

Abstract
γ-Secretase is an unusual protease with an intramembrane catalytic site that cleaves many type I membrane proteins, including the amyloid β-protein (Aβ) precursor (APP) and the Notch receptor. Genetic and biochemical studies have identified four membrane proteins as components of γ-secretase: heterodimeric presenilin composed of its N- and C-terminal fragments, nicastrin, Aph-1, and Pen-2. Here we demonstrated that certain compounds, including protein kinase inhibitors and their derivatives, act directly on purified γ-secretase to selectively block cleavage of APP- but not Notch-based substrates. Moreover, ATP activated the generation of the APP intracellular domain and Aβ, but not the generation of the Notch intracellular domain by the purified protease complex, and was a direct competitor of the APP-selective inhibitors, as were other nucleotides. In accord, purified γ-secretase bound specifically to an ATP-linked resin. Finally, a photoactivable ATP analog specifically labeled presenilin 1-C-terminal fragments in purified γ-secretase preparations; the labeling was blocked by ATP itself and APP-selective γ-secretase inhibitors. We concluded that a nucleotide-binding site exists within γ-secretase, and certain compounds that bind to this site can specifically modulate the generation of Aβ while sparing Notch. Drugs targeting the γ-secretase nucleotide-binding site represent an attractive strategy for safely treating Alzheimer disease.
Alzheimer disease is characterized by the progressive accumulation of amyloid β-protein (Aβ)3 in brain regions subserving memory and cognition (1). Sequential proteolytic cleavages of the amyloid β-protein precursor (APP) by the β- and γ-secretases generate the amyloid β-protein (Aβ) (1). β-Secretase is a single membrane-spanning aspartyl pro-tease expressed at high levels in neurons (2). γ-Secretase is also an aspartyl protease but with an unprecedented intramembranous catalytic site (3, 4) that is required for the cleavage of a wide range of type I membrane proteins that include APP and the Notch receptors (for a review see Ref. 5). We recently reported a specific and reproducible procedure for the high grade purification of active human γ-secretase and characterized various factors that affect its activity in vitro (6). In further investigating the properties of the purified enzyme, we have observed that ATP can activate purified γ-secretase in vitro by up to 2-fold. This observation is in agreement with the recent report of Netzer et al. (7) that γ-secretase-mediated generation of Aβ in a mouse N2a neuroblastoma cell-free system is ATP-dependent. These authors also found that imatinib mesylate (Gleevec, formerly STI571), a selective Abl kinase inhibitor approved to treat chronic myelogenous leukemia (8–10), inhibited γ-secretase cleavage of APP without affecting Notch processing in an N2a cell-free system, in intact N2a cells expressing human APP, and in rat primary neurons (7). Another compound with a pyrido-(2,3-d)pyrimidine structure (called inhibitor 2 (7) or PD173955 (11)) showed a similar effect (7). Both Gleevec and inhibitor 2 are known to inhibit Abl kinase by targeting its ATP-binding site (12–16), but cells deficient in this enzyme were still sensitive to Gleevec with respect to lowering Aβ production (7). The mechanism by which these two compounds affect γ-secretase cleavage of APP is unknown. Because Gleevec and inhibitor 2 target several protein-tyrosine kinases besides Abl, Netzer et al. (7) suggested that platelet-derived growth factor receptor, Src kinase, or c-kit might be involved. Another proposed mechanism involves an effect on the localization of γ-secretase or APP in a way that prevents interaction of enzyme with substrate.
A central concern about inhibiting γ-secretase to lower Aβ production in AD is that this should also interfere with Notch processing and lead to severe toxicity because of interference with cell differentiation. Indeed, significant adverse effects of γ-secretase inhibitors caused by blocking Notch signaling have been described in preclinical animal studies (17–20). Because Netzer et al. (7) showed that Gleevec and inhibitor 2 inhibited APP but not Notch cleavage in their systems, we investigated the effects of selected protein-tyrosine kinase inhibitors on Aβ production and on Notch substrate cleavage using isolated, purified γ-secretase.
MATERIALS AND METHODS
Cell Lines and Cultures
HeLa S3 cells, the Chinese hamster ovary (CHO) γ-30 cell line (co-expressing human PS1, FLAG-Pen-2, and Aph1α2-HA), and the S-1 CHO cell line (co-expressing human PS1, FLAG-Pen-2, Aph1α2-HA, and NCT-GST) were cultured as described previously (6, 21, 22).
Purification of γ-Secretase and in Vitro γ-Secretase Assays
The multistep procedure for the high grade purification of human γ-secretase from the S-1 cells was performed as described previously (6). In vitro γ-secretase assays using the recombinant APP-based substrate C100FLAG and the recombinant Notch-based substrate N100FLAG were performed as reported previously (4, 21). Basically, the proteolytic reaction mixtures contained C100FLAG and N100FLAG substrate at a concentration of 1 μm, purified γ-secretase solubilized in 0.2% CHAPSO/HEPES, pH 7.5, at 10-fold dilution from stock (stock = the M2 anti-FLAG-eluted fraction in the purification protocol from S-1 cells (6)), 0.025% phosphatidylethanolamine (PE), and 0.10% phosphatidylcholine (PC). All the reactions were stopped by adding 0.5% SDS, and the samples were assayed for Aβ40 and Aβ42 by ELISA as described (23). The capture antibodies were 2G3 (to Aβ residues 33–40) for the Aβ40 species and 21F12 (to Aβ residues 33–42) for the Aβ42 species.
Inhibitors
Powder containing Gleevec (Novartis) was dissolved from capsules or tablets in a mixture composed of ethyl acetate and aqueous saturated sodium bicarbonate solution. The organic layer was washed several times with brine, dried on sodium sulfate, and evaporated under vacuum. Gleevec was purified and analyzed by reverse phase-HPLC using a Vydac C18 preparative column (10 μm, 2.2 × 25 cm) and a C18 analytical column (5 μm, 0.46 × 25 cm), respectively. Chromatographic separations were performed at a flow of 1.5 ml/min, with a gradient of 0–100% MeOH in water over 30 min. This isolated material is referred to as “Gleevec extract.” Purified Gleevec was from Sequoia Research Products, UK. Final purity and characterization of the two Gleevec extracts (from capsules and tablets, respectively) and the purified Gleevec (Sequoia Research Products, UK) were performed by MALDI-TOF mass spectroscopy (Applied Biosystems Voyager System 4036). Gleevec was detected with a m/z of (M + H)+ = 494 g/mol (expected m/z of (M + H)+ = 493 g/mol). The well characterized γ-secretase inhibitor III-31C was prepared as described previously (4). Bryostatin-1 was purchased from Biomol. All compounds listed in Fig. 4 were purchased from TOCRIS-UK; the structure and action of these compounds are described in TABLE ONE (modified from TOCRIS-UK in the Supplemental Material). All inhibitors were added to the reaction mixtures from a Me2SO stock (final Me2SO concentration of 1%, which alone did not affect γ-secretase activity).
FIGURE 4. ZM39923 (1367), a potent Janus kinase 3 inhibitor, preferentially blocks the generation of Aβ by purified γ-secretase.
