

Abstract
High-throughput screening (HTS) campaigns can be dominated by hits that ultimately turn out to be non-drug-like. These ‘nuisance’ compounds often behave strangely, with steep dose-response curves, absence of clear structure-activity relationships, and high sensitivity to assay conditions. Several mechanisms contribute to these artifacts, including chemically reactive molecules, those that absorb light in assays and those that affect redox conditions. One of the most common mechanisms behind artifactual inhibition is discussed in this review: at micromolar concentrations organic molecules can aggregate to form particles in aqueous buffers, and these aggregates can sequester and thereby inhibit protein targets. Aggregation-based inhibition is baffling from a chemical perspective, but viewed biophysically such behavior is expected. The range of molecules that behave this way, their rapid detection in a screening environment and their possible biological implications will be considered here.
If high-throughput screening (HTS) has changed drug discovery, it has also introduced into it a bestiary of peculiar molecules. Some of these have turned out to be interesting and important; others have proven to be ‘nuisance’ compounds with strange properties. Steep dose-response curves, flat structure-activity relationships and high sensitivity to assay conditions are unusual with classic, well-behaved drugs and reagents, but are common among nuisance hits. These are rarely suited for development, but much time and passion can be wasted chasing them before they are abandoned. Their prevalence has contributed to the evolution of screening practices towards high-quality compound libraries, the maintenance of dry stocks of pure compounds and ever-lower concentrations of compound in initial screens.
‘Nonsense is always nonsense, but the study of nonsense can be scholarship’, said Saul Lieberman of the Kabbalist Gershom Scholem. Much scholarly ink has been spilled on compounds in screening decks that are prone to artifactual inhibition. Lipinski’s now famous rules [1] focused on the physical properties of drugs, reacting to an early tendency in HTS libraries toward large and hydrophobic molecules that were unlikely to be orally bioavailable. Subsequent studies, typically using retrospective analysis of hit lists, have focused on chemical reactivity [2], assay interference [3], high flexibility [4], oxidation potential [5], formal molecular charge [6], or liability to degradation and precipitation [7]. Indeed, these characteristics have been incorporated at most pharmaceutical companies using computational filters that flag likely nuisance compounds in screening collections, so that they can be scrutinized when reviewing screening hit lists. Whereas these filters have been implemented since the late 1990s, identifying pathological hits unambiguously using these criteria has proven difficult. As one class of nuisance inhibitor is identified, another emerges Hydralike. This is partly a problem of the apparent specificity of nuisance compounds for particular assay conditions-a promiscuous hit in one assay can behave demurely in another, conferring on it a cruel imitation of fidelity. But there were also screening hits that did not obviously manifest the nuisance properties identified in the initial studies. These molecules did not appear to be chemically reactive, were not obviously interfering spectrally, passed internal filters and Lipinski rules and had little in common other than their similar behavior in assays. This assay behavior was unusual: many compounds had steep dose-response curves [3] and many series, on investigation, led to flat structure-activity relationships (i.e. when analogs were made around the initial hit, only small changes in affinity were observed). Such compounds were widely known among screeners but the mechanism and properties that related them were obscure. They were not publicly discussed.
This review describes a single mechanism that explains the behavior of apparently unrelated nuisance hits and is consistent with their sensitivities to assay conditions and perverse variability. At micromolar and sometimes submicromolar concentrations, many drug-like organic molecules aggregate into colloid-like particles in aqueous media. These aggregates can sequester protein targets, thereby inhibiting them. Aggregating inhibitors are often unrelated chemically, although they typically share certain physical properties. Like colloids and vesicles, they are sensitive to assay conditions and target concentration. This contributes to their haunted, skittish behavior. From a chemical perspective this can be baffling, but from a biophysical point of view, such behavior is expected. Indeed, based on these features, aggregation-based ‘promiscuous’ inhibitors can be rapidly detected and controlled for. In this article, I summarize the range of molecules now known to behave this way, their mechanism of action, their rapid detection in a screening environment and their possible effects in biological environments.
Phenomenology of aggregation
We encountered nuisance compounds by accident, while looking for inhibitors of the enzyme β-lactamase. We had tested tens of compounds predicted by virtual screening, finding many apparent inhibitors (Table 1). All had strange properties: they were non-competitive, time-dependent and inhibited not only β-lactamase, but also dihydrofolate reductase (DHFR), chymotrypsin, β-galactosidase, and malate dehydrogenase (MDH) [8]. They also had unusually steep dose-response curves. Although there are reasons that can explain such curves, such as a high enzyme to Ki ratio [9], such dose-response curves are unusual. We initially thought that these compounds might be covalent inhibitors, but inhibition was reversible by dilution, inconsistent with such a mechanism of action. We then wondered if these inhibitors, so dissimilar structurally, were acting as denaturants. If this was true, we might have expected inhibition to be increased by guanidinium, urea or temperature. Instead, when we tested this, the opposite was true, inhibition was attenuated. Intriguingly, the potency of these compounds was strangely sensitive to protein concentration, diminishing considerably on addition of large amounts of bovine serum albumin or even increased amounts of the target enzyme [8]. Thus, increasing the concentration of β-lactamase in the assay ten-fold diminished potency dramatically. Correspondingly, increasing inhibitor concentration by a similar amount would return efficacy. This was difficult to reconcile with any classical mechanism of enzyme inhibition of which we were then aware. Rather, it seemed to point to a stoichiometric mechanism of inhibition, except that stoichiometries would not be 1:1 or even 10:1, but more like thousands of inhibitor molecules to one enzyme molecule. The only mechanism that we could think of with such high molar ratios was one where the inhibitors acted as colloid-like aggregates that somehow sequestered and inhibited enzyme targets without specificity.
TABLE 1.
Early inhibitors found to act through aggregationa
| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| structure | Original target(s) | β-Lactamase | Chymotrypsin | cDHFR | β-Gal | |
 |
0.5 β-Lactamaseb | 0.5 | 2.5 | 5 | 15 | |
| 5 β-Lactamaseb | 5 | 25 | 35 | 90 | ||
 |
5 β-Lactamaseb | 5 | 15 | N.D. | N.D. | |
| 8 Malarial protease | 10 | 55 | 70 | 180 | ||
| 7 pDHFR | 10 | 50 | 60 | 300 | ||
 |
80 pDHFR | 50 | 25 | N.D. | 600 | |
 |
50 HIV Tar RNA | 10 | 90 | N.D. | 600 | |
 |
3 TS | 30 Kinesin | 3 | 11 | 20 | 200 |
 |
20cInsulin receptor | 7.5 Kinesin | 16 | 50 | N.D. | 80 |
 |
5.2 VEGF | 10.0 IGF-1 | 6 | 30 | 30 | 55 |
 |
25 Farnesyltransferase | 3 | 9 | 25 | 150 | |
 |
15dGyrