

Abstract
Objective
Infiltrating inflammatory cells within the myocardium have been shown to be apoptotic, but the significance of apoptotic inflammatory cells to the development of cardiomyopathy remains undefined. Transgenic mice with cardiac-specific expression of MCP-1 exhibit extensive apoptosis of infiltrating mononuclear cells and develop heart failure. Here, we tested the hypothesis that in vivo selective inhibition of apoptosis of infiltrating mononuclear cells would preserve cardiac structure and function and improve survival in this murine model.
Methods
Mice with cardiac-specific expression of MCP-1 and monocyte-specific expression of Bcl-2 were generated by cross-breeding MCP-1 transgenic mice with hMRP8-Bcl-2 mice that over-express Bcl-2 in the monocytes. Structural and functional parameters and the inflammatory response of the heart were evaluated and compared between the wild-type and transgenic mice.
Results
Expression of Bcl-2 in monocytes results in superior preservation of myocardial structure, cardiac function and a significant prolongation of survival of MCP-1 transgenic mice. The beneficial effects of monocyte-specific Bcl-2 expression are associated with inhibition of apoptosis of infiltrating mononuclear cells, normalization of circulating C-reactive protein levels, attenuation of cellular infiltrates, macrophage activation and production of proinflammatory cytokines, tumor necrosis factor (TNF-α), interleukin (IL)-1β and IL-6 in the hearts.
Conclusions
These results demonstrate that apoptosis of infiltrating mononuclear cells plays a detrimental role in the development of heart failure in this murine model, suggesting that modulation of apoptosis of infiltrating mononuclear cells may be of clinical benefit in heart failure.
Keywords: Inflammation, Monocyte chemoattractant protein-1, Apoptosis, Cardiomyopathy, Transgenic animal model
1. Introduction
Inflammation contributes to the pathogenesis of heart disease, and the pathophysiological basis for the initiation and maintenance of inflammation in this setting has been related to the recruitment and activation of monocytes/macrophages within the myocardium [1–3]. Monocyte chemoattractant protein-1 (MCP-1) is the chief chemokine responsible for the recruitment of monocytes to sites of inflammation [4]. Its expression and functional significance have been shown in atherosclerosis, myocardial infarction and remodeling, and congestive heart failure [5–8]. To mimic the inflammatory component implicated in the heart disease, we developed transgenic mice that express MCP-1 in the heart under the control of α-myosin heavy chain promoter [9]. These mice exhibit mononuclear cells infiltrating into the myocardium, ventricular hypertrophy and dilatation, interstitial fibrosis, impaired cardiac function and increased mortality with heart failure by 6 months of age [9,10]. A number of infiltrating mononuclear cells in the hearts of these mice also display apoptotic cell death features.
Apoptotic cell death is increasing recognized as a potentially important pathophysiological process that occurs in a broad variety of disease conditions [11]. There is increasing evidence for association of apoptosis with the pathological states of cardiovascular diseases [11–13]. The presence of apoptotic cells in these setting is associated mostly with infiltrating mononuclear cells or non-cardiomyocytes [12,14–16]. However, the actual role of apoptosis of infiltrating mononuclear cells in the development of cardiomyopathy remains obscure. Recent studies of in vivo experimental models and clinical studies have demonstrated that failure to remove apoptotic cells from inflammatory foci contributes to a persistent state of inflammation in various organs and tissues [17,18]. Thus, it is postulated that the extensive apoptosis of infiltrating mononuclear cells in the hearts of MCP-1 transgenic mice would enhance myocardial inflammation and trigger apoptosis of vascular cells and cardiomyocytes resulting in the progression of heart failure. To test this hypothesis, we attempted to prevent the death of infiltrating mononuclear cells in MCP-1 transgenic mice by targeted over-expression of Bcl-2 in monocytes since over-expression of Bcl-2 in monocytic cell lineages has been shown to protect monocytes from apoptosis [19]. To achieve this, we crossed MCP-1 transgenic mice with hMRP8-Bcl-2 mice that over-express human Bcl-2 in monocytes [19] and assessed whether Bcl-2 expression affected monocytes survival and the disease progression in MCP-1 transgenic mice. We report here that Bcl-2 over-expression in the monocytes of MCP-1 transgenic mice inhibited apoptosis of infiltrating mononuclear cells, attenuated myocardial inflammatory response by suppressing cellular infiltrates, macrophage activation and inflammatory cytokine production in the hearts, and ameliorated cardiomyopathy resulting in a dramatically preserved cardiac structure, function and improved survival in this murine model of cardiomyopathy.
2. Methods
2.1. Animals
Experimental procedures in mice and protocols used in this study were approved by Animal Care and Use Committee of the University of Central Florida and the Ohio State University, conforming with the Guide for the care and use of laboratory animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Male mice were used for all experiments. The generation of hMRP8-Bcl-2 transgenic mice that over-express Bcl-2 in monocytes under the control of the human MRP8 promoter has been described previously [19]. MCP-1 transgenic (MCP) mice were generated by expressing the murine MCP-1(JE) gene under the control of α-myosin heavy chain promoter [9]. Cross-breeding generated double homozygous transgenic mice (MCP/Bcl-2) that over-express MCP-1 in heart and Bcl-2 in monocytes. The genotypes of MCP/Bcl-2 mice were identified by polymerase chain reaction (PCR) with genomic DNA from tail clips. Quantitative real-time PCR was performed to confirm expression of the transgenes. Primer pairs used were as follows: MCP-1, forward 5′-CAGGTCCCTGTCATGCTTCT-3′, reverse 5′-CACTGTCACACTGGTCACT-3′; Bcl-2, forward 5′-GGGAGGATTGTGGCCTTCTT-3′, reverse 5′-ATCC-CAGCCTCCGTTATCCT-3′. Normal FVB/N strain mice purchased from Harlan (Indianapolis, Indiana) were used as wild-type controls.