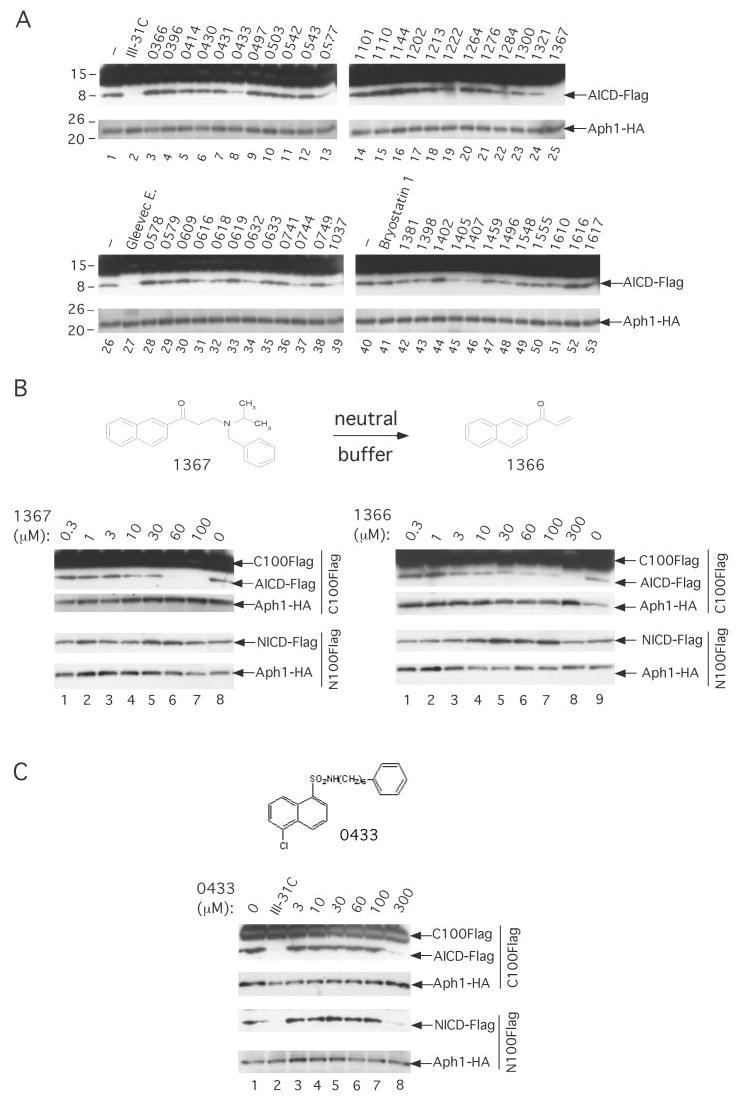
A, effect of a large number of protein kinase/phosphatase inhibitors or activators on purified γ-secretase activity. γ-Secretase diluted in 0.2% CHAPSO/HEPES, pH 7.5, was incubated at 37 °C for 16 h in the presence of 1 μm C100FLAG, 0.1% PC, 0.025% PE, and 100 μm of the indicated compounds, except for III-31C (10 μm). The generation of AICD-FLAG was probed by Western blotting with M2 anti-FLAG antibody. B, effect of ZM39923 (1367) and its break-down product ZM449829 (1366) (structures shown) on the cleavage by purified γ-secretase of C100FLAG and N100FLAG. Activity assays were performed as described above in the presence of the indicated concentrations of 1367 and 1366, and the generation of AICD-FLAG and NICD-FLAG was probed by Western blotting with M2 and Notch Ab1744 antibody, respectively. In all figures, levels of Aph1-HA are shown as equal loading controls. C, similarly, the effect of SC-9 (0433) (structure shown) on the cleavage by purified γ-secretase of C100FLAG and N100FLAG was probed.
Western Blotting and Antibodies
For Western analysis of PS1-NTF, PS1-CTF, Aph1α2-HA, FLAG-Pen-2, and NCT-GST, the samples were run on 4–20% Tris-glycine polyacrylamide gels, transferred to polyvinylidene difluoride, and probed with Ab14 (for PS1-NTF, 1:2000; a gift of S. Gandy), 13A11 (for PS1-CTF, 5 μg/ml; a gift of Elan Pharmaceuticals), 3F10 (for Aph1α2-HA, 50 ng/ml; Roche Applied Science), anti-FLAG M2 (for FLAG-Pen-2, 1:1000; Sigma), or αGST antibodies (for NCT-GST, 1:3000; Sigma). Samples from the γ-secretase activity assays (above) were run on 4–20% Tris-glycine gels and transferred to polyvinylidene difluoride membranes to detect AICD-FLAG with anti-FLAG M2 antibodies (1:1000, Sigma) and NICD-FLAG with Notch Ab1744 antibody (1:1000, Cell Signaling Technology), which is selective for the N terminus of NICD; the same samples were transferred to nitrocellulose membranes to detect Aβ with the anti-Aβ 6E10 antibody. Levels of AICD-FLAG and NICD-FLAG were estimated by densitometry using AlphaEase/Spot Denso (Alpha Innotech Corp.).
Purified γ-Secretase and Binding to ATP-immobilized Resins
The purified γ-secretase was diluted 10-fold from stock (6) in 50 mm HEPES buffer, pH 7.0, containing 0.2 or 1% CHAPSO, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2 and incubated overnight, in the presence or absence of 50 mm ATP (Sigma), with ATP-agarose (ATP attached to agarose through the ribose hydroxyls, Sigma catalog number A-4793) or ATP-acrylamide (ATP attached to acrylamide through the γ-phosphate; Novagen catalog number 71438-3). Each resin was washed three times with 0.2 or 1% CHAPSO/HEPES buffer, and the bound proteins were collected in 2× Laemmli sample buffer, resolved on 4–20% Tris-glycine gels, and transferred to polyvinylidene difluoride membranes to detect NCT-GST, PS1-NTF, Aph1-HA, PS1-CTF, and FLAG-Pen2 as described above.
Photoaffinity Labeling Experiments
8-Azido-[γ-32P]ATP (18 Ci/ mmol) was purchased from Affinity Labeling Technology (Lexington, KY). For the photoaffinity labeling of the purified γ-secretase, the enzyme was diluted 10-fold from stock (6) in 50 mm HEPES buffer, pH 7.0, containing 0.2% CHAPSO, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2, 0.025% PE, and 0.10% PC. The samples were exposed to UV light for 5 min (hand-held UV lamp at 254 nm; UVP model UVGL-25) on ice, and the reaction was quenched with 1 mm dithiothreitol. The proteins were diluted in 0.5% CHAPSO/HEPES buffer and incubated overnight for affinity precipitation with GSH resin as described previously (6, 22). The unbound nucleotides were removed by washing the resin three times and then the washed proteins were resuspended in Laemmli sample buffer. For the photoaffinity labeling of the purified γ-secretase followed by the BN-PAGE analysis, the enzyme was diluted in 0.1% digitonin/ TBS, exposed to UV light for 5 min, and directly loaded onto a 5–13.5% BN-polyacrylamide gel. For the photoaffinity labeling of endogenous γ-secretase, HeLa S3 membranes (the equivalent of 3.0 × 108 cells) were incubated with 22.5 μm 8-azido-[γ-32P]ATP (10 μCi per reaction), 50 mm HEPES, pH 7.0, 150 mm NaCl, 5 mm MgCl2, and 5 mm CaCl2 in a total volume of 60 μl for 10 min at 37 °C. The resuspended membranes were exposed to UV light as described above. The unbound nucleotides were removed by washing the membranes three times and then the washed membranes were resuspended for 1 h in 0.5 ml of 1% CHAPSO/ HEPES, pH 7.4. The solubilized proteins were diluted 1:2 in HEPES buffer (final CHAPSO concentration = 0.5%) and incubated overnight with X81 antibody for immunoprecipitation, as described previously (6, 22). Samples were electrophoresed on 4–20% Tris-glycine gels and autoradiographed (BioMax MS films used with BioMax Transcreen HE (Eastman Kodak Co.)).
ATPase Assays
[α-32P]ATP (11.9 Ci/mmol) was purchased from Affinity Labeling Technology (Lexington, KY). The purified γ-secretase was prepared as described for the photoaffinity labeling experiments; 5 μCi of [α-32P]ATP was added; the reactions were incubated at 37 °C, and at the indicated time points aliquots were removed and reactions stopped by addition of 10% SDS. A total of 2 μl of each stopped reaction was analyzed by TLC on polyethyleneimine cellulose plastic sheets (Baker-Flex, Germany) with 0.75 m KH2PO4, pH 3.5, as the running buffer to separate ATP from ADP. To identify hydrolysis products, a reaction of [α-32P]ATP incubated in the presence of 0.005 units of canine kidney phosphatase (Sigma) was loaded. Samples were autoradiographed as described above.
