

Abstract
We used ethylcyclopentane (ECP) as a model alicyclic hydrocarbon and investigated its metabolism by a sulfate-reducing bacterial enrichment obtained from a gas condensate-contaminated aquifer. The enrichment coupled the consumption of ECP with the stoichiometrically expected amount of sulfate reduced. During ECP biodegradation, we observed the transient accumulation of metabolite peaks by gas chromatography-mass spectrometry, three of which had identical mass spectrometry profiles. Mass-spectral similarities to analogous authentic standards allowed us to identify these metabolites as ethylcyclopentylsuccinic acids, ethylcyclopentylpropionic acid, ethylcyclopentylcarboxylic acid, and ethylsuccinic acid. Based on these findings, we propose a pathway for the degradation of this alicyclic hydrocarbon. Furthermore, a putative metabolite similar to ethylcyclopentylsuccinic acid was also found in samples of contaminated groundwater from the aquifer. However, no such finding was evident for samples collected from wells located upgradient of the gas condensate spill. Microbial community analysis of the ECP-degrading enrichment by denaturing gradient gel electrophoresis revealed the presence of at least three different organisms using universal eubacterial primers targeting 550 bp of the 16S rRNA gene. Based on sequence analysis, these organisms are phylogenetically related to the genera Syntrophobacter and Desulfotomaculum as well as a member of the Cytophaga-Flexibacter-Bacteroides group. The evidence suggests that alicyclic hydrocarbons such as ECP can be anaerobically activated by the addition to the double bond of fumarate to form alkylsuccinate derivatives under sulfate-reducing conditions and that the reaction occurs in the laboratory and in hydrocarbon-impacted environments.
Alicyclic hydrocarbons can comprise a substantial fraction of organic molecules in petroleum mixtures, such as gas condensate, gasoline, and crude oil. In the former two mixtures, alicyclic hydrocarbons typically represent 11 to 12% (wt/wt) of the total hydrocarbons (certificate of analysis of unleaded gasoline, 1991 [American Petroleum Institute]; also unpublished data). In crude oil, this fraction can represent up to 12% (wt/wt), depending on the origin of the petroleum formation (20). Not surprisingly, complex petroleum mixtures containing alicyclic hydrocarbons find their way into the environment and pollute aquifers and various water bodies. As of 2001, more than 418,000 underground fuel tanks were found to be leaking hydrocarbons into the environment within the United States (31).
Despite the frequency of environmental contamination with petroleum mixtures and the quantitative importance of alicyclic hydrocarbons, the biological fate of the latter group of compounds under aerobic conditions has only rarely been addressed (28, 29) and, to our knowledge has not been documented in the absence of molecular oxygen. In contrast, the anaerobic biodegradation of petroleum constituents such as aromatic and normal paraffin hydrocarbons has been demonstrated under nitrate-reducing (1, 6, 10, 17, 19, 26, 27, 35), sulfate-reducing (2, 7, 18, 26), and methanogenic conditions (4, 36). Under anaerobic conditions these hydrocarbons are activated by a novel enzymatic mechanism that employs the common tricarboxylic acid cycle intermediate fumarate (1, 2, 4, 6, 11, 13, 17, 18, 21, 26, 27, 35). This fumarate addition mechanism produces a succinyl derivative of a hydrocarbon, such as benzylsuccinic acid from toluene, dodecylsuccinic acid from dodecane, and (1-methylpentyl)-succinic acid from hexane. In the case of toluene, the addition across the double bond of fumarate results in the production of near-optically pure (R)-(+)-benzylsuccinic acid (2, 19) and is catalyzed by the enzyme benzylsuccinate synthase. Furthermore, partially purified benzylsuccinate synthase catalyzed the addition of fumarate to the methyl groups of xylenes and 1-methyl-1-cyclohexene but failed to react with 4-methyl-1-cyclohexene or methylcyclohexane (3). These results led researchers to conclude that an aromatic ring or a conjugated double bond was necessary to stabilize the methyl radical produced during the fumarate addition reaction (3). In contrast, the activation of alkanes by this strategy does not require the formation of the alkene prior to fumarate addition (18, 27, 35).
We studied the anaerobic biodegradation of ethylcyclopentane (ECP), a model alicyclic hydrocarbon, under sulfate-reducing conditions and enriched for a bacterial consortium capable of mineralizing this substrate. The enrichment was obtained from a gas condensate-contaminated aquifer where the intrinsic bioremediation of hydrocarbons under sulfate reducing conditions was previously documented (11, 13). Ethylcyclopentane was chosen as a model compound because gas chromatographic (GC) analysis of contaminated core samples from the aquifer suggested that it was depleted relative to other hydrocarbons present in the gas condensate (unpublished results). This suggested that ECP might have been biologically attenuated in this aquifer in a manner similar to that previously demonstrated for BTEX hydrocarbons (13). In sediment-free laboratory enrichments, various putative metabolites were identified during the course of ECP degradation. Based on the identification of these metabolites, we propose a pathway for the degradation of ECP. In addition, GC-mass spectrometry (MS) analysis of extracted and derivatized groundwater samples suggested that the same metabolic strategy was used to activate alicyclic hydrocarbons in situ. To our knowledge, this is the first report of alicyclic hydrocarbon biodegraded under anaerobic conditions.
MATERIALS AND METHODS
Sample collection.