2.2. Cardiac morphology and histomorphometric analysis
Wild-type and transgenic mice of 2, 4 and 6 months of age were examined. After removing blood samples, the heart was removed, weighed and fixed in 10% phosphate-buffered formaldehyde or snap-frozen in liquid nitrogen for RNA and protein analysis. Photographs of whole hearts were taken with a Spot Insight digital camera (Diagnostic Instrument, Inc., Sterling Heights, MI) with a computerized Nikon SMZ1000 dissecting microscope. The hearts fixed in 10% buffered formalin were routinely processed and paraffin embedded. Sections (5μm) were stained with hematoxylin and eosin, and Masson’s trichrome for histopathological analysis. Quantitative assessments of interstitial fibrosis were performed on five randomly selected fields per section and expressed as interstitial collagen volume fraction. The collagen volume fraction was calculated as a percentage of the sum of all blue-stained areas to the total ventricular areas by using the Metamorph Series 6.2 image analysis program (Universal Imaging, West Chester, PA).
2.3. Detection of infiltrated inflammatory cells by immunohistochemistry
Infiltration of inflammatory cells was detected by immunohistochemical staining. Sections were incubated with anti-CD45 antibody, anti-Mac-3 antibody (Pharmingen, San Diego, CA), anti-Mac-1 (CD11b, clone M1/70) antibody (Abcam, Inc., Cambridge, UK) or isotype control overnight at 4 °C, then incubated with horseradish peroxidase-conjugated goat anti-rat antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Peroxidase activity was visualized with diaminobenzidine and sections were counterstained with haematoxylin. Quantitative assessments for CD45-, Mac-1- and Mac-3-positive cells were performed using the Metamorph Series 6.2 image analysis program, and the number of the labeled inflammatory cells on tissue sections was expressed as the number per field of view under ×200 magnification (0.22 mm2).
2.4. Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end-labeling (TUNEL) staining
TUNEL assay was performed in sections with Cardio-TACS in situ Apoptosis Detection Kit (R&D Systems, Minneapolis, USA) according to the manufacturer’s recommendations. In this procedure, nuclei undergoing apoptosis were stained blue. Only cells with clear striations were scored as cardiac myocytes. The number of TUNEL-positive infiltrating mononuclear cell was manually counted and expressed as a percentage of the total amount of infiltrating cells in five randomly selected fields of view (magnification, × 200) (0.22 mm2) for each animal. Five animals were studied per group.
2.5. Real-time PCR assay for mRNA quantification
Total RNA was isolated from left ventricle tissues of transgenic mice and age-matched wild-type controls by use of TRIzol Reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized from 1 μg RNA using the SuperScript First-Strand Synthesis System (Invitrogen). Relative levels of mRNA transcripts for cytokines, tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6 were quantified by real-time PCR. As a control, the quantity of an endogenous ‘‘housekeeping’’ messenger RNA (mouse β -actin) was measured and used to normalize the data. A total of five samples were run for each experimental group and all PCR samples were in duplicate. The expression levels of each targeted gene were normalized by subtracting the corresponding mouse β -actin threshold cycle (CT) values by using the ΔΔCT comparative method [20]. Primer pairs used were as follows: TNF-α, forward 5′-ACTCAACAAACTGCCCTTCTGAG-3′, reverse 5′-TTACAGCTGGTTTCGATCCATTT-3′; IL-1β , forward 5′-TGTGGCTGTGGAGAAGCTGT-3′, reverse 5′-CA-GCTCATATGGGTCCGAGA-3′; IL-6, forward 5′-CA-CGGCCTTCCCTACTTCAC-3′, reverse 5′-TGCA-AGTGCATCATCGTTGT-3′; β -actin, forward 5′-GAAATCGTGCGTGACATCAAAG-3′, reverse 5′-TGTAGTTTCATGGATGCCACAG-3′.
2.6. ELISA for circulating C-reactive protein
The circulating levels of circulating C-reactive protein (CRP) in plasma were measured with mouse high-sensitive CRP ELISA kit (Kamiya Biomedical Company, Seattle, WA), which specifically detects mouse CRP.
2.7. Echocardiographic measurements
Echocardiographic studies were performed with an Agilent Technologies SONOS 4500 ultrasonography system equipped with 15-MHz high frequency transducer (Philips Medical System, Germany). Mice were lightly anesthetized via a nose cone and maintained with 0.5–2.0% isoflurane (AErrane, Baxter, USA) mixed with oxygen. A two-dimensional short-axis view of left ventricle was obtained at the level of the papillary muscles and two-dimensionally targeted M-mode tracings were recorded at a sweep speed of 100 mm/s. The percentage of left ventricular fractional shortening (%FS) was calculated as: FS (%)=[(LVEDD − L-LVESD)/LVEDD] × 100, where LVEDD and LVESD indicate left ventricular end-diastolic and end-systolic diameter, respectively. Data from three to five consecutive selected cardiac cycles were analyzed and averaged.
2.8. Survival studies
Additional groups of mice, wild-type (n =28), MCP (n =36) and MCP/Bcl-2 (n =32) mice were included in survival analysis. During the study period of 28 weeks, the mice were allowed free access to food and water, and cages were inspected daily. Animals that developed a clinical heart failure consisting of decreased activity, tachypnea, hunched posture and poor grooming were euthanized by CO2 asphyxiation.
2.9. Statistical analysis
All values are presented as means ± standard errors. Results of functional and biochemical tests were compared between groups by a nonparametric one-way analysis of variance (ANOVA) followed by t-tests. Survival analysis was performed by using a Kaplan–Meier method and between-group difference in survival was tested by the log-rank test. Differences were considered significant at P-value of <0.05.
3. Results
3.1. Over-expression of Bcl-2 in monocytes of MCP mice
Targeted over-expression of Bcl-2 in monocytes of MCP mice was achieved by cross-breeding homozygous MCP mice [9] with homozygous hMRP8-Bcl-2 transgenic mice that over-express Bcl-2 in monocytes [19]. The transcript levels of MCP-1 and Bcl-2 transgenes were measured in whole heart by real-time PCR. Both MCP-1 and Bcl-2 transcripts were undetectable in wild-type controls, whereas nearly equal expression of the transgene was found in mice expressing only one transgene versus mice expressing both transgenes (Fig. 1A,B).