RESULTS
Nonhydrolyzed ATP Can Activate the Generation of AICD and Aβ but Not the Generation of NICD by Purified γ-Secretase
We took advantage of our highly purified γ-secretase complexes to investigate factors that might affect the cleavage of APP, using a C100FLAG substrate consisting of the β-CTF (C99) portion of APP (amino acids 596–695) plus a Met at the N terminus and a FLAG tag at the C terminus (4, 24). We observed that increased concentrations of ATP promoted a dose-dependent rise in the production of both AICD-FLAG (Fig. 1A, compare lanes 2–6 with lane 1) and Aβ (both Aβ40 and Aβ42), reaching an ~1.75-fold increase at 1–5 mm as estimated by Aβ ELISA (Fig. 1B). Densitometry of the AICD-FLAG bands (Fig. 1A) showed that the fold increases were similar to those seen for Aβ by ELISA in Fig. 1B. These findings are in good agreement with the reported data of Netzer et al. (7), which showed that the addition of 1–3 mm ATP in a cell-free membrane preparation (derived from mouse N2a cells expressing human APP-695) results in an ~2-fold increase in Aβ production. Next, we addressed whether ATP can stimulate the cleavage of a Notch-based substrate (N100FLAG) in a similar manner and under the same conditions as for C100FLAG. N100FLAG is an analogous Notch-based substrate corresponding to residues Val-1711 to Glu-1809 from the mouse Notch-1 receptor (plus a Met and a FLAG tag at the N and C termini, respectively, just like C100) (21). We found that increased concentrations of ATP did not alter the generation of NICD-FLAG (Fig. 1A, compare lanes 2–6 with lane 1), and this was confirmed by densitometry.
FIGURE 1. Nonhydrolyzed ATP can activate the in vitro generation of Aβ40 and Aβ42.
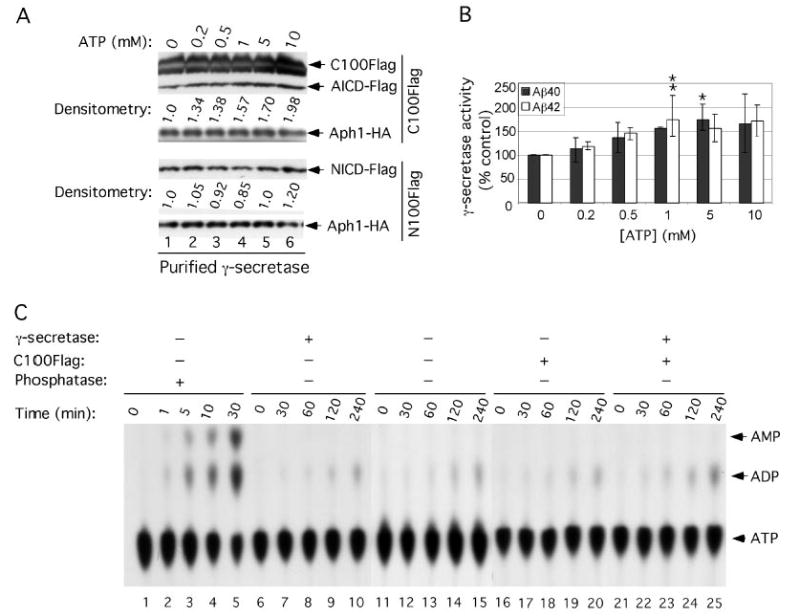
A, effect of ATP on purified γ-secretase activity. γ-Secretase diluted in 0.2% CHAPSO/HEPES, pH 7.5, was incubated at 37 °C for 4 h in the presence of 0.1% PC, 0.025% PE, the indicated concentrations of ATP, and 1 μm C100FLAG (an APP-based substrate) or 1 μm N100FLAG (a Notch-based substrate); both substrates were adjusted to 0.5% SDS prior to addition to the reactions (6). The reactions were Western-blotted for AICD-FLAG (with M2 anti-FLAG antibody) and for NICD-FLAG (with Notch Ab1744 antibody). Levels of Aph1-HA serve as equal loading controls. Aβ40 and Aβ42 were measured by ELISA (B, means ± S.D.; n = 3). Levels of AICD-FLAG and NICD-FLAG were estimated by densitometry (values are single determinations from the blot shown). Asterisks indicate significant differences in Aβ40 (*, p < 0.01) and Aβ42 (**, p < 0.01) production compared with samples without ATP. C, ATPase assays on purified γ-secretase. [α-32P]ATP was incubated at 37 °C for the indicated times in the reaction buffer (0.2% CHAPSO/HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2, 0.025% PE, and 0.10% PC) alone (lanes 11–15) or in the presence of purified γ-secretase (lanes 6 –10), C100FLAG substrate (lanes 16 –20), or both purified γ-secretase and C100FLAG substrate (lanes 21–25). Two μl of each reaction were then analyzed by TLC to separate ATP from ADP. As a positive control to show ATP hydrolysis products, [α-32P]ATP was incubated at the indicated times in the presence of canine kidney phosphatase (lanes 1–5).
ATP stores energy in the form of a chemical bond and releases it in the process of hydrolysis, providing a readily available energy supply for many enzymatic reactions. Thus, we intended to determine whether the peptidase activity of γ-secretase is associated with ATP hydrolysis. As shown in Fig. 1C, the purified γ-secretase incubated in the presence (lanes 21–25) or absence (lanes 6–10) of C100FLAG substrate did not increase the hydrolysis of [α-32P]ATP into [α-32P]ADP when compared with the hydrolysis occurring in the reaction buffer alone (lanes 11–15). This result indicates that ATP hydrolysis is not required for the peptidase activity of γ-secretase. We further found that 1 mm levels of the nonhydrolyzable ATP analog, ATPγS, also resulted in an ~1.35 increase in the generation of Aβ40 and Aβ42 from C100FLAG substrate by purified γ-secretase (data not shown), supporting our observation that ATP hydrolysis is not required for the effect.
Gleevec Itself Is Not a Direct γ-Secretase Inhibitor; However, a Gleevec Extract Inhibits the Generation of Aβ by Purified γ-Secretase Without Affecting the Cleavage of a Notch-based Recombinant Substrate
Because ATP activated the purified γ-secretase complex (Fig. 1, A and B), we used this preparation to examine the effects of a Gleevec extract (prepared from capsules and characterized as described in detail under “Materials and Methods”) on the cleavage efficiency of C100FLAG substrate. We first confirmed that III-31C (4) and DAPT (25), two well characterized inhibitors of γ-secretase, inhibited both C100FLAG and N100FLAG cleavage by our purified γ-secretase with similar potencies (III-31C, IC50 of ~50 nM for cleavage of C100FLAG, as estimated by ELISA and densitometry, and ~100 nm for cleavage of N100FLAG, as estimated by densitometry; DAPT, IC50 of ~100 nM for cleavage of C100FLAG, as estimated by ELISA and densitometry, and ~100 nm for cleavage of N100FLAG, as estimated by densitometry) (Fig. 2A, and ELISA data not shown). We then probed the effects of our Gleevec extract (extracted from capsules and isolated by HPLC as described under “Materials and Methods”) on Aβ production by the purified γ-secretase. The cleavage products, Met-Aβ40 and Met-Aβ42, (which we designate Aβ40 and Aβ42 for simplicity) were quantified by ELISA (Fig. 2B) and also detected by blotting with the 6E10 anti-Aβ antibody (Fig. 2C). The other proteolytic product, FLAG-tagged AICD, was detected with anti-FLAG M2 antibodies (Fig. 2C). Our Gleevec extract inhibited the generation of Aβ40, Aβ42, and AICD in a concentration-dependent fashion and with a similar potency, yielding an approximate IC50 value (estimated by Aβ ELISA) of ~75 μm (Fig. 2, B and C). Next, we examined the effects of our Gleevec extract on the cleavage of N100FLAG by the purified protease. The Gleevec extract did not inhibit the generation of NICD-FLAG, even at concentrations >10-fold the IC50 value for the generation of Aβ40 and Aβ42 from C100FLAG substrate (i.e. at 1 mm) (Fig. 2C, lane 7). Similarly, the Gleevec extract was found to inhibit the generation of Aβ by endogenous γ-secretase solubilized from HeLa cell membranes without affecting the cleavage of N100FLAG substrate (Fig. 2D). Because pH is an important factor modulating the activity of the purified γ-secretase (6), the pH of all the above reactions was checked and found to be consistently at pH 7.4, even at high concentrations (1 mm) of the Gleevec extract.
FIGURE 2. Gleevec itself is not a direct γ-secretase inhibitor, but a Gleevec extract inhibits the generation of Aβ by purified γ-secretase without affecting the cleavage of a Notch-based substrate.