Sediments collected from a gas condensate-contaminated aquifer near Fort Lupton, Colo., were used to evaluate the anaerobic biodegradation of ECP. Uncontaminated and hydrocarbon-laden sediments were collected at the water table in jars that were filled to capacity, sealed without a headspace, stored on ice, and transported back to the laboratory. One-liter samples of groundwater were collected in sterile glass bottles from five monitoring wells within the hydrocarbon-impacted aquifer and from another well located upgradient of the contamination (13). The latter sample served as a background comparison and as incubation medium. The groundwater samples were used for DNA extraction or to search for putative hydrocarbon metabolites. The former samples were preserved on ice, while the latter samples were acidified in the field with sulfuric acid to a pH of <2. Aquifer sediments and groundwater were used for experiments immediately upon return to the laboratory.
Biodegradation experiments.
Background and hydrocarbon-contaminated sediments and groundwater were amended with ethylcyclopentane (1 μl undiluted, 7.8 μmol or ∼1 mM) or benzoate (1 mM) as electron donors and 10 mM sulfate as the electron acceptor. Benzoate was used as a positive control, since this compound is readily degraded under anaerobic conditions. Typically, 10 g of sediment and 20 ml of Na2S-reduced (0.1 ml/10 ml of a 1.25% solution), autoclaved pristine groundwater were placed in sterile 40-ml serum bottles while all materials were inside an anaerobic glove box containing 5% H2 in N2. The bottles were sealed with sterile composite stoppers (15a) and aluminum crimp seals. After the bottles were removed from the glove box, the headspaces of the incubations were exchanged with 20% CO2 in N2. Substrate-unamended and autoclaved samples served as controls. All incubations were at 30°C in the dark. The rate of sulfate depletion was monitored by ion chromatography as previously reported (7). Ethylcyclopentane concentrations in the incubations were monitored by headspace analysis on a Hewlett Packard model 5890 GC equipped with a flame ionization detector. Ten-microliter headspace samples were injected at 250°C using a gas-tight glass syringe onto a Carbograph-VOC column (30 m by 0.25 mm; Alltech, Deerfield, Ill.) held isothermally at 150°C with the flame ionization detector heated at 250°C. Helium was used as the carrier gas at a flow of 0.8 ml/min. Once ECP degradation was evident, 10% transfers of the culture into sulfate-medium containing ECP as the sole carbon and energy source were made repeatedly until a sediment-free enrichment was obtained.
Ethylcyclopentane metabolites.
To assay for putative ECP metabolites in the sediment-free enrichments, 30 ml of culture fluids were periodically taken and treated with base and then acid as previously reported (18). The pooled extracts were dried over anhydrous Na2SO4, and an internal standard (hexadecane) was added before concentration on a rotary evaporator and subsequently under a flow of N2 gas. The extracts were derivatized with N,O-bis(trimethylsilyl)trifluoroacetamide (Pierce, Rockford, Ill.) to form trimethylsilyl (TMS) esters. Groundwater samples, acidified on-site with sulfuric acid, were extracted with ethyl acetate, dried over Na2SO4, concentrated, and derivatized in the same manner. Authentic standards of 3-cyclopentylpropionic acid and cyclopentanecarboxylic acid were acquired from Aldrich (Milwaukee, Wis.), while n-propylsuccinic acid was a gift from the laboratory of Frank Abbott, Faculty of Pharmaceutical Science, University of British Columbia. These authentic standards were analyzed as TMS esters.
One microliter of the derivatized culture or groundwater extracts was analyzed using a HP Model 5890 GC connected to a HP Model 5970 MS detector. Injector and detector temperatures were held at 250°C. Compounds were separated using a DB-5 column (30 m by 0.25 mm; thickness, 0.1 μm; Alltech). The temperature program for the oven began at 65°C, was held for 3 min, and then was increased by 10°C/min to 155°C and held for 5 min. The oven temperature was further increased by 5°C/min to 185°C and then ramped by 20°C/min to 220°C with a final hold time of 5 min. Helium was used as the carrier gas at a flow of 0.8 ml/min.
DNA extraction.
Samples of the enrichment culture (1 to 2 ml) were placed in sterile polypropylene screw-cap tubes (2 ml) containing 1 g of 0.1-mm zirconia beads (BioSpec Products, Bartlesville, Okla.). Samples were centrifuged at 18,000 × g for 5 min, and 750 μl of the supernatants was discarded. The tubes were then stored at −20°C until DNA extraction could be performed. After the samples were thawed, 300 μl of each of the following liquids was added: 100 mM phosphate buffer (pH 8), lysis buffer (100 mM NaCl, 500 mM Tris (pH 8), 10% sodium dodecyl sulfate), and chloroform-isoamyl alcohol (24:1). The cells were physically disrupted in a mini-bead beater at 3,800 rpm for 1 min (BioSpec Products, Bartlesville, Okla.) (22). The DNA was isolated by phenol, chloroform-isoamyl alcohol (24:1) extraction and then precipitated with sodium acetate (3 M; pH = 7) and 100% cold ethanol and centrifuged at 18,000 × g for 20 min. The pellet was washed twice, first with 70% and then with 100% cold ethanol. The resulting DNA was resuspended in 20 μl of TE buffer (10 mM Tris [pH 8], 1 mM EDTA) or sterile water, and the nucleic acid concentration was estimated by density in an ethidium bromide-stained, 0.8% agarose gel.
The cells present in the groundwater samples were collected by centrifugation, resuspended in 1 ml of TE buffer (10 mM Tris [pH 8], 1 mM EDTA), and serially diluted prior to DNA extraction. The DNA was extracted using the procedure described above.
PCR amplification of 16S rRNA gene.