Fig. 1.
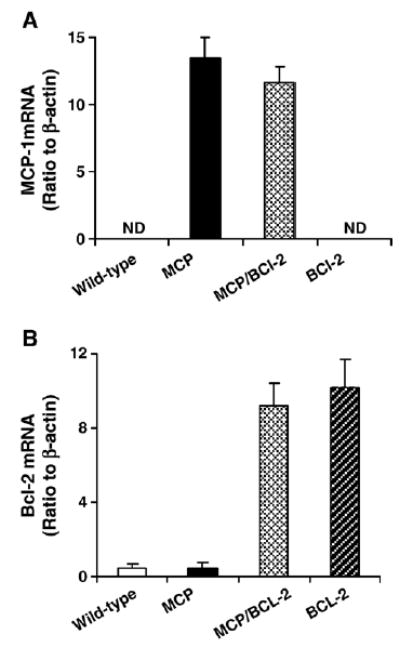
Real-time PCR demonstrates transgene mRNA expression in the hearts. Histograms showing the levels of MCP-1 (A) and Bcl-2 (B) mRNA expression in the hearts of wild-type and transgenic mice of 4 months of age (n =3 per group). Equal expression of transgene was found in mice expressing only one transgene versus mice expressing both transgenes. ND, not detectable.
3.2. Monocyte-specific expression of Bcl-2 in MCP mice attenuates cardiac deterioration
As shown in Fig. 2, cardiac deterioration and increased heart/body weight (HW/BW) ratio in MCP mice were clearly attenuated by targeted Bcl-2 expression in monocytes (Fig. 2A,B). Such Bcl-2 expression in the wild-type background did not alter the HW/BW ratio (4.41 ± 0.06, 4.36 ± 0.19 mg/g at 2 and 6 months of age, respectively; P >0.05 versus age-matched wild-type controls, n =6 per time point). Histological examination showed that the MCP/Bcl-2 mice had much less interstitial inflammatory cells infiltration, myocardial degeneration (vacuolized cardiomyocytes) and interstitial collagen deposition when compared with age-matched MCP mice (Fig. 2C). Cardiac interstitial collagen volume fraction in 6-month-old MCP mice was 24.1 ± 1.6%. In the age-matched MCP/Bcl-2 mice, there was a significant decrease in interstitial collagen volume fraction (10.8 ± 1.2%, P <0.001 versus MCP mice) although the levels did not reach exactly the levels found in age-matched wild-type controls (2.3 ± 0.6%, P <0.05 versus MCP/Bcl-2 mice) (Fig. 2D).
Fig. 2.
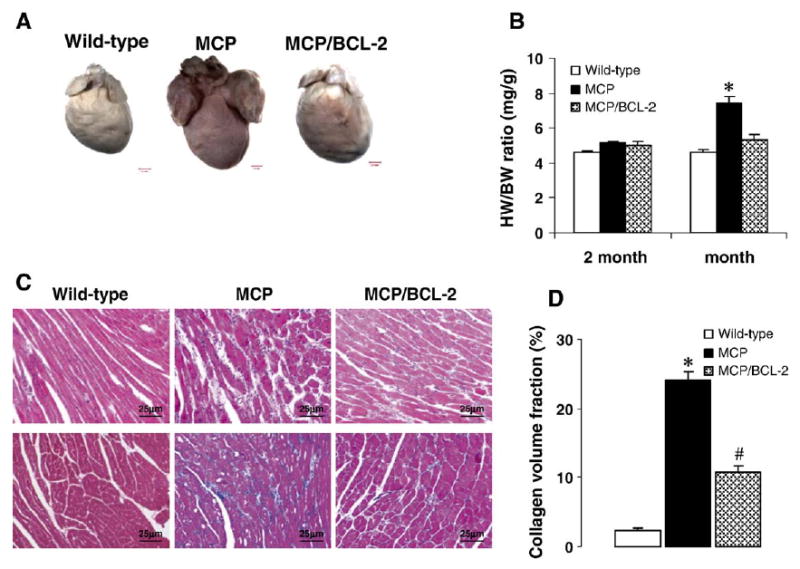
Monocyte-specific Bcl-2 expression in MCP mice attenuates cardiac deterioration. (A) Representative photographs showing the whole hearts from the wild-type, MCP and MCP/Bcl-2 mouse at 6 months of age. The heart from MCP mouse is significantly larger and noticeably discolored when compared to the wild-type and MCP/Bcl-2 mouse. (B) Heart weight to body weight (HW/BW) ratio was depicted from 2 to 6 months of age. *P <0.001 versus MCP/Bcl-2 mice and wild-type mice, n =6 per group per time point. (C) Representative photomicrographs demonstrating morphology of heart sections from 6-month-old wild-type and transgenic mice stained with H&E (upper panel) and Masson’s trichrome (lower panel). Blue staining represents collagen deposition. (D) Collagen volume fraction in the hearts of 6-month-old wild-type, MCP and MCP/Bcl-2 mice. n =6 per group. *P <0.001 versus wild-type and MCP/Bcl-2 mice; #P <0.05 versus wild-type mice.
