanakawa Y, Amagai M, Stanley JR (2004) Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol 16: 536–543 [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Littlewood T, Khan M, Elia G, Evan G (1999) Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell 3: 565–577 [DOI] [PubMed] [Google Scholar]
- Posthaus H, Williamson L, Baumann D, Kemler R, Caldelari R, Suter MM, Schwarz H, Müller E (2002) β-Catenin is not required for proliferation and differentiation of epidermal mouse keratinocytes. J Cell Sci 115: 4587–4597 [DOI] [PubMed] [Google Scholar]
- Sato M, Aoyama Y, Kitajima Y (2000) Assembly pathway of desmoglein 3 to desmosomes and its perturbation by pemphigus vulgaris-IgG in cultured keratinocytes, as revealed by time-lapsed labeling immunoelectron microscopy. Lab Invest 80: 1583–1592 [DOI] [PubMed] [Google Scholar]
- Shirakata Y, Amagai M, Hanakawa Y, Nishikawa T, Hashimoto K (1998) Lack of mucosal involvement in pemphigus foliaceous may be due to low expression of desmoglein 1. J Invest Dermatol 110: 76–78 [DOI] [PubMed] [Google Scholar]
- Suter MM, Pantano DM, Flanders JA, Augustin-Voss HG, Dougherty EP, Varvayanis M (1991) Comparison of growth and differentiation of normal and neoplastic canine keratinocyte cultures. Vet Pathol 28: 131–138 [DOI] [PubMed] [Google Scholar]
- Tao L, Kramer PM, Wang W, Yang S, Lubet RA, Steele VE, Pereira MA (2002) Altered expression of c-myc, p16 and p27 in rat colon tumors and its reversal by short-term treatment with chemopreventive agents. Carcinogenesis 23: 1447–1454 [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426 [DOI] [PubMed] [Google Scholar]
- Teuliere J, Faraldo MM, Shtutman M, Birchmeier W, Huelsken J, Thiery JP, Glukhova MA (2004) Beta-catenin-dependent and -independent effects of DeltaN-plakoglobin on epidermal growth and differentiation. Mol Cell Biol 24: 8649–8661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis A, Amsterdam A, Belanger C, Grosschedl R (1991) LEF-1, a gene encoding a lymphoid-specific protein with an HMG domain, regulates T-cell receptor alpha enhancer function [corrected] [published erratum appears in Genes Dev 1991 Jun; 5(6):following 1113]. Genes Dev 5: 880–894 [DOI] [PubMed] [Google Scholar]
- Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M (2003) Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol 170: 2170–2178 [DOI] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR (2001) Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 28: 165–168 [DOI] [PubMed] [Google Scholar]
- Waikel RL, Wang XJ, Roop DR (1999) Targeted expression of c-Myc in the epidermis alters normal proliferation, differentiation and UV-B induced apoptosis. Oncogene 18: 4870–4878 [DOI] [PubMed] [Google Scholar]
- Wan H, Stone MG, Simpson C, Reynolds LE, Marshall JF, Hart IR, Hodivala-Dilke KM, Eady RA (2003) Desmosomal proteins, including desmoglein 3, serve as novel negative markers for epidermal stem cell-containing population of keratinocytes. J Cell Sci 116: 4239–4248 [DOI] [PubMed] [Google Scholar]
- Watt FM (2001) Stem cell fate and patterning in mammalian epidermis. Curr Opin Genet Dev 11: 410–417 [DOI] [PubMed] [Google Scholar]
- Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Muller EJ, Green KJ (2005a) Mechanisms of Plakoglobin-dependent adhesion: desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem 280: 40355–40363 [DOI] [PubMed] [Google Scholar]
- Yin T, Getsios S, Caldelari R, Kowalczyk AP, Muller EJ, Jones JC, Green KJ (2005b) Plakoglobin suppresses keratinocyte motility through both cell–cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci USA 102: 5420–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Giap C, Lazo JS, Prochownik EV (2003) Low molecular weight inhibitors of Myc–Max interaction and function. Oncogene 22: 6151–6159 [DOI] [PubMed] [Google Scholar]
- Zhurinsky J, Shtutman M, Ben-Ze'ev A (2000a) Plakoglobin and β-catenin: protein interactions, regulation and biological roles. J Cell Sci 113 (Part 18): 3127–3139 [DOI] [PubMed] [Google Scholar]
- Zhurinsky J, Shtutman M, Ben-Ze'ev A (2000b) Differential mechanisms of LEF/TCF family-dependent transcriptional activation by beta-catenin and plakoglobin. Mol Cell Biol 20: 4238–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Table 1