PCR amplifications were performed with a Techne Genius temperature cycler (Techne, Cambridge, United Kingdom). Approximately 25 ng of purified genomic DNA, 10 pmol each of the appropriate primers, 250 μmol of each deoxyribonucleoside triphosphate, 10 μl of 10× PCR buffer B (Fisher Scientific, Fair Lawn, N.J.), 10 μl of MgCl2 (25 mM), 2.5 U of Taq polymerase (Fisher Scientific), and sterile water were mixed to a final volume of 100 μl. The samples were amplified using a touchdown PCR protocol (8). This was carried out as follows: the samples were first denatured (94°C/4 min) and then subjected to 35 cycling steps of denaturing (94°C/45 s), annealing (45 s), and elongation (72°C/2 min). The process was completed following one final elongation step (72°C/10 min). The touchdown protocol started with an annealing temperature of 10°C above the expected annealing temperature (63°C) and was decreased by 1°C every two cycles until 53°C, the temperature at which 15 additional cycles were carried out (8). The two universal eubacterial primers GM5F and DS907R were used for amplification (23). These primers amplify a 550-bp fragment within the 16S rRNA gene (bp 341 to 907, Escherichia coli). All PCR product sizes were confirmed by agarose-gel electrophoresis, stained with ethidium bromide, and visualized by UV transillumination.
DGGE analysis.
The PCR products were separated by denaturing gradient gel electrophoresis (DGGE) using a D-code universal mutation detection system (Bio-Rad, Hercules, Calif.) as described previously (23). The PCR samples were loaded directly onto a 6% polyacrylamide gel with a 30 to 60% built-in denaturant gradient (100% is equal to 7 M urea and 40% formamide) in 1× TAE (40 mM Tris [pH = 7], 20 mM acetate, 1 mM EDTA). The gels were loaded with 12 to 20 μl of PCR sample, and electrophoresis was performed at a constant voltage of 130 V and a temperature of 60°C for 6 h or at 60 V for 16 h at the same temperature. After electrophoresis, gels were stained with ethidium bromide for 5 to 10 min and destained for 10 min in nanopure water. The gels were then photographed under UV transillumination (302 nm) using a Kodak DC120 digital camera and analyzed with the NucleoTech GelExpert-Lite software.
Sequencing of fragments.
The 550-bp fragments of interest were excised from the DGGE gels using a sterile razor blade and pipette tip, immediately placed in 200-μl sterile polypropylene tubes containing 36 μl of sterile water, and then stored at 4°C overnight. One microliter of supernatant was used as the DNA template in a PCR reamplification using the same primers, which was then run in a DGGE gel following the protocols described above. Once the purity of a given fragment was confirmed by DGGE, the PCR product was cleaned and concentrated by one of the following methods: Wizard PCR Preps DNA purification system (Promega, Madison, Wis.), UltraClean 15 (Mo Bio), or UltraFree-MC 30,000 NMWL filter unit (Millipore) following the manufacturer's instructions. If the fragment could not be purified by the above method, it was used as a template for cloning using the TOPO TA Cloning kit following manufacturer's instructions (Invitrogen, Carlsbad, Calif.).
Once the fragments were deemed pure, they were sequenced using the BigDye terminator cycle sequencing ready reaction kit (PE Applied Biosystems, Foster City, Calif.) according to the manufacturer's instructions. The primers used in the sequencing reactions were GM5F, DS907R, and the M13 vector primers (forward, GTAAAACGACGGCCAG; reverse, CAGGAAACAGCTATGAC; both 5′ to 3′); this resulted in the twofold coverage of the sequenced area.
Sequence analysis.
The sequences were aligned and corrected using the computer program Sequencer. Related sequences were obtained from the GenBank database by using the Blastn search program of the National Center for Biotechnology Information.
RESULTS
Response of microflora to substrate exposure.
In sediment-containing incubations amended with ECP and sulfate, the initial amount of ECP added was consumed in 150 days with an approximate lag period of 50 days. Sulfate consumption paralleled the ECP degradation activity observed in these incubations (data not shown). Ethylcyclopentane decay was not observed in the incubations consisting of uncontaminated sediments, although a positive control substrate (benzoate) was metabolized (data not shown). Neither ECP nor benzoate decay was detected in the autoclaved controls. Subsequent additions of ECP to the sediment-containing incubations were utilized without a lag phase and at higher rates of degradation. This sulfidogenic, ECP-degrading activity was transferable to sediment-free medium, since upon transfer the culture continued to consume ECP and sulfate (Fig. 1).
FIG. 1.

Ethylcyclopentane degradation by a sediment-free sulfate-reducing microbial enrichment derived from sediments impacted with gas condensate hydrocarbons. Open symbols, sulfate; closed symbols, ethylcyclopentane.
The amount of sulfate consumption theoretically expected, assuming complete ethylcyclopentane mineralization, was determined according to the following equation:
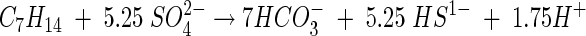 |
After correction for sulfate reduction in the substrate-unamended controls, the concentration of sulfate reduced in the sediment-free incubations was 3.51 mM. This amount represents 94% of the theoretically expected amount, assuming the complete mineralization of ECP.
Metabolites of ethylcyclopentane degradation.
After extraction, derivatization and GC-MS analysis of the ECP-degrading culture fluids, we detected four transient metabolites relative to a hexadecane internal standard in nonsterile incubations exposed to ECP. These peaks were not observed in the corresponding controls.