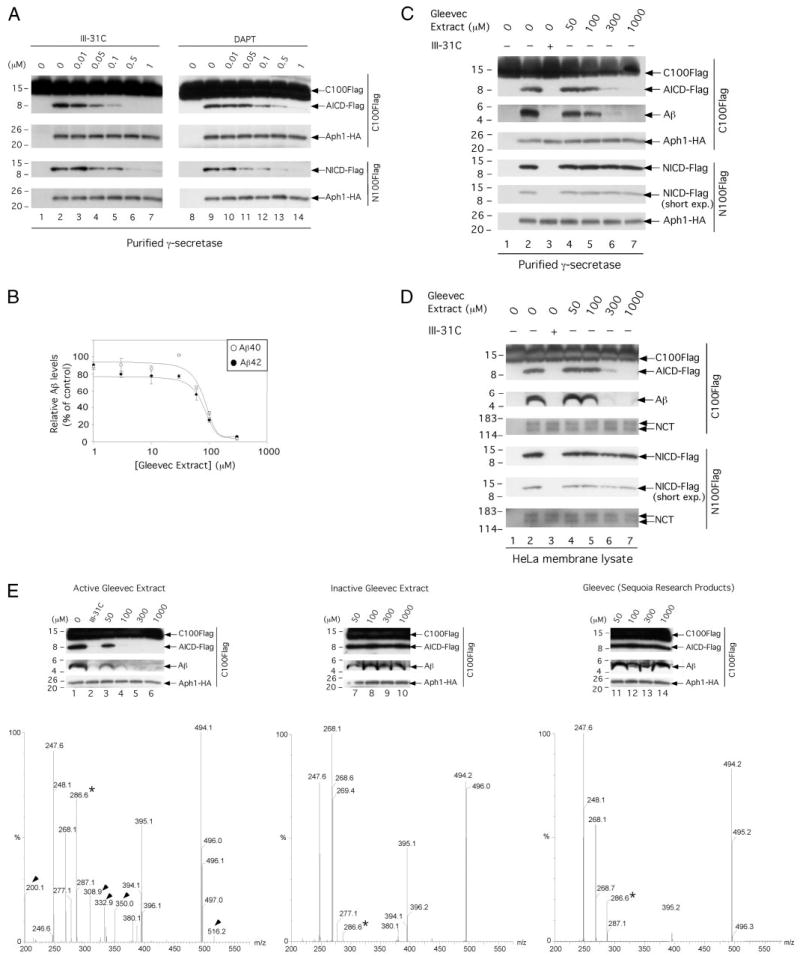
A, effect of III-31C and DAPT on the cleavage by purified γ-secretase of C100FLAG and N100FLAG. γ-Secretase diluted in 0.2% CHAPSO/HEPES, pH 7.5, was incubated at 37 °C for 4 h with 1 μm C100FLAG or N100FLAG substrate, 0.1% PC, 0.025% PE, and the indicated concentrations of III-31C and DAPT. The reactions were Western-blotted for AICD-FLAG (M2 antibody) and NICD-FLAG (Ab1744 antibody). Levels of Aph1-HA serve as equal loading controls. B, effect of Gleevec extract on Aβ40 and Aβ42 generation by purified γ-secretase. γ-Secretase diluted in 0.2% CHAPSO/HEPES, pH 7.5, was incubated at 37 °C for 4 h with 1 μm C100FLAG substrate, 0.1% PC, 0.025% PE, and the indicated concentrations of Gleevec extract. Aβ40 and Aβ42 were measured by ELISA (means ± S.D., n = 3). C, effects of Gleevec extract on C100FLAG and N100FLAG cleavage by purified γ-secretase. Parallel reaction mixtures in the presence of 1 μm C100FLAG or N100FLAG, 10 μm III-31C, or the indicated concentrations of the Gleevec extract were incubated at 37 °C for 4 h and Western-blotted for AICD-FLAG (M2), Aβ (6E10), and for NICD-FLAG (Ab1744); short exposures (short exp.) are also shown to validate the lack of effect on NICD-FLAG levels. Levels of Aph1-HA serve as equal loading controls. D, effect of Gleevec extract on C100FLAG and N100FLAG cleavage by endogenous γ-secretase solubilized from HeLa membranes. γ-Secretase assays and the generation of AICD-FLAG, Aβ, and NICD-FLAG were performed as in C. Levels of NCT serve as loading controls. E, effect of three different Gleevec samples on C100FLAG cleavage by purified γ-secretase. γ-Secretase was incubated as described in B with C100FLAG and the indicated concentrations of Gleevec extracted from capsules (left panel), Gleevec extracted from tablets (middle panel), or purified Gleevec (right panel). The reactions were Western-blotted for AICD-FLAG and Aβ. Levels of Aph1-HA serve as equal loading controls. Final purity and characterization of the three Gleevec samples was performed by MALDI-TOF mass spectroscopy; the compounds identified specifically in the active Gleevec extract (left panel) are labeled with arrowheads. Note that a very minor peak at 286.6 in the inactive extract is a major peak in the active extract (asterisk).
Because several other compounds (impurities or degradation products probably generated during the extraction and purification procedures, m/z of 200.1, 247.6, 268.2, 277.1, 286.6, 308.9, 332.9, 350.0, 380.1, 395.1, and 516.2) were detected in the Gleevec preparation (Fig. 2E, left panel and data not shown), we decided to examine the effect on the purified γ-secretase of two additional Gleevec samples prepared from two different sources as follows: Gleevec extracted from tablets as described in detail under “Materials and Methods” and purified Gleevec (Sequoia Research Products, UK). Final purity of the two Gleevec samples described above was addressed by MALDI-TOF mass spectroscopy (Fig. 2E, middle and right panels, respectively). We found that these two Gleevec samples did not inhibit the generation of AICD, Aβ40, and Aβ42 by the purified γ-secretase (Fig. 2E, middle and right panels, respectively, and ELISA data not shown). Because Gleevec (m/z of (M + H)+ = 494) was found in the three different Gleevec samples, our data strongly suggest that Gleevec is not the active compound found in the Gleevec preparation extracted from capsules. Most interestingly, the analyses by mass spectroscopy of the three Gleevec samples revealed several compounds (m/z of 200.1, 308.9, 332.9, 350.0, and 516.2) that were only present in the active Gleevec preparation (Fig. 2E, the compounds identified specifically in the active Gleevec preparation are labeled with arrowheads), leaving open the possibility that one or more of those compounds are active toward γ-secretase. Also, a very minor peak at 286.6 in the inactive extract is a major peak in the active extract (Fig. 2E, asterisk). Although purification and characterization of each of the compounds found in our active Gleevec extract will now be necessary for the identification of the active compound, our data show the following: 1) Gleevec itself is not a direct γ-secretase inhibitor, and 2) the generation of Aβ from γ-secretase can be inhibited directly and selectively without affecting Notch proteolysis.
Next, we analyzed extracellular Aβ40 and Aβ42 levels after treatment of CHO cells stably expressing human APP with purified Gleevec (Sequoia Research Products, UK). Consistent with the previous report of Netzer et al. (7), treatment of cells with 10 μm purified Gleevec reduced secreted Aβ40 and Aβ42 by ~50% (data not shown). Based on this observation, an attractive hypothesis is that Gleevec breaks down in cells to form the active compound found in the Gleevec preparation extracted from capsules.