3.3. Protection of infiltrating cells, vascular cells and cardiomyocytes from apoptosis by monocyte-specific Bcl-2 expression
To determine whether monocyte-specific Bcl-2 expression prevents apoptosis of infiltrating mononuclear cells within the myocardium, TUNEL staining was performed. The majority of infiltrating mononuclear cells were TUNEL-positive in the hearts of 6-month-old MCP mice manifesting heart failure. Cells infiltrating in the hearts of MCP/Bcl-2 mice showed few TUNEL-positive cells with many fields entirely negative, and the incidence of TUNEL-positive cells were similar to that seen in both age-matched wild-type controls (Fig. 3A–C). Quantitative analysis showed that the majority of infiltrating mononuclear cells in the hearts of MCP mice were apoptotic, whereas only a small fractions of the infiltrating cells were TUNEL-positive in the hearts of MCP/Bcl-2 mice; there was no significant difference in the percentage of TUNEL-positive infiltrating cells between MCP/Bcl-2 mice and age-matched wild-type controls (Fig. 3F). TUNEL-positive nuclei were also observed in scattered cardiac myocytes of 6-month-old MCP mice (Fig. 3B). There was an almost 8-fold increase in the number of TUNEL-positive myocytes in MCP mice hearts (737 ± 110 per 105 cells, n =6) compared with age-matched wild-type controls (98 ± 36 per 105 cells, n =5). In contrast, TUNEL-positive myocytes were comparable in wild-type controls and the age-matched MCP/Bcl-2 mice (101 ± 16 per 105 cells, n =5). Vascular smooth muscle cells and endothelial cells in the hearts of MCP mice were TUNEL-positive but in the hearts of age-matched MCP/Bcl-2 mice and the wild-type controls were TUNEL-negative (Fig. 3D,E). The number of TUNEL-positive cells in the hearts of hMRP8-Bcl-2 transgenic mice was similar to those in the age-matched wild-type controls (data not shown).
Fig. 3.
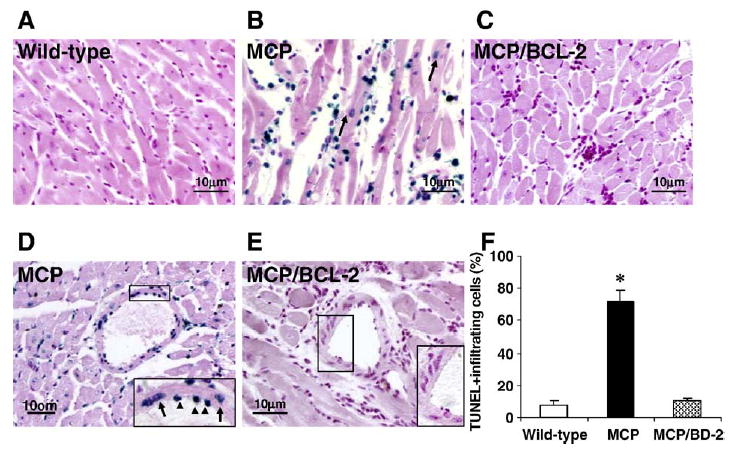
Bcl-2 expression in monocytes prevents apoptotic cell death in the hearts of MCP mice. (A–E) Representative photomicrographs demonstrating TUNEL staining of heart sections from 6-month-old wild-type, MCP and MCP/Bcl-2 mice. Blue staining indicates TUNEL-positive cells and the TUNEL-positive cardiac myocytes are indicated by arrows in B. The insert in D shows TUNEL-positive vascular endothelial (arrowheads) and smooth muscle cells (arrows) in the heart of MCP mouse. The insert in E shows no TUNEL-positive vascular cells in the heart of MCP/Bcl-2 mouse. (F) Histogram showing the quantitative analysis of TUNEL-positive infiltrating cells in the hearts of mice at 6 months of age. n =5 per group. *P <0.001 versus wild-type mice and MCP/Bcl-2 mice.
3.4. Attenuation of myocardial inflammation by monocyte-specific Bcl-2 expression
Apoptotic cells induce inflammation by the release of inflammatory cytokines [21,22]. To determine whether inhibition of apoptosis of infiltrating mononuclear cells by monocyte-specific Bcl-2 expression would attenuate myocardial inflammation in MCP mice, the cellular infiltrate was characterized by immunohistochemical analyses. Sections from left ventricles of MCP, MCP/Bcl-2 and age-matched wild-type mice were immunostained for CD45, Mac-1 and Mac-3 (Fig. 4A). Whereas CD45 is a pan-leukocyte marker which labels neutrophils and weakly detects hematogenous macrophages, Mac-1 is expressed on both granulocytes and monocytes/macrophages and Mac-3 specifically is expressed in activated macrophages [23]. There were no significant differences in the number of infiltrating inflammatory cells between the hearts of the hMRP8-Bcl-2 mice and the age-matched wild-type controls (data not shown). The number of positive cells for CD45, Mac-1 and Mac-3 was clearly less in the hearts of MCP/Bcl-2 animals compared to age-matched MCP mice, but was higher than that in the age-matched wild-type controls (Fig. 4B–D), suggesting that cellular infiltration and macrophage activation in the hearts of MCP mice were attenuated by monocyte-specific expression of Bcl-2.
Fig. 4.