The first transient metabolite eluted as a cluster of three peaks which shared identical mass spectral profiles (Fig. 2). The ion at m/z 343 likely represents the molecular ion less 15 mass units, a common fragment observed with TMS-derivatized compounds, suggesting the loss of a methyl group from one TMS substituent (25). Thus, the ECP metabolite presumably has a mass of 358, which is consistent with the structure of the diTMS ester of ethylcyclopentylsuccinic acid. In addition, the mass spectra contained the key fragment ions m/z 73, 117, 147, and 204, which are associated with a molecule containing two TMS-derivatized carboxylic acids (25). The other ions observed in the MS profile are distinctive of and have been used to characterize the presumed fumarate addition metabolite of dodecane, namely dodecylsuccinic acid (TMS ester; at m/z 262, 217, 172, 147, and 73) (18). Thus, we propose that these ECP metabolites are ethylcyclopentylsuccinic acids. The GC cluster of three peaks could be further resolved into five peaks (using a different temperature program), all of which had the same MS profile. This observation may be a reflection of the maximum number of stereoisomers resolved by our analytical method.
FIG. 2.

Mass-spectral profiles of presumed ethylcyclopentylsuccinic acid isomers (analyzed as TMS esters). (A and B) Mass profiles of two of the three metabolites found in laboratory enrichment culture degrading ECP. (C) Putative metabolite found in all contaminated groundwater samples analyzed having the same GC retention time as one of the metabolite peaks in the microbial enrichment.
Another putative metabolite found in the ECP culture extracts had a presumed M+-15 of m/z 227, suggesting a molecular weight of 242 (Fig. 3A). The mass spectrum of this metabolite showed predominant ions at m/z 145, 129, 117, 75, 73, and 55. This compound was tentatively identified as ethylcyclopentylpropionic acid by comparison of its MS profile to that of an authentic standard of 3-cyclopentylpropionic acid (Fig. 3B). The mass spectra of these two compounds were nearly identical with the exception that the M+-15 ion of the metabolite was 28 mass units higher (m/z 227 versus m/z 199). This difference is consistent with the mass of the ethyl group, absent from the TMS-derivatized authentic standard. Similarly, a third metabolite was identified as ethylcyclopentylcarboxylic acid. This compound had an M+-15 ion of m/z 199, suggesting a molecular weight of 214, and predominant ions at m/z 145, 129, 117, 96, 75, 73, and 55 (Fig. 4A). The mass spectrum of this metabolite was compared with that of the authentic standard cyclopentanecarboxylic acid (Fig. 4B). Again, the MS profiles of these two compounds were nearly identical with the exception that the M+-15 ion of the formed metabolite was 28 mass units higher (m/z 199 versus m/z 171) than the standard. Finally, the fourth metabolite was tentatively identified as ethylsuccinic acid. This compound had an M+-15 ion of m/z 275, suggesting a molecular weight of 290, and predominant ions at m/z 55, 73, 75, 117, 129, 147, 172, 204, 217, and 262 (Fig. 5A). The mass spectrum of this metabolite was compared with that of an authentic standard of n-propylsuccinic acid (Fig. 5B) and was identical except for the M+-15 ion, which was 14 mass units less than that of the standard. In addition, this metabolite shared many of the predominant ions with the ethylcyclopentylsuccinic acids (Fig. 3) and dodecylsuccinic acid (18).
FIG. 3.

(A) Mass spectrum of a metabolite tentatively identified as ethylcyclopentylpropionic acid (as TMS ester) in ECP-degrading cultures. (B). Mass spectrum of an authentic standard of 3-cyclopentylpropionic acid (as TMS ester).
FIG. 4.

(A) Mass spectrum of a metabolite tentatively identified as ethylcyclopentylcarboxylic acid (as TMS ester) in ECP-degrading cultures. (B) Mass spectrum of an authentic standard of cyclopentylcarboxylic acid (as TMS ester).
FIG. 5.

(A) Mass spectrum of a metabolite tentatively identified as ethylsuccinic acid (as TMS ester) in ECP-degrading cultures. (B) Mass spectrum of an authentic standard of n-propylsuccinic acid (as TMS ester).
A putative hydrocarbon metabolite with the same GC retention time and MS features as one of the anaerobic fumarate addition intermediates produced by the ECP-degrading culture was also detected in groundwater from the gas condensate-contaminated aquifer from which the laboratory culture was derived (Fig. 2C). This metabolite could not be detected in the groundwater samples from the background location but was detected in all five contaminated locations.
DGGE findings.
The amplifiable eubacterial microbial community in the sediment-free ECP-degrading enrichment was assayed by DGGE. We were able to purify and sequence three predominant bands present in this microbial culture (Fig. 6). One of the bands showed 97% sequence homology to the uncultured eubacterial clone OCG6 (accession no. AB047117) in the CFB group (Fig. 6). The other organisms in the enrichment were similarly sequenced to reveal the presence of organisms phylogenetically related to sulfate reducers belonging to the genera Desulfotomaculum (96%) (accession no. AB074935.1) and Syntrophobacter (92%) (accession no. X82875) species (Fig. 6).
FIG. 6.

DGGE of PCR-amplified 16S rRNA genes and phylogenetic characterization of the microorganisms amplified from the ECP-degrading culture using the partial sequence of the 16S rRNA gene. Lanes: 1, 97% similar to uncultured eubacterial clone OCG6; 2, 92% similar to a syntrophic propionate-oxidizing bacterium, KoProp1; 3, 96% similar to a clone, KB13; 4, ECP-degrading culture. Numbers in parentheses indicate GenBank accession numbers.