Nucleotides Are Direct Competitors of the Active Compound in the Gleevec Extract on γ-Secretase
Because ATP activated the cleavage of an APP-based but not a Notch-based substrate and because Gleevec binds to the ATP-binding site of protein kinases (26, 27), we compared ATP and other nucleotides (ADP, AMP, GTP, CTP, and TTP) as potential competitors with respect to the active Gleevec extract, reasoning that the active Gleevec derivative may interact with a nucleotide-binding site on γ-secretase. Again, all nucleotide stock solutions were neutralized to pH 7.4 before use. We found that the active Gleevec extract at 100 μm inhibited ~70% of the generation of both Aβ40 and Aβ42 and accordingly decreased AICD-FLAG generation, when compared with levels observed in the absence of compound (Fig. 2B and Fig. 3A, compare lanes 1 and 2 (top panel) and B and C). In the presence of the active Gleevec extract at 100 μm, increasing concentrations of ATP (from 0.1 to 5 μm) restored γ-secretase activity in a concentration-dependent fashion to ~100% of that observed without the Gleevec extract (Fig. 3, A, compare lanes 2–6 with lane 1 in the top panel; also, B and C). These highly consistent results suggest that ATP is a direct competitor of the active compound present in the Gleevec extract on purified γ-secretase. Most interestingly, ADP and AMP also prevented the inhibitory effect of the Gleevec extract, but with slightly less potency than ATP. For example, in the presence of 1 mm ATP, ADP, or AMP, the Gleevec extract at 100 μm inhibited ~25, 35, and 50% of the generation of Aβ40, respectively, and ~25, 42, and 62% of the generation of Aβ42, respectively (Fig. 3, B and C). Next, we assessed whether GTP, CTP, and TTP can act as direct competitors with respect to the Gleevec extract. We found that both GTP and TTP were potent Gleevec extract competitors, whereas CTP was not, even at 5 mm (data not shown). Taken together, our results yield the following order of potency of competition against Gleevec extract: ATP = GTP > ADP = TTP > AMP > CTP. Moreover, they suggest a common binding site on γ-secretase for the active compound in the Gleevec extract and these nucleotides.
FIGURE 3. Nucleotides prevent the inhibitory effect of Gleevec extract on purified γ-secretase.

A, ATP, ADP, and AMP are direct competitors with respect to the Gleevec extract. γ-Secretase diluted in 0.2% CHAPSO/HEPES, pH 7.5, was incubated at 37 °C for 16 h in the absence (lane 1) or presence (lanes 2– 6) of 100 μm Gleevec extract and the indicated concentrations of ATP, ADP, or AMP. The generation of AICD-FLAG was probed by Western blotting with M2 anti-FLAG. Levels of Aph1-HA served as equal loading controls. Note that the lane 1 control without ATP is the same control for the absence of ADP or AMP. B and C, the effects of increasing concentrations of ATP, ADP, and AMP on Aβ40 (B) and Aβ42 (C) generation by purified γ-secretase in the presence of 100 μm Gleevec extract (reactions performed at 37 °C for 4 h) were quantified by ELISA (n = 3).
ZM39923 (1367), a Potent Janus Kinase 3 Inhibitor, Preferentially Blocks APP Cleavage by Purified γ-Secretase
On the basis of the results described above, the Gleevec extract contains a direct competitive inhibitor of γ-secretase with respect to the cleavage of APP to Aβ that does not noticeably affect Notch cleavage. Because the active compound is apparently an impurity or degradation product of Gleevec (a potent tyrosine kinase inhibitor known to compete with ATP-binding sites (26, 27)) and because ATP is a direct competitor of the active compound in the Gleevec extract, we decided to screen ~50 well characterized protein kinase inhibitors (most of them known to compete with the ATP-binding site on their cognate targets) for their ability to inhibit C100FLAG cleavage by purified γ-secretase. All of the reactions contained 1 μm C100FLAG substrate, purified γ-secretase solubilized in 0.20% CHAPSO/HEPES, pH 7.5, at 10-fold dilution from stock (6), 0.025% phosphatidylethanolamine (PE), 0.10% phosphatidylcholine (PC), and 100 μm of the respective compound. The concentration of 100 μm for each compound was chosen to attempt to identify inhibitors more potent than the active Gleevec extract (IC50 value for the cleavage of C100FLAG is ~75 μm). III-31C (10 μm) or the Gleevec extract (100 μm) were used as positive controls to inhibit γ-secretase; bryostatin 1 (100 μm), a protein kinase C activator that has been reported to increase APP processing by the α-secretase pathway (28), was used as a negative control for the inhibition of γ-secretase. After 4 h of incubation at 37 °C, the reactions were probed for the generation of AICD-FLAG as described above. We found one compound (1367) that by Western blotting had a potency at least equal to that of the Gleevec extract (Fig. 4, compare lanes 25–27). Several other compounds (0433, 0577, 1321, 0744, 1037, 1405, and 1407) also inhibited purified γ-secretase (Fig. 4A, compare lanes 8, 13, and 24 with lane 1; lanes 37 and 39 with lane 26; and lanes 45 and 46 with lane 40) but with less potency than the Gleevec extract (lane 27). Because 1367 was the most potent compound when compared with the Gleevec extract, we further characterized this inhibitor. 1367 (also called ZM39923 or compound 7) is a potent inhibitor of the tyrosine kinase, Janus kinase 3 (29). 1367, which belongs to the class of 2-aminoethyl ketones (or naphthyl ketones; Fig. 4B), binds to the ATP-binding site of Janus kinase 3 (29). We found that 1367 inhibited the generation of AICD-FLAG and both Aβ40 and Aβ42 in a concentration-dependent fashion with an approximate IC50 of ~20 μm (Fig. 4B, left panel, and ELISA data not shown). Next, we examined the effects of 1367 on the cleavage efficiency of the purified enzyme on N100FLAG, and we found that this compound does not inhibit the generation of NICD-FLAG at a concentration of 100 μm (roughly 4–5-fold the IC50 value for generation of Aβ40 and Aβ42; Fig. 4B, left panel, compare lane 7 with lane 8) but does so at very high concentration (250 μm, data not shown). These data indicate that 1367, like the Gleevec extract, preferentially inhibits the cleavage by purified γ-secretase of the APP-based substrate over the Notch-based substrate. 1367 has been reported to act as a prodrug and break down in neutral buffer (t½ = 36 min, pH 7.43, 25 °C) to form ZM449829 (1366; Ref. 29) (Fig. 4B). Because the breakdown product 1366 is a Janus kinase 3 inhibitor with similar potency when compared with 1367 (29), we decided to investigate the effect of 1366 on the cleavage efficiency of C100FLAG and N100FLAG substrates by purified γ-secretase. We found that 1366 inhibited the generation of AICD-FLAG and both Aβ40 and Aβ42 in a concentration-dependent fashion (IC50 of ~60 μm) (Fig. 4B, right panel, and ELISA data not shown). Like 1367, 1366 does not inhibit the generation of NICD-FLAG, even at a concentration of 300 μm (roughly 5-fold the IC50 value for generation of Aβ40 and Aβ42) (Fig. 4B, right panel, compare lanes 8 and 9). Most interestingly, both 1367 and 1366 consistently displayed some stimulatory effect on NICD-FLAG production in the 30–100 μm range (Fig. 4B, left panel, compare lanes 5, 6, and 8, and Fig. 4B, right panel, compare lanes 5–7 and 9). Still another compound, the sulfonamide 0433, behaved differently from the active Gleevec extract, 1367 and 1366, inhibiting the cleavage of C100FLAG and N100FLAG with a similar potency (Fig. 4C and ELISA data not shown).
Purified γ-Secretase Binds to an ATP-immobilized Resin
In view of all of the above results, we examined the ability of γ-secretase to bind ATP. Purified γ-secretase (prepared as described under “Materials and Methods” and diluted in 0.2% CHAPSO/HEPES) was incubated overnight with ATP-immobilized resins (ATP attached to agarose through the ribose hydroxyls, or else ATP attached to acrylamide through the γ-phosphate) in the absence or presence of excess (50 mm) ATP. We observed that purified γ-secretase bound to the resin that had ATP attached to acrylamide through the γ-phosphate (Fig. 5, lanes 1–3), and this binding was specific in that 50 mm ATP fully prevented the complex from binding (Fig. 5, lanes 4–6). Most interestingly, purified γ-secretase diluted in 0.2% CHAPSO/HEPES bound poorly to the resin in which ATP was attached to agarose through the ribose hydroxyls (Fig. 5, lanes 10–12), suggesting a lesser accessibility of the nucleotide to the binding pocket. We also found that γ-secretase present in crude whole lysates of γ-30 cells binds very poorly to the resin with ATP attached to acrylamide through the γ-phosphate, in contrast to the efficient binding of the purified enzyme (Fig. 5, compare lanes 7–9 to 1–3). The following two hypotheses could explain this observation: (a) the whole cell lysate contains ATP, which acts as a competitor for the γ-secretase nucleotide-binding site, consistent with the fact that the intracellular concentration of ATP can exceed 5 mm (30); and (b) the lysate is enriched in ATP-binding proteins that act as competitors for the ATP bound to the resin. Yet we cannot exclude that purified γ-secretase adopts subtle conformational changes that differ from γ-secretase present in crude whole lysates, thus promoting the binding to the ATP resin. In any event, these observations suggest that such a resin would not be useful as an early step for the purification of endogenous γ-secretase.