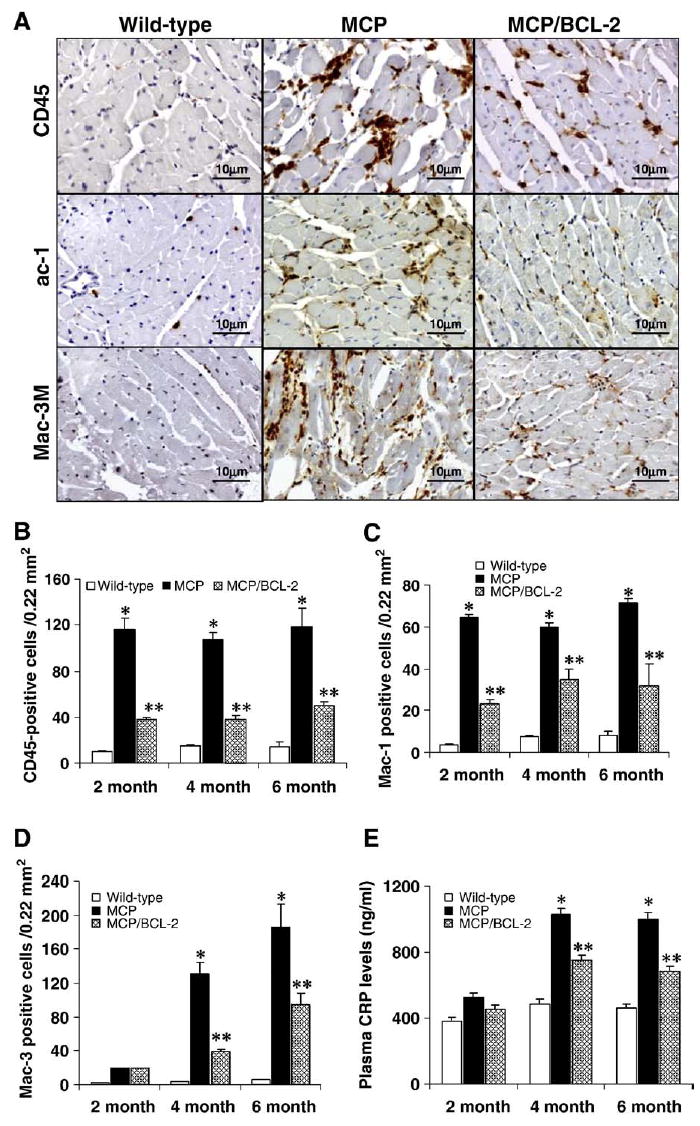
Bcl-2 expression in monocytes results in reduced inflammatory cells infiltration in the hearts of MCP mice. (A) Representative photomicrographs demonstrating immunohistochemical staining with anti-CD45, -Mac-1 and -Mac-3 antibodies in heart sections from 6-month-old wild-type and transgenic mice. Positive-stained cells were visualized with diaminobenzidine (brown). (B–D) Histograms showing the number of CD45-, Mac-1- and Mac-3-positive cells in the hearts of wild-type, MCP and MCP/Bcl-2 mice. *P <0.001 versus wild-type mice; #P <0.05 versus wild-type and MCP mice; n =6 per group per time point. (E) Circulating levels of CRP assayed by ELISA. n =6 per group per time point. *P <0.05 versus wild-type and MCP/Bcl-2 mice; #P <0.05 versus wild-type mice.
CRP, a marker of inflammation, has been consistently found to be associated with coronary heart disease and heart failure [24]. Circulating levels of CRP were measured by ELISA to obtain a quantitative estimate of extent of inflammation in both MCP and MCP/Bcl-2 mice. The levels of circulating CRP were significantly lower in the MCP/Bcl-2 mice compared with age-matched MCP mice (Fig. 4E).
Production of proinflammatory cytokines TNF-α, IL-6 and IL-1β in the hearts of wild-type, MCP and MCP/Bcl-2 mice were also tested by measuring the levels of their mRNA transcripts by real-time PCR. The hearts of MCP/Bcl-2 mice showed greatly reduced levels of transcripts of these inflammatory cytokines when compared with those of MCP mice, although the levels did not reach exactly the levels found in age-matched wild-type controls (Fig. 5A–C). Clearly, the up-regulated expression of proinflammatory cytokines in the hearts of MCP mice was significantly suppressed by monocyte-specific Bcl-2 expression. Taken together, these results suggest that monocyte-specific Bcl-2 expression attenuated myocardial inflammatory response in MCP mice.
Fig. 5.
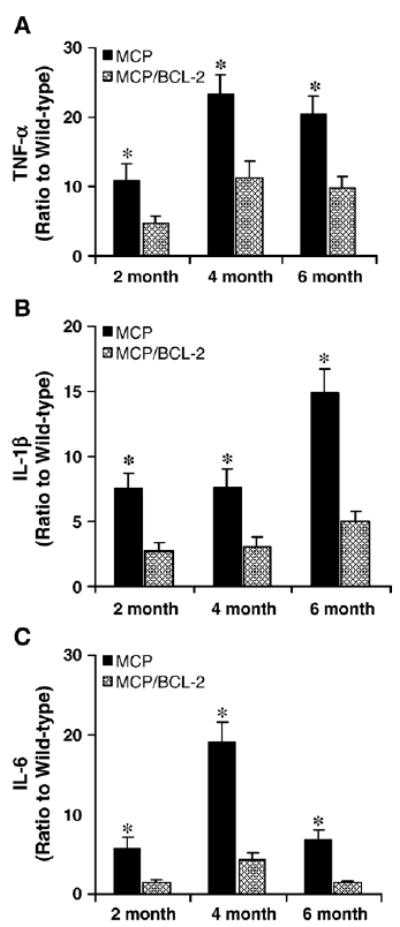
Bcl-2 expression in monocytes inhibits proinflammatory cytokines mRNA expression in the hearts of MCP mice. Relative levels of TNF-α (A), IL-1β (B) and IL-6 (C) mRNA expression in the hearts of wild-type and transgenic mice were assayed by real-time PCR. The expression of TNF-α, IL-1β and IL-6 in the hearts was expressed as a ratio relative to age-matched wild-type controls. Over-expression of Bcl-2 in monocytes resulted in decreased TNF-α, IL-1β and IL-6 mRNA expression in the hearts of MCP/Bcl-2 mice compared to age-matched MCP mice. *P <0.05, n =6 per group per time point.
3.5. Improvement of cardiac function of MCP mice by monocyte-specific Bcl-2 expression
To determine whether protection of infiltrating mononuclear cells from apoptosis combined with suppression of myocardial inflammation by the monocyte-targeted expression of Bcl-2 would benefit cardiac structure and function of MCP mice, serial echocardiographic measurements were performed in lightly anesthetized wild-type and transgenic mice. Representative M-mode of echocardiogram obtained from 6-month-old wile-type, MCP and MCP/Bcl-2 mice are presented in Fig. 6A. The MCP mice showed enlarged left ventricle chambers, as shown by progressive increase in left ventricle end-diastolic dimensions (LVEDD) (Fig. 6B), and severe cardiac dysfunction, as shown by the markedly reduced fractional shortening (FS) with age (Fig. 6C). Monocyte-specific Bcl-2 expression in MCP mice resulted in significant preservation of left ventricle geometry with less ventricular dilatation and slightly decreased fractional shortening (Fig. 6A–C). This observation exhibited good correlation with the pathological findings presented in Fig. 2.
Fig. 6.