DISCUSSION
To our knowledge, this is the first study to report that a saturated alicyclic hydrocarbon can serve as an electron donor for sulfate-reducing bacteria. The fact that this activity was observed with contaminated sediments and not with pristine sediments might suggest an adaptation by the microbial community to the selective pressure imposed by the gas condensate contaminants. The possibility that the organisms within the pristine sediments were killed or inactivated by the manipulation of the sample could be ruled out, since microorganisms from these sediments were able to degrade benzoate. This indicates that the microorganisms were viable and that the incubations were anaerobic, since sulfate was reduced in stoichiometrically expected amounts. However, it is also possible that the organisms responsible for the degradation of ECP have a lower tolerance to oxygen than the organisms responsible for the degradation of benzoate. If this is correct, then the highly reduced contaminated sediments may have protected the ECP degraders from the possible exposure to molecular oxygen, while the pristine sediments were incapable of providing the same protection.
Anaerobic ethylcyclopentane biodegradation under sulfate-reducing conditions appears to be mediated by an anaerobic fumarate addition mechanism. This activation mechanism was previously demonstrated for alkylbenzenes (1-3, 6, 17, 19, 26), straight chain alkanes (18, 27), and for methylnaphthalene (21) occurring under nitrate- and/or sulfate-reducing conditions and for toluene under methanogenic conditions (4). The mass spectra of the observed ECP metabolites formed by microorganisms in the sediment-free incubations (analyzed as TMS esters) contained key fragment ions associated with two TMS-derivatized functional groups (m/z 73, 117, 147, and 204) and a succinyl moiety (m/z 172, 217, and 262) (Fig. 2A). These mass-spectral features were also prominent in TMS-derivatized dodecylsuccinic acid, a metabolite associated with anaerobic dodecane biodegradation (18). The fact that we observed up to five chromatographic components with identical MS profiles suggests that the attack of ECP is most likely at positions 2 or 3 of the alicyclic ring with respect to the ethyl group (Fig. 2). The presumed addition of fumarate to ECP at either of these positions would result in a metabolite with three chiral centers, assuming that the enzyme responsible for such an addition reaction is not stereoselective. This assumption is in contrast to studies performed with benzylsuccinate synthase characterized from Thauera aromatica that has been shown to be highly stereoselective (2, 19). The production of the three chiral centers within the diTMS ester of ethylcyclopentylsuccinic acid would theoretically result in the formation of eight possible stereoisomers (M = 2n), producing four pairs of enantiomers, which results in four diastereomers. The latter could be resolved with our GC conditions, since they have different physical properties, producing four distinct peaks with identical MS profiles (although we observed five peaks). The addition of fumarate at other positions within the ECP molecule would produce fewer chiral centers and correspondingly fewer diastereomers than we have successfully resolved. It is possible that more than one organism in the enrichment or the same organism could activate ECP at different positions (carbons 2 or 3 of the ring) with respect to the methine carbon in the succinyl moiety, producing four stereoisomers and two diastereomers from each position. This could feasibly produce four distinct GC peaks that correspond to the four diastereomers. However, the mass-spectral profile of these metabolites will most likely be different, due to the proximity of the succinyl moiety to the ethyl group (attack to carbon 2), not to mention the steric effect that this group will cause if both groups are positioned simultaneously to the front or back of the cyclopentane plane. At present, the lack of an authentic standard precludes a more rigorous identification of these metabolites.
Interestingly, we identified a similar putative metabolite (ethylcyclopentylsuccinic acid) in all contaminated groundwater samples obtained from the gas condensate impacted aquifer from which the ECP-degrading culture was derived. This metabolite had the same GC retention time and MS profile as one of the three peaks produced by the enrichment culture. Importantly, this metabolite was not found in the pristine portion of the aquifer. The fact that we can only identify one comigrating peak does not indicate that the other two peaks formed in the enrichment culture are not formed in situ; this observation may just reflect our analytical limitations. It is possible that these metabolites are formed and degraded simultaneously in situ by organisms that we were unable to culture in the laboratory. It is also possible that the putative metabolite found in native groundwater is an intermediate of another alicyclic compound, such as methylcyclohexane or dimethylcyclopentane, since both of these chemicals are present at higher concentrations within the gas condensate mixture (unpublished data). Nevertheless, these findings suggest that alicyclic hydrocarbons like ECP could be subject to biodegradation by anaerobic activation mechanisms only in the contaminated portion of the aquifer. This suggests that the anaerobic metabolites of alicyclic hydrocarbons could be used as indicators of in situ anaerobic biodegradation, as has been found with alkylbenzenes and alkanes (5, 11, 14). It should be noted that we have previously detected succinic acid analogs in this gas condensate-contaminated aquifer resulting from ethylbenzene and xylene decay under sulfate-reducing conditions (11) and, most recently, from alkane degradation (14). The finding of a putative anaerobic metabolite of an alicyclic compound at this site extends our knowledge of the biodegradation capabilities of the native microbial populations at hydrocarbon-impacted sites.
The other metabolites produced during ECP degradation were identified based on comparison with analogous authentic standards. Ethylcyclopentylpropionic acid and ethylcyclopentylcarboxylic acid were compared to their ethyl-free counterparts, differing only in their M+ and M+-15 fragments by 28 mass units (Fig. 3 and 4). The common ion observed at m/z 145 (Fig. 3 and 4) is produced by a rearrangement of the derivatized propyl tail, which undergoes another rearrangement to lose a methane equivalent (16 mass units), producing the ion at m/z 129 (30). The abundant ion at m/z 117 is thought to be the result of a methyl group loss from ion m/z 132 (9). The ion at m/z 73 indicates that a functional group has been derivatized as a TMS ester (25), while the ion at m/z 75 is the result of a rearrangement and the loss of a methylene group from an ion at m/z 89 (30).