FIGURE 5. Purified γ-secretase binds specifically to an ATP resin.

Purified γ-secretase (St. indicates starting material) was incubated overnight in the presence or absence of 50 mm ATP with two different ATP-immobilized resins as follows: ATP attached to acrylamide through the γ-phosphate (lanes 1–9) or ATP attached to agarose through the ribose hydroxyls (lanes 10 –12). The unbound fractions (Unb.) were recovered, and the resins washed three times in 0.2% CHAPSO/HEPES and resuspended in Laemmli sample buffer to recover the bound proteins (P. indicates precipitate). All samples were electrophoresed on 4 –20% Tris-glycine gels and transferred to polyvinylidene difluoride membranes to detect NCT-GST (αGST), PS1-NTF (Ab14), Aph1-HA (3F10), PS1-CTF (13A1), and FLAG-Pen2 (M2-anti FLAG). Note that purified γ-secretase binds specifically to the resin in which ATP is attached to acrylamide through the γ-phosphate (lanes 1– 6), whereas γ-secretase from a crude lysate (of γ-30 cells) is unable to bind to the same resin (lanes 7–9). Starting material (St.) and unbound (Unb.) lanes were each loaded with the equivalent of 25% of the material that was bound to the resins (P.) so that the unbound and bound protein levels can be compared directly.
Photoaffinity Labeling of the γ-Secretase Component PS1-CTF with the ATP Analog 8-Azido-[γ-32P]ATP
In an effort to identify the component(s) of the purified complex that mediates the interaction with the Gleevec extract, ZM39923 (1367) and ATP, we used the ATP analog 8-azido-[γ-32P]ATP for attempted site-specific photolabeling of γ-secretase. First, we observed that like ATP itself, unlabeled 8-azido-ATP (5 mm) was able to rescue the inhibition of purified γ-secretase activity by 100 μm Gleevec extract (data not shown), indicating that azido-modified ATP still binds to the apparent nucleotide binding domain. Next, purified γ-secretase was incubated with 8-azido-[γ-32P]ATP (Fig. 6A, lane 4) in the presence of 10 mm ATP (Fig. 6A, lane 5) or 1 mm Gleevec extract (Fig. 6A, lane 6) and was subjected to BN-PAGE analysis after UV irradiation. The 32P labeling assessed by autoradiography revealed a high molecular weight complex (HMWC, Fig. 6A, lane 4) comigrating with the well characterized active γ-secretase complex (31) that is detected by BN-PAGE analysis and Western blotting (Fig. 6A, lane 1). The intensity of this band was lost in the presence of excess cold ATP or Gleevec extract (Fig. 6A, compare lanes 5 and 6 with lane 4), indicating that purified and active γ-secretase binds specifically 8-azido-[γ-32P]ATP. Next, purified γ-secretase was incubated with 8-azido-[γ-32P]ATP in the absence or presence of 10 mm ATP, 1 mm Gleevec extract, 1 μm III-31C, 1 μm C100FLAG, 1 μm N100FLAG, or 300 μm 1367; as a control for the specificity of photolabeling, purified γ-secretase was also incubated with [γ-32P]ATP (without the 8-azido group). After UV irradiation, γ-secretase was affinity-precipitated with GSH resin (which binds nicastrin-GST in the complex), and its 32P labeling was assessed by SDS-PAGE and autoradiography. A prominent ~18-kDa band consistent with the known position of PS1-CTF was found to be photoaffinity-labeled in the purified γ-secretase (Fig. 6B, lanes 1 and 2). Next, the sample loaded in lane 1 (Fig. 6B) was probed simultaneously for PS1-CTF (13A11) and Aph1-HA (3F10); the photoaffinity-labeled ~18-kDa band was found to comigrate with PS1-CTF but not with Aph1-HA (Fig. 6B, compare lane 10 with lanes 1 and 2), strongly suggesting that the ~18-kDa protein is PS1-CTF. The intensity of this band decreased markedly or was lost in the presence of excess cold ATP, Gleevec extract, or 1367 (Fig. 6B, compare lanes 1 and 2 with lanes 3, 4 and 9), indicating that azido-[γ-32P]ATP bound to this protein specifically. In contrast, the presence of 1 μm III-31C, a transition state analog inhibitor, did not affect the labeling of this protein (Fig. 6B, lane 5), suggesting that azido-[γ-32P]ATP does not bind to the active site of γ-secretase. The presence of 1 μm C100FLAG or 1 μm N100FLAG also did not affect the labeling of this protein (Fig. 6B, lanes 7 and 8, respectively), suggesting that azido-[γ-32P]ATP does not bind to a putative common substrate docking site of γ-secretase (4, 32). Most interestingly, no labeling was observed with purified γ-secretase incubated with azido-[γ-32P]ATP (without the 8-azido group) (Fig. 6B, lane 6), supporting the specific photoaffinity labeling of PS1-CTF. Moreover, this observation indicates that the γ-32P does not transfer from azido-[γ-32P]ATP to PS1-CTF. Together with our observation that ATP is not hydrolyzed when incubated with purified γ-secretase (Fig. 1B), these findings indicate that intact azido-[γ-32P]ATP binds directly to PS1-CTF. To confirm the interaction of ATP with this component of the γ-secretase complex when it is located within membranes, we incubated membranes from HeLa cells (expressing endogenous γ-secretase) with azido-[γ-32P]ATP. After removing unbound nucleotide by washing the membranes with excess buffer and then co-immunoprecipitating solubilized γ-secretase with antibodies directed against PS1-NTF, we observed a specifically photoaffinity-labeled ~18-kDa protein migrating at the position of PS1-CTF (Fig. 6C, lane 1). This labeling was abolished in the presence of excess cold ATP (Fig. 6C, lane 2).
FIGURE 6. Specific labeling of the γ-secretase component PS1-CTF with 8-azido-[γ-32P]ATP, a photoaffinity ATP analog.