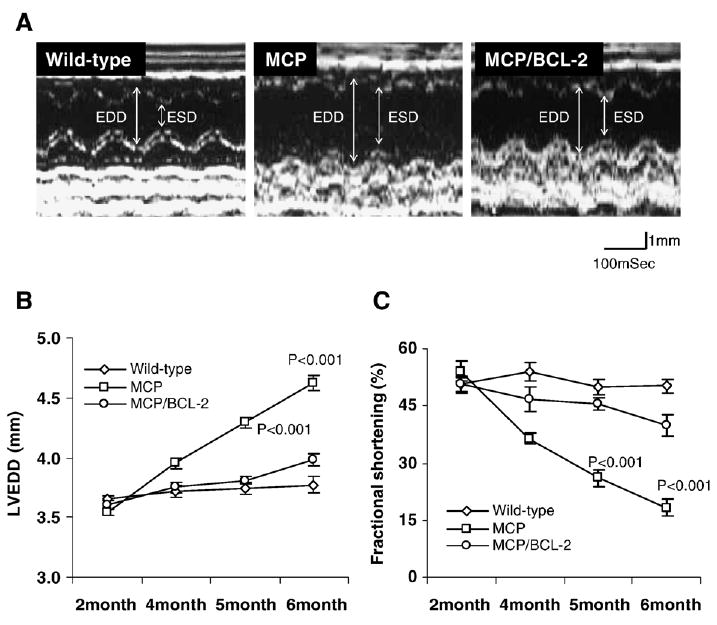
Bcl-2 expression in monocytes prevents progression of left ventricle (LV) dilatation and preserves cardiac function. (A) Representative M-mode tracing showing changes in left ventricle dimensions at 6 months of age in wild-type and transgenic mice. EDD and ESD indicate LV end-diastolic dimension and end-systolic dimension, respectively. (B–C) LV end-diastolic dimension (LVEDD) and percent functional shortening (FS) were depicted from 2 to 6 months of age. MCP mice had progressive LV dilatation and cardiac dysfunction compared to wild-type and MCP/Bcl-2 mice, whereas MCP/Bcl-2 mice had slightly increased LVEDD and reduced FS, but were not significantly different from LVEDD and FS in wild-type mice, P <0.001 versus wild-type and MCP/Bcl-2 mice, n =6 per group per time point.
3.6. Monocyte-specific Bcl-2 expression prolongs survival of MCP mice
As shown in Fig. 7, a Kaplan–Meier analysis of survival showed that cumulative survival probability for MCP mice was greatly shortened. Over a 28-week follow-up period, the probability of survival for MCP mice was approximately 7.36% compared with 91.4% for wild-type controls. Over the same period, MCP/Bcl-2 mice showed a probability of survival of 76.2%. These data clearly demonstrate that prevention of apoptosis of infiltrating mononuclear cells by monocyte-specific Bcl-2 expression drastically improved survival of MCP mice.
Fig. 7.
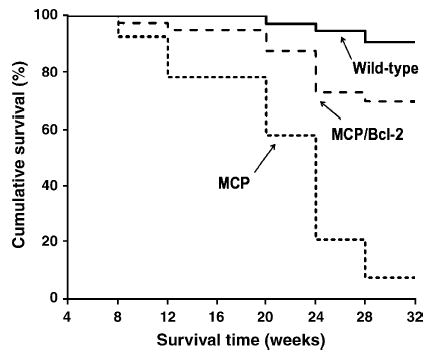
Kaplan–Meier survival analysis of wild-type (n =28), MCP (n =36) and MCP/Bcl-2 (n =32) mice showing improvement of survival profile of MCP/Bcl-2 mice compared with MCP mice ( P <0.001).
4. Discussion
The major purpose of this study was to assess the contribution of apoptosis of infiltrating inflammatory cells to the development of cardiomyopathy and heart failure. Results presented here show that inhibition of apoptosis of infiltrating mononuclear cells by targeted monocyte-specific Bcl-2 expression resulted in reduced cellular infiltration, macrophage activation and production of proinflammatory cytokines in the hearts of MCP mice. These improvements in the pathophysiology led to superior preservation of cardiac structure and function, and significant prolongation of survival of MCP-1 transgenic mice, suggesting that apoptotic cell death of infiltrating mononuclear cells is a key event that mediates signaling pathways contributing to the development of cardiomyopathy and heart failure in this murine model.
Apoptotic inflammatory cells are found to be associated with many cardiovascular diseases such as myocardial infarction, heart failure and myocarditis [12,14–16]. However, the extent to which apoptosis of the infiltrating cells contribute to the progression of these diseases has not been elucidated. Transgenic mice with cardiac-specific expression of MCP-1 display mononuclear cell infiltration and apoptosis within myocardium, and develop cardiomyopathy and heart failure. Thus, this model provides a system to test the role of apoptosis of infiltrating mononuclear cells in the development of heart disease. The Bcl-2 gene is known to block apoptosis [25], and Bcl-2 expression is low in the infiltrating mononuclear cells in human infarcted myocardium [26]. Therefore, the endogenous Bcl-2 levels may not be adequate to protect against apoptosis of the large number of infiltrated mononuclear cells in the myocardium of MCP mice. To test this hypothesis, we investigated whether monocyte-specific expression of Bcl-2 can prevent the death of the infiltrating mononuclear cells and thus affect the disease progression in this mouse model of cardiomyopathy. We observed that increased levels of Bcl-2 in monocytes protected infiltrating mononuclear cells from apoptotic cell death. Similarly, the death of cardiomyocytes and vascular cells that occurred in the hearts of MCP mice was also prevented by monocyte-specific Bcl-2 expression. This observation is consistent with the fact that apoptotic monocytes/macrophages are the main source of Fas ligand that would cause death in the cardiac cells, specifically vascular cells [27,28]. Thus, inhibition of apoptosis of infiltrating cells resulted in preservation of cardiac structure, function and, therefore, improvement of survival in MCP/Bcl-2 mice. Our observation suggests that apoptotic cell death of infiltrating mononuclear cells is a key event that contributes to the development of cardiomyopathy and heart failure in this murine model.