Ethylsuccinic acid (Fig. 5) was identified by comparison to the authentic standard of n-propylsuccinic acid. In this particular case the standard was 14 mass units heavier than the metabolite, because it had an additional methylene (CH2) group. Notably, the predominant ions present in the MS profile of ethylsuccinic acid were present in those from n-propylsuccinic acid, ethylcyclopentylsuccinic acid, and all alkylsuccinates derivatized with TMS (14, 18). The majority of the ions present in these profiles are highly indicative of a succinyl moiety.
Based on the metabolite evidence presented herein, we propose a pathway for the anaerobic biodegradation of ECP under sulfate-reducing conditions (Fig. 7). This pathway is analogous to the pathway outlined by Wilkes et al. (35) for the degradation of hexane by a denitrifying organism (HxN1) that uses fumarate addition to activate alkanes. Our pathway also includes the regeneration of the presumed cosubstrate fumarate (Fig. 7, P) for the activation of ECP (Fig. 7, A) to ethylcyclopentylsuccinic acid (Fig. 7, B).
FIG. 7.

Proposed pathway for the anaerobic biodegradation of the hydrocarbon ethylcyclopentane by a sulfate-reducing enrichment culture. In the following list of depicted compounds, those that have been identified as TMS esters in culture supernatants are in boldface. A, ethylcyclopentane; B, ethylcyclopentylsuccinic acid; C, ethylcyclopentylmethylmalonic acid; D, ethylcyclopentylpropionic acid; E, ethylcyclopentanecarboxylic acid; F, ethylcyclopent-1-enecarboxylic acid; G, 2-hydroxyethylcyclopentanecarboxylic acid; H, 2-oxoethylcyclopentanecarboxylic acid; I, β-ethyladipic acid; J, ethylsuccinic acid; K, propylmalonic acid; L, valerate; M, propionate; N, methylmalonic acid; O, succinic acid; P, fumarate. Dashed lines indicate alternate possible bioconversions.
In the pathway, ethylcyclopentylsuccinic acid is presumably decarboxylated to produce the ethylcyclopentylpropionic acid (Fig. 7, D). We do not know if the removal of the CO2 is directly from the succinyl moiety or if a carbon rearrangement producing ethylcyclopentylmethylmalonic acid (Fig. 7, C) is necessary prior to the loss of CO2 as proposed by Wilkes et al. (35). The next metabolite identified in our proposed pathway is ethylcyclopentanecarboxylic acid (Fig. 7, E). This metabolite would presumably result from β-oxidation of the propionic acid substituent, a step which would also produce acetate and reducing equivalents. The cyclopentyl ring of this metabolite may then become unsaturated (Fig. 7, F) and be sequentially oxidized to an alcohol (Fig. 7, G) and a ketone (Fig. 7, H), followed by ring cleavage, producing the proposed metabolite β-ethyladipic acid (Fig. 7, I). These series of reactions are similar to the well-characterized reactions carried out by the phototrophic anaerobe Rhodopseudomonas palustris as well as Syntrophus acidotrophicus when grown on benzoate (12, 24). β-Ethyladipic acid (Fig. 7, I) is postulated to then undergo classical β-oxidation of the longer fatty-acid chain to produce a second molecule of acetate, reducing equivalents, and the identified metabolite ethylsuccinic acid (Fig. 7, J). This metabolite is a key intermediate in this pathway since it would be produced regardless of the actual position (carbon 2 or 3) at which the initial attack of the fumarate takes place. Ethylsuccinic acid may be further decarboxylated to the fatty acid valerate (Fig. 7, L). Again, this decarboxylation could occur directly from the succinyl derivative or after a carbon rearrangement by an unknown mechanism analogous to the decarboxylation of ethylcyclopentylsuccinic acid (Fig. 7, steps B to D). Thus, valerate may be produced directly or via the presumed metabolite propylmalonic acid (Fig. 7, K). Valerate may then undergo β-oxidation to produce propionate (Fig. 7, M), which may be carboxylated to methylmalonic acid (Fig. 7, N) and further metabolized to succinic acid (Fig. 7, O). These series of reactions would be similar to those used by propionate oxidizers represented by the genera Desulfobulbus (16) and Syntrophobacter (33). Finally, succinic acid is then dehydrogenated, regenerating fumarate.