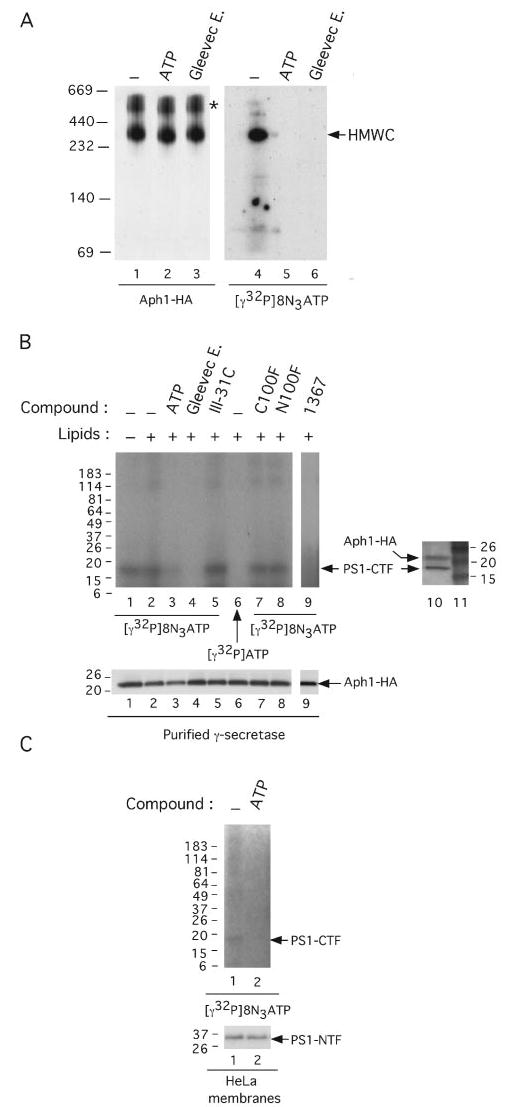
A, photolabeling of γ-secretase with 8-azido-[γ-32P]ATP as revealed by nondenaturing BN-PAGE. Purified γ-secretase solubilized in 0.1% digitonin/Tris-buffered saline was incubated with 22.5 μm of 8-azido-[γ-32P]ATP (10 μCi per reaction) in the presence of 10 mm ATP (lanes 2 and 5) or 1 mm Gleevec extract (Gleevec E., lanes 3 and 6), exposed to UV light for 5 min, and subjected to BN-PAGE analysis as described (31). 32P labeling was assessed by autoradiography (lanes 4 – 6) (BioMax MS films used with BioMax Transcreen HE; Kodak). As a control for the migration of γ-secretase, the purified complex solubilized in 0.1% digitonin/Tris-buffered saline (lane 1), in the presence of 10 mm ATP (lane 2) or 1 mm Gleevec extract (lane 3), was exposed to UV light for 5 min, subjected to BN-PAGE analysis on the same gel as above, and probed for the γ-secretase complex using 3F10 antibody to the Aph1-HA component (lanes 1–3). The asterisk denotes nonspecific aggregates containing Aph1-HA (P. C. Fraering, W. Ye, M. J. LaVoie, B. L. Ostaszewski, D. J. Selkoe, and M. S. Wolfe, unpublished data). HMWC, the high molecular weight γ-secretase complex (31). B, photolabeling of γ-secretase with 8-azido-[γ-32P]ATP as revealed by denaturing SDS-PAGE. Purified γ-secretase was incubated with 22.5 μm of 8-azido-[γ-32P]ATP (10 μCi per reaction) in the absence (lane 1) or presence (lane 2) of PC and PE (lipids) or in the presence of 10 mm ATP (lane 3), 1 mm Gleevec extract (lane 4), 1 μm III-31C (lane 5), 1 μm C100FLAG (lane 7), 1 μm N100FLAG (lane 8), or 300 μm 1367 (lane 9). As a control for the specificity of photolabeling, purified γ-secretase was also incubated with [γ-32P]ATP (without the 8-azido group) (lane 6). All samples were exposed to UV light for 5 min, and the reactions were quenched with 1 mm dithiothreitol. The samples were diluted and were incubated overnight at 4 °C with GSH resin for the affinity precipitation of the NCT-GST component and its associated proteins (6). The resins were then washed three times, and all precipitated proteins were electrophoresed on 4 –20% Tris-glycine gels and the photolabeled proteins autoradiographed as in A. As a control for the mobility of the photolabeled proteins, the same sample as shown in lane 1 was electrophoresed on a 4 –20% Tris-glycine gel and Western-blotted simultaneously for PS1-CTF and Aph1-HA with 13A11 and 3F10 antibodies, respectively (lane 10). Molecular weight markers are in lane 11. The levels of the Aph1-HA on the gel in B are shown to demonstrate equal protein loading. C, photolabeling of endogenous γ-secretase with 8-azido-[γ-32P]ATP. Membranes from untransfected HeLa cells were incubated with 8-azido-[γ-32P]ATP (10 μCi per reaction) in the absence (lane 1) or presence (lane 2) of 10 mm ATP and exposed to UV light as described above. The membranes were washed; the proteins were solubilized in 1% CHAPSO/HEPES, and γ-secretase was co-immunoprecipitated with anti-PS1-NTF antibodies. The photolabeled proteins were detected as described above, and levels of PS1-NTF (Western-blotted with mAb1563 antibody) serve as co-immunoprecipitated controls.
DISCUSSION
Here we demonstrate that ATP can activate the generation of AICD and Aβ by purified γ-secretase from an APP-based substrate without affecting the generation of NICD from a Notch-based substrate. We also demonstrate that Gleevec itself is not a direct γ-secretase inhibitor but a Gleevec extract from capsules that contain a direct γ-secretase inhibitor, which is competitive with ATP and has no effect on the cleavage of a Notch-based substrate. We further identify ZM39923 (1367) and ZM449829 (1366), two tyrosine kinase inhibitors known to bind to the ATP-binding site of Janus kinase 3 (29), as direct γ-secretase inhibitors that block preferentially the cleavage of the APP-based substrate compared with the Notch-based substrate. ZM39923, ZM449829, and Gleevec are tyrosine kinase inhibitors that bind to the ATP-binding sites of their corresponding targets. Accordingly, ATP is able to rescue the γ-secretase activity inhibited by the Gleevec extract, suggesting a potential nucleotide-binding domain on γ-secretase. In support of this hypothesis, we found that purified γ-secretase binds to a resin having ATP attached to acrylamide through the γ-phosphate. Moreover, γ-secretase and the PS1-CTF are specifically photolabeled with the ATP analog 8-azido-[γ-32P]ATP in reactions containing either purified γ-secretase from cells overexpressing all components of the protease or endogenous γ-secretase in membranes from untransfected HeLa cells. Finally, photolabeling of γ-secretase and PS1-CTF with the ATP analog 8-azido-[γ-32P]ATP was lost in the presence of excess cold ATP, Gleevec extract, or ZM39923. Taken together, our data strongly suggest the following: 1) that γ-secretase cleavage of an APP-based substrate (C100FLAG), but not a Notch-based substrate (N100FLAG), can be regulated in vitro by direct binding of nucleotide to the enzyme; and 2) that binding to this site by certain compounds, including kinase inhibitors and/or their derivatives, results in the selective inhibition of γ-secretase cleavage of APP vis à vis Notch. Most intriguingly, binding to this site by ZM39923 or ZM449829 can even stimulate N100FLAG cleavage, suggesting that this site allows tunable conformational changes for allosteric control of substrate selectivity.
ATP hydrolysis is likely not to be required for γ-secretase activity, suggesting that this enzyme does not belong to the Walker family of ATPases, a vast and functionally diverse collection of enzymes that includes ATP-dependent proteases (33, 34). Supporting this assumption, no conserved Walker consensus sequence motif A (GXGXXGK(T/ S)) associated with ATPase activity (33, 35) is present in PS, NCT, Aph-1, or Pen-2. One hypothetical model to explain the direct ATP effects on purified γ-secretase involves nucleotide binding to a hydrophobic pocket shaped by an integral protein component of the complex, namely PS1-CTF, in a way that effects a conformational change of the enzyme, perhaps promoting the opening of a “trap door” into the catalytic site that allows the substrate to enter and be processed. Other models could be proposed, and precisely how ATP acts on the purified γ-secretase requires further work. Nevertheless, we speculate that nucleotides and the inhibitors have different conformational effects upon binding to this site on the protease complex, in a manner analogous to the differential effects of agonists and antagonists on receptors.
Accumulating data have demonstrated that presenilin/γ-secretase is involved in many crucial physiological functions in virtually all cells. For example, genetic deletion has revealed that presenilin/γ-secretase is essential for regulation of the immune system, for memory, and for synaptic plasticity (for a review see Ref. 36). To chronically prevent the accumulation of Aβ deposits by inhibiting γ-secretase without adversely affecting its other physiological functions, it has been a major goal to identify γ-secretase inhibitors that discriminate between APP and the processing of Notch and other substrates. Weggen et al. (37) reported that certain nonsteroidal anti-inflammatory drugs can subtly alter the γ-secretase-mediated generation of Aβ, selectively lowering the production of Aβ42 over that of Aβ40 without significantly decreasing Notch cleavage. More recently, Netzer et al. (7) reported that Gleevec, an approved Abl kinase inhibitor used to treat chronic myelogenous leukemia, inhibits γ-secretase cleavage of APP without affecting Notch processing. Our work shows that Gleevec itself is not a direct γ-secretase inhibitor but supports this key finding in that certain kinase inhibitors (1367 and 1366) can act directly on γ-secretase via a mechanism that allows the protease to distinguish between the cleavages of APP and Notch. Gleevec may be metabolized or degraded under physiological conditions, thus leading to the generation of the active compound(s) found in our active Gleevec extract. Purification and characterization of each of the compounds found in our active Gleevec extract will now be necessary to address this question.