Classically, apoptosis is regarded as a type of cell death that does not induce significant inflammation and immune response. However, apoptotic cells have been shown to induce inflammation by the release of inflammatory cytokines such as Fas ligand [21,22]. Fas ligand also has a proinflammatory function, and the ligation of Fas to circulating monocytes and tissue macrophages may induce proinflammatory cytokine responses that can initiate and amplify the inflammatory response [29]. TNF-α, IL-1β and IL-6 are also known to contribute to cardiac disease and heart failure [30,31]. Our previous study showed that neutralization of Fas ligand by targeted cardiac-specific expression of soluble Fas attenuated myocardial inflammation and rescued MCP mice from the development of heart failure [32]. In the current study, our findings show that inhibition of apoptosis of infiltrating mononuclear cells by over-expression of Bcl-2 resulted in reduced production of TNF-α, IL-1β and IL-6 in the hearts of MCP/Bcl-2 mice compared to age-matched MCP mice. This effect correlated with the decrease in the number of CD45, Mac-1 and Mac-3 positive cells in the hearts of MCP/Bcl-2 mice. Thus, prevention of apoptosis of inflammatory cells resulted in the decreased production of inflammatory cytokines in the hearts and inhibited further cellular infiltration. The decreased levels of circulating CRP in MCP/Bcl-2 mice may also contribute to the reduction in cellular infiltration because CRP is known to up-regulate monocyte chemotaxis receptor CCR2 and thus would promote infiltration [33]. These findings were consistent with the previous reports that over-expression of Bcl-2 in endothelial cells inhibits nuclear factor kappaB (NF-kB) translocation [34] and thus down-regulates of NF-kB-dependent genes that are involved in immune and inflammatory response [35]. In fact, over-expression of Bcl-2 family members has also been shown to inhibit lipopolysaccharide (LPS)-induced production of cytokines such as TNF-α and IL-1β [36].
The prolonged survival of infiltrating mononuclear cells in the hearts of MCP/Bcl-2 mice suggests that this could contribute to the protection of cardiac function. Over-expression of Bcl-2 may create the milieu necessary for monocyte survival and differentiation. Differentiation of monocytes into macrophages is important for them to actively participate in the repair process through the production of cytokines and growth factors. Previous studies have shown that an appropriate recruitment of inflammatory cells and the production of cytokines have beneficial effects to cardiac repair [37]. Transplantation of bone marrow mononuclear cells has been shown to limit myocardial infarct size through the release of vascular endothelial growth factor [38]. Thus, the prolonged survival of infiltrating mononuclear cells may help limit myocardial remodeling and preserve cardiac function in MCP/Bcl-2 mice. Monocyte survival and differentiation into macrophage could be important in regulating fibroblast proliferation and extracellular matrix deposition through expression of fibrogenic growth factors and production of metal-loproteinases and cytokines. Consistent with the reduced number of macrophages and decreased production of TNF-α, IL1β and IL-6 in the heart, collagen deposition in the hearts was also reduced in MCP/Bcl-2 animals. In fact, the 6-month-old MCP/Bcl-2 mice showed less ventricular dilatation and slightly decreased fractional shortening compared to wild-type controls. All of these results demonstrated that over-expression of Bcl-2 in monocytes may attenuate the progression of cardiomyopathy but does not completely rescue the degree of pathology in MCP mice.
In summary, our findings demonstrate that apoptosis of infiltrating inflammatory cells contributes to the development of cardiovascular disease. Inhibition of apoptosis of infiltrating mononuclear cells by monocyte-specific Bcl-2 expression attenuates myocardial inflammation by inhibition of cell infiltration, macrophage activation and production of proinflammatory cytokines, preserves cardiac structure and function, and improves survival of MCP mice. Our findings raise the interesting possibility that the physiological regulation of apoptosis of infiltrating inflammatory cells may represent a potential target for therapeutic intervention, which may also be relevant to other chronic inflammatory conditions in which monocyte/macrophage accumulation is essential, such as transplant arteriosclerosis.
Acknowledgments
This work was supported by grant HL-69458 from the National Institutes of Health. The authors have no conflicting financial interests.
Footnotes
This work was presented in part at the 2005 Scientific Sessions of the American Heart Association, Dallas, Texas, USA, in November 2005.
References
- 1.Ikeda U. Inflammation and coronary artery disease. Curr Vasc Pharmacol. 2003;1:65–70. doi: 10.2174/1570161033386727. [DOI] [PubMed] [Google Scholar]
- 2.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 4.Dawson J, Miltz W, Mir AK, Wiessner C. Targeting monocyte chemoattractant protein-1 signaling in disease. Expert Opin Ther Targets. 2003;7:35–48. doi: 10.1517/14728222.7.1.35. [DOI] [PubMed] [Google Scholar]
- 5.Sheikine Y, Hansson GK. Chemokines and atherosclerosis. Ann Med. 2004;36:98–118. doi: 10.1080/07853890310019961. [DOI] [PubMed] [Google Scholar]
- 6.Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:1–7. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- 7.Aukrust P, Dam äs JK, Gullestad L, Fröland SS. Chemokines in myocardial failure—pathogenic importance and potential therapeutic targets. Clin Exp Immunol. 2001;124:343–5. doi: 10.1046/j.1365-2249.2001.01527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker LC. Yin and Yang of MCP-1. Circ Res. 2005;96:812–4. doi: 10.1161/01.RES.0000165652.82726.d9. [DOI] [PubMed] [Google Scholar]
- 9.Kolattukudy PE, Quach T, Bergese S, Breckenridge S, Hensley J, Altschuld R, et al. Myocarditis induced by targeted expression of the MCP-1 gene in murine cardiac muscle. Am J Pathol. 1998;152:101–11. [PMC free article] [PubMed] [Google Scholar]
- 10.Moldovan NI, Goldschmidt-Clermont PJ, Parker-Thornburg J, Shapiro SD, Kolattukudy PE. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metal-loelastase-positive tunnels in ischemic myocardium. Circ Res. 2000;87:378–84. doi: 10.1161/01.res.87.5.378. [DOI] [PubMed] [Google Scholar]
- 11.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 12.Kang PM, Izumo S. Apoptosis and heart failure: a critical review of the literature. Circ Res. 2000;86:1107–13. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 13.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115:565–71. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, et al. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ Res. 1998;82:1130–8. doi: 10.1161/01.res.82.11.1130. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda S, Hamada M, Hiwada K. Contribution of non-cardiomyocyte apoptosis to cardiac remodeling that occurs in the transition from compensated hypertrophy to heart failure in spontaneously hypertensive rats. Clin Sci. 1999;97:239–46. [PubMed] [Google Scholar]
- 16.Seko Y, Kayagaki N, Seino KI, Yagita H, Okumura K, Nagai R. Role of Fas/FasL pathway in the activation of infiltrating cells in murine acute myocarditis caused by coxsackievirus B3. J Am Coll Cardiol. 2002;39:1399–403. doi: 10.1016/s0735-1097(02)01776-x. [DOI] [PubMed] [Google Scholar]
- 17.Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta. 2003;1639:141–51. doi: 10.1016/j.bbadis.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-dificient mice. Science. 2004;304:1147–50. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 19.Lagasse E, Weissman IL. Enforced expression of Bcl-2 in monocytes rescue macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997;89:1021–31. doi: 10.1016/s0092-8674(00)80290-1. [DOI] [PubMed] [Google Scholar]
- 20.Muller PY, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Bio-techniques. 2002;32:1372–9. [PubMed] [Google Scholar]
- 21.Brown SB, Savill J. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J Immunol. 1999;162:2110–9. [PubMed] [Google Scholar]
- 22.Stuart LM, Savill J, Lacy-Hulbert A. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. J Immunol. 2003;171:2610–5. doi: 10.4049/jimmunol.171.5.2610. [DOI] [PubMed] [Google Scholar]
- 23.Lecnen PJ, De Bruijn MF, Voerman JS, Campbell PA, Van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:5–10. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 24.Yeh ET, Willerson JT. Coming of age C-reactive protein: using inflammation markers in cardiology. Circulation. 2003;107:730–2. doi: 10.1161/01.cir.0000053731.05365.5a. [DOI] [PubMed] [Google Scholar]
- 25.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–20. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 26.Misao J, Hayakawa Y, Ohno M, Kato S, Fujiwara T, Fujiwara H. Expression of Bcl-2 protein, an inhibitor of apoptosis, and bax, an accelerator of apoptosis, in ventricular myocytes of humans with myocardial infarction. Circulation. 1996;94:1506–12. doi: 10.1161/01.cir.94.7.1506. [DOI] [PubMed] [Google Scholar]
- 27.Imanishi T, Han DKM, Hofstra L, Hano T, Nishio I, Liles WC, et al. Apoptosis of vascular smooth muscle cells is induced by Fas ligand derived from monocytes/macrophages. Atherosclerosis. 2002;161:143–51. doi: 10.1016/s0021-9150(01)00631-1. [DOI] [PubMed] [Google Scholar]
- 28.Lee P, Sata M, Lefer DJ, Factor SM, Walsh K, Kitsis RN. Fas pathway is a critical mediator for cardiac myocyte death and MI during ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol. 2003;184:H456–63. doi: 10.1152/ajpheart.00777.2002. [DOI] [PubMed] [Google Scholar]
- 29.O’Connel J. Role of Fas-FasL in inflammatory disease. Expert Rev Mol Med. 2001;10:1–18. [Google Scholar]
- 30.Andreotti F, Porto I, Crea F, Maseri A. Inflammation gene polymorphisms and ischemic heart disease: review of population association studies. Heart. 2002;87:107–12. doi: 10.1136/heart.87.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson L. Candidate-based proteomics in the search for biomarkers of cardiovascular disease. J Physiol. 2005;563:23–60. doi: 10.1113/jphysiol.2004.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu J, Azfer A, Deucher MF, Goldschmidt-Clermont PJ, Kolattukudy PE. Targeted myocardial expression of soluble Fas prevents the development of heart failure in mice with cardiac-specific expression of monocyte chemoattractant protein-1. Circulation. 2004;110(Suppl III):III-30. [Abstract] [Google Scholar]
- 33.Han KH, Hong KH, Park JH, Ko J, Kang DH, Choi KJ, et al. C-reactive protein promotes monocyte chemoattractant protein-1-mediated chemotaxis through upregulation CC chemokine receptor 2 expression in human monocytes. Circulation. 2004;109:2566–71. doi: 10.1161/01.CIR.0000131160.94926.6E. [DOI] [PubMed] [Google Scholar]
- 34.Badrichani AZ, Stroka DM, Bilbao G, Curiel DT, Bach FH, Ferran C. Bcl-2 and Bcl-XL serve an anti-inflammatory function in endothelial cells through inhibition of NF-kB. J Clin Invest. 1999;103:543–53. doi: 10.1172/JCI2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Z, Andersson R. NF-kappaB activation and inhibition: a review. Shock. 2002;18:99–106. doi: 10.1097/00024382-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Lakics V, Medvedev AE, Okada S, Vogel SN. Inhibition of LPS-induced cytokines by BCL-xl in a murine macrophage cell line. J Immunol. 2000;165:2729–37. doi: 10.4049/jimmunol.165.5.2729. [DOI] [PubMed] [Google Scholar]
- 37.Wilson EM, Diwan A, Spinale FG, Mann DL. Duality of innate stress responses in cardiac injury, repair, and remodeling. J Mol Cell Cardiol. 2004;37:801–11. doi: 10.1016/j.yjmcc.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 38.Hiasa KI, Egashira K, Kitamoto S, Ishibashi M, Inoue S, Ni W, et al. Bone marrow mononuclear cell therapy limits myocardial infarct size through vascular endothelial growth factor. Basic Res Cardiol. 2004;99:165–72. doi: 10.1007/s00395-004-0456-9. [DOI] [PubMed] [Google Scholar]