The DGGE profile of the sediment-free ECP-degrading culture shows the presence of three predominant bands representing the dominant amplifiable eubacterial microorganisms in the culture (Fig. 6). The associated sequences suggest that the organisms are phylogenetically related to the genus Desulfotomaculum within the Bacillus-Clostridium group, the genus Syntrophobacter within the δ subdivision of the Proteobacteria, and to the members of the Bacteroidaceae within the CFB group (Fig. 6). The sum of these organisms, represented by their sequences, constitutes the predominant members of the ECP-degrading culture and have been deposited in GenBank as AF529223, AF529224, and AF529225, respectively. The sequence belonging to the Bacteroidaceae was 97% homologous to an uncultured clone obtained from groundwater contaminated with oil from an underground storage cavity (GenBank accession no. AB047117). It is very difficult to discuss the niche of this particular organism within our enrichment without knowing the metabolic ability of its closest relative, but their respective habitats are clearly related to hydrocarbon contamination. In contrast, we found a sequence that is related to the genus Syntrophobacter, comprised of organisms which are best known for their ability to degrade fatty acids and other simple organic molecules by syntrophic association or using sulfate as a terminal electron acceptor (15, 32, 33). Interestingly, various metabolites that might serve as substrates for these organisms are proposed in the ECP pathway (Fig. 7). Unfortunately, we were unable to detect the transient accumulation of fatty acids, such as acetate or propionate, in the cultures actively degrading ECP. It is possible that these fatty acids were consumed as rapidly as they were produced, maintaining their concentration lower than 10 μM, our detection limit for these compounds. Furthermore, we were able to isolate this organism, and it is not able to degrade ECP as a pure culture or in coculture with a hydrogen-consuming methanogen (data not shown). A brief characterization of its degradative range revealed its ability to degrade acetate, propionate, isovalerate, and crotonate under sulfate-reducing conditions (data not shown). These findings suggest that this organism might not be the ECP degrader in our ECP-degrading enrichment but might play an important role in the degradation of low-molecular-weight fatty acids. The other organism capable of using sulfate as a terminal electron acceptor in our enrichment based on our sequence data is an organism related to the genus Desulfotomaculum obtained from oil-contaminated groundwater (GenBank sequence accession no. AB074935.1). The organisms in this genus are not known for their ability to degrade hydrocarbons, but alcohols, fatty acids, other aliphatic monocarboxylic or dicarboxylic acids among other compounds are used as electron donors for dissimilatory sulfate reduction (34). The fact that ECP degradation is coupled to sulfate reduction in our enrichment and the inability of the Syntrophobacter sp. isolate to degrade ECP support our working hypothesis that the Desulfotomaculum sp. is responsible for the degradation of ECP.
Acknowledgments
This research was supported in part through grants from the Integrated Petroleum Environmental Consortium (IPEC) and EPSCoR.
We thank Frank Abbott for supplying n-propylsuccinic acid and Kathleen Duncan and Mostafa S. Elshahed for critical comments and discussions on the manuscript.
REFERENCES
- 1.Beller, H. R., and A. M. Spormann. 1997. Anaerobic activation of toluene and o-xylene by addition to fumarate in denitrifying strain T. J. Bacteriol. 179:670-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beller, H. R., and A. M. Spormann. 1997. Benzylsuccinate formation as a means of anaerobic toluene activation by sulfate-reducing strain PRTOL1. Appl. Environ. Microbiol. 63:3729-3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beller, H. R., and A. M. Spormann. 1999. Substrate range of benzylsuccinate synthase from Azoarcus sp. strain T. FEMS Microbiol. Lett. 178:147-153. [DOI] [PubMed] [Google Scholar]
- 4.Beller, H. R., and E. A. Edwards. 2000. Anaerobic toluene activation by benzylsuccinate synthase in a highly enriched methanogenic culture. Appl. Environ. Microbiol. 66:5503-5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beller, H. R. 2000. Metabolic indicators for detecting in situ anaerobic alkylbenzene degradation. Biodegradation 11:125-139. [DOI] [PubMed] [Google Scholar]
- 6.Biegert, T., G. Fuchs, and J. Heider. 1996. Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur. J. Biochem. 238:661-668. [DOI] [PubMed] [Google Scholar]
- 7.Caldwell, M. E., R. M. Garrett, R. C. Prince, and J. M. Suflita. 1998. Anaerobic biodegradation of long-chain n-alkanes under sulfate-reducing conditions. Environ. Sci. Technol. 32:2191-2195. [Google Scholar]
- 8.Don, R. H., P. T. Cox, B. Wainwright, K. Baker, and J. S. Mattick. 1991. “Touchdown” PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 19:4008-4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draffan, G. H., R. N. Stillwell, and J. A. McCloskey. 1968. Electron impact-induced rearrangement of trimethylsilyl groups in long chain compounds. Org. Mass Spectrom. 1:669. [Google Scholar]
- 10.Ehrenreich, P., A. Behrends, J. Harder, and F. Widdel. 2000. Anaerobic oxidation of alkanes by newly isolated denitrifying bacteria. Arch. Microbiol. 173:58-64. [DOI] [PubMed] [Google Scholar]
- 11.Elshahed, M. S., L. M. Gieg, M. J. McInerney, and J. M. Suflita. 2000. Signature metabolites attesting to the in situ attenuation of alkylbenzenes in anerobic environments. Environ. Sci. Technol. 35:682-689. [DOI] [PubMed] [Google Scholar]
- 12.Elshahed, M. S., M. A. Nanny, N. Q. Wofford, V. K. Bhupathiraju, and M. J. McInerney. 2001. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus acidotrophicus” strain SB in syntrophic association with H2-using microorganisms. Appl. Environ. Microbiol. 67:1728-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gieg, L. M., R. V. Kolhatkar, M. J. McInerney, R. S. Tanner, S. H. Harris, Jr., K. L. Sublette, and J. M. Suflita. 1999. Intrinsic bioremediation of petroleum hydrocarbons in a gas condensate-contaminated aquifer. Environ. Sci. Technol. 33:2550-2560. [Google Scholar]
- 14.Gieg, L. M., and J. M. Suflita. 2002. Detection of anaerobic metabolites of saturated and aromatic hydrocarbons in petroleum-contaminated aquifers. Environ. Sci. Technol. 36:3755-3762. [DOI] [PubMed] [Google Scholar]
- 15.Harmsen, H. J. M., B. L. M. Van Kuijk, C. M. Plugge, A. D. L. Akkermans, W. M. De Vos, and J. M. Stams. 1998. Syntrophobacter fumaroxidans sp. nov., a syntrophic propionate-degrading sulfate-reducing bacterium. Int. J. Syst. Bacteriol. 48:1383-1387. [DOI] [PubMed] [Google Scholar]
- 15a.Kazumi, J., M. E. Caldwell, J. M. Suflita, D. R. Lovley, and L. Y. Young. 1997. Anaerobic degradation of benzene in diverse anoxic environments. Environ. Sci. Technol. 31:813-818. [Google Scholar]
- 16.Kremer, D. R., and T. A. Hansen. 1988. Pathway of propionate degradation in Desulfobulbus propionicus. FEMS Microbiol. Lett. 49:273-277. [Google Scholar]
- 17.Krieger, C. J., H. R. Beller, M. Reinhard, and A. M. Spormann. 1999. Initial reactions in anaerobic oxidation of m-xylene by the denitrifying bacterium Azoarcus sp. strain T. J. Bacteriol. 181:6403-6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kropp, K. G., I. A. Davidova, and J. M. Suflita. 2000. Anaerobic oxidation of n-dodecane by an addition reaction in a sulfate-reducing bacterial enrichment culture. Appl. Environ. Microbiol. 66:5393-5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leutwein, C., and J. Heider. 1999. Anaerobic toluene-catabolic pathway in denitrifying Thauera aromatica: activation and β-oxidation of the first intermediate, (R)-(+)-benzylsuccinate. Microbiology 145:3265-3271. [DOI] [PubMed] [Google Scholar]
- 20.Malins, D. C. 1977. Effects of petroleum on arctic and subarctic marine environments and organics, vol. 1, p. 24. Academic Press, New York, N.Y.
- 21.Meckenstock, R. U., E. Annweiler, A. Materna, M. Safinowski, A. Kappler, H. H. Richnow, and W. Michaelis. 2000. Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl. Environ. Microbiol. 66:5329-5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller, D. N., J. E. Bryant, E. L. Madsen, and W. C. Ghiorse. 1999. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 65:4715-4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muyzer, G., A. Teske, C. O. Wirsen, and H. W. Jannasch. 1995. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164:165-172. [DOI] [PubMed] [Google Scholar]
- 24.Pelletier, D. A., and C. S. Harwood. 1998. 2-Ketocyclohexanecarboxyl coenzyme A hydrolase, the ring cleavage enzyme required for anaerobic benzoate degradation by Rhodopseudomonas palustris. J. Bacteriol. 180:2330-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pierce, A. E. 1968. Silylation of organic compounds. Pierce Chemical Co., Rockford, Ill.
- 26.Rabus, R., and J. Heider. 1998. Initial reactions of anaerobic metabolism of alkylbenzenes in denitrifying and sulfate-reducing bacteria. Arch. Microbiol. 170:377-384. [Google Scholar]
- 27.Rabus, R., H. Wilkes, A. Behrends, A. Armstroff, T. Fischer, A. J. Pierik, and F. Widdel. 2001. Anaerobic initial reaction of n-alkanes in a denitrifying bacterium: evidence for (1-methylpentyl)succinate as initial product and for involvement of an organic radical in n-hexane metabolism. J. Bacteriol. 183:1707-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solano-Serena, F., R. Marchal, J. M. Lebeault, M. Ropars, and J. P. Vandecasteele. 1999. Biodegradation of gasoline: kinetics, mass balance and fate of individual hydrocarbons. J. Appl. Microbiol. 86:1008-1016. [DOI] [PubMed] [Google Scholar]
- 29.Solano-Serena, F., R. Marchal, S. Casaregola, C. Vasnier, J. M. Lebeault, and J. P. Vandecasteele. 2000. A Mycobacterium strain with extended capacities for degradation of gasoline hydrocarbons. Appl. Environ. Microbiol. 66:2392-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tulloch, A. P. 1985. Mass spectra of TMS esters of deuterated decanoic acids and of TMS ethers of deuterated decanols. Lipids 20:404-411.
- 31.U.S. Environmental Protection Agency. 2002. Office of Underground Storage Tanks: Cleaning up UST system releases. [Online.] http://www.epa.gov/swerust1/cat/index.htm.
- 32.Wallrabenstein, C., E. Hauschild, and B. Schink. 1994. Pure culture and cytological properties of Syntrophobacter wolinii. FEMS Microbiol. Lett. 123:249-254. [Google Scholar]
- 33.Wallrabenstein, C., E. Hauschild, and B. Schink. 1995. Syntrophobacter pfennigii sp. nov., new syntrophically propionate-oxidizing anaerobe growing in pure culture with propionate and sulfate. Arch. Microbiol. 164:346-352. [Google Scholar]
- 34.Widdel, F. 1992. The genus Desulfotomaculum, p. 1793-1799. In A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K.-H. Schleifer (ed.), The procaryotes, vol. 2. Springer-Verlag, Inc., New York, N.Y.
- 35.Wilkes, H., R. Rabus, T. Fischer, A. Armstroff, A. Behrends, and F. Widdel. 2002. Anaerobic degradation of n-hexane in a denitrifying bacterium: further degradation of the initial intermediate (1-methylpentyl)succinate via C-skeleton rearrangement. Arch. Microbiol. 177:235-243. [DOI] [PubMed] [Google Scholar]
- 36.Zengler, K., H. H. Richnow, R. Rossello-Mora, W. Michaelis, and F. Widdel. 1999. Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401:266-269. [DOI] [PubMed] [Google Scholar]