Most intriguingly, while our manuscript was in preparation, PS2 and PS1 missense mutations causing cases of familial AD that are located within the transmembrane domain 5 of PS were reported to reduce production of the AICD fragment of APP without affecting NICD levels (38). This observation suggests that transmembrane domain 5 of both PS1 and PS2 is more important for γ-secretase cleavage of APP than of Notch. Most interestingly, the apparently selective effect of these PS mutations on APP processing is mimicked pharmacologically by the Gleevec extracts, ZM39923 (1367) and ZM449829 (1366), apparently via direct interaction with the protease complex. One plausible hypothesis is that these familial Alzheimer disease mutations affect the conformation (either directly or allosterically) of a domain involved in the regulation of APP but not Notch cleavage. The binding to this domain by the tyrosine kinase inhibitors described in this study could result in a similar modulation of the protease, with selective inhibition of γ-secretase cleavage of APP vis à vis Notch. Another plausible but speculative explanation is that these kinase inhibitors may bind to a site on PS that interacts with APP substrates but not with Notch substrates; the binding of these compounds to this site would compete selectively with the binding of C83/C99 and result in the inhibition of γ-secretase cleavage of APP vis à vis Notch.
The physiological relevance of nucleotide binding and regulation of γ-secretase activity remains unknown. Nevertheless, whatever the precise biochemical mechanism, our findings are pharmacologically relevant and could have major therapeutic implications. Apparently, the γ-secretase complex can be directly affected in a way that allows selective inhibition of Aβ production without interfering with Notch proteolysis. Thus, the disturbing specter of Notch-associated toxicities previously reported upon in vivo administration of γ-secretase inhibitors may be avoidable, making this protease a much more attractive therapeutic target for the prevention and/or treatment of AD.
Supplementary Material
Acknowledgments
We thank S. Gandy for PS1 antibody Ab14. We also thank J. Sears, J. Strahle, and W. Xia for ELISA analysis; F. Bihel, A. Kuppanna, and E. Spooner for the preparation and analysis of Gleevec extracts; and A. Saci for helpful discussions about protein kinase/phosphatase assays.
Footnotes
This work was supported by Swiss National Science Foundation Grant 81FR-68842 (to P. C. F.), NIA Grant AG15379 from the National Institutes of Health, a Pioneer award from the Alzheimer Association (to D. J. S.), and by National Institutes of Health Grants AG17574, AG15379, and NS41355 (to M. S. W.).
The on-line version of this article (available at http://www.jbc.org) contains Table S1.
The abbreviations used are: Aβ, amyloid β-protein; AD, Alzheimer disease; APP, amyloid β-protein precursor; AICD, APP intracellular domain; CHO, Chinese hamster ovary; CTF, C-terminal fragment; ELISA, enzyme-linked immunosorbent assay; GST, glutathione S-transferase; NCT, nicastrin; NTF, N-terminal fragment; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PS, presenilin; CHAPSO, 3-[(3-cholamido-propyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid; ATPγS, adenosine 5′-O-(thiotriphosphate); BN, blue native; HPLC, high pressure liquid chromatography; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight.
References
- 1.Selkoe DJ. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Vassar R, Citron M. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 3.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 4.Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS. Proc Natl Acad Sci U S A. 2002;99:2720–2725. doi: 10.1073/pnas.052436599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopan R, Ilagan MX. Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 6.Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, Van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS. Biochemistry. 2004;43:9774–9789. doi: 10.1021/bi0494976. [DOI] [PubMed] [Google Scholar]
- 7.Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Proc Natl Acad Sci U S A. 2003;100:12444–12449. doi: 10.1073/pnas.1534745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 9.Druker BJ, Lydon NB. J Clin Investig. 2000;105:3–7. doi: 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mauro MJ, O’Dwyer M, Heinrich MC, Druker BJ. J Clin Oncol. 2002;20:325–334. doi: 10.1200/JCO.2002.20.1.325. [DOI] [PubMed] [Google Scholar]
- 11.Moasser MM, Srethapakdi M, Sachar KS, Kraker AJ, Rosen N. Cancer Res. 1999;59:6145–6152. [PubMed] [Google Scholar]
- 12.Wisniewski D, Lambek CL, Liu C, Strife A, Veach DR, Nagar B, Young MA, Schindler T, Bornmann WG, Bertino JR, Kuriyan J, Clarkson B. Cancer Res. 2002;62:4244–4255. [PubMed] [Google Scholar]
- 13.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller W, Clarkson B, Kuriyan J. Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 14.Nagar B, Hantschel O, Young M, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 15.Strife A, Wisniewski D, Liu C, Lambek CL, Darzynkiewicz Z, Silver RT, Clarkson B. Mol Cancer Res. 2003;1:176–185. [PubMed] [Google Scholar]
- 16.Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, Superti-Furga G. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 17.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, Zhang L, Higgins GA, Parker EM. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 18.Harrison T, Churcher I, Beher D. Curr Opin Drug Discovery Dev. 2004;7:709–719. [PubMed] [Google Scholar]
- 19.Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. J Biol Chem. 2003;278:46107–46116. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- 20.Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Greenberg B, Ciaccio PJ. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 21.Kimberly WT, Esler WP, Ye W, Ostaszewski BL, Gao J, Diehl T, Selkoe DJ, Wolfe MS. Biochemistry. 2003;42:137–144. doi: 10.1021/bi026888g. [DOI] [PubMed] [Google Scholar]
- 22.Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, Wolfe MS. Biochemistry. 2004;43:323–333. doi: 10.1021/bi035748j. [DOI] [PubMed] [Google Scholar]
- 23.Xia W, Zhang J, Ostaszewski BL, Kimberly WT, Seubert P, Koo EH, Shen J, Selkoe DJ. Biochemistry. 1998;37:16465–16471. doi: 10.1021/bi9816195. [DOI] [PubMed] [Google Scholar]
- 24.Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Proc Natl Acad Sci U S A. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dovey HF, John V, Anderson J, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN, Mutter LC, Nietz J, Quinn KP, Sacchi KL, Seubert PA, Shopp GM, Thorsett ED, Tung JS, Wu J, Yang S, Yin CT, Schenk DB, May PC, Altstiel LD, Bender MH, Boggs LN, Britton TC, Clemens JC, Czilli DL, Dieckman-McGinty DK, Droste JJ, Fuson KS, Gitter BD, Hyslop PA, Johnstone EM, Li WY, Little SP, Mabry TE, Miller FD, Audia JE. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 26.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 27.Cowan-Jacob SW, Guez V, Fendrich G, Griffin JD, Fabbro D, Furet P, Liebetanz J, Mestan J, Manley PW. Mini Rev Med Chem. 2004;4:285–299. doi: 10.2174/1389557043487321. [DOI] [PubMed] [Google Scholar]
- 28.Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc Natl Acad Sci U S A. 2004;101:11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown GR, Bamford AM, Bowyer J, James DS, Rankine N, Tang E, Torr V, Culbert EJ. Bioorg Med Chem Lett. 2000;10:575–579. doi: 10.1016/s0960-894x(00)00051-2. [DOI] [PubMed] [Google Scholar]
- 30.Morin MJ. Oncogene. 2000;19:6574–6583. doi: 10.1038/sj.onc.1204102. [DOI] [PubMed] [Google Scholar]
- 31.LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ. J Biol Chem. 2003;278:37213–37222. doi: 10.1074/jbc.M303941200. [DOI] [PubMed] [Google Scholar]
- 32.Kornilova AY, Bihel F, Das C, Wolfe MS. Proc Natl Acad Sci U S A. 2005;102:3230–3235. doi: 10.1073/pnas.0407640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker JE, Saraste M, Runswick MJ, Gay NJ. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Confalonieri F, Guguet M. BioEssays. 1995;17:639–650. doi: 10.1002/bies.950170710. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida M, Amano T. FEBS Lett. 1995;359:1–5. doi: 10.1016/0014-5793(94)01438-7. [DOI] [PubMed] [Google Scholar]
- 36.Marjaux E, Hartmann D, De Strooper B. Neuron. 2004;42:189–192. doi: 10.1016/s0896-6273(04)00218-1. [DOI] [PubMed] [Google Scholar]
- 37.Weggen S, Eriksen J, Das P, Sagi S, Wang R, Pietrzik C, Findlay K, Smith T, Murphy M, Bulter T, Kang D, Marquez-Sterling N, Golde T, Koo E. Nat Neurosci. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 38.Walker ES, Martinez M, Brunkan AL, Goate A. J Neurochem. 2005;92:294–301. doi: 10.1111/j.1471-4159.2004.02858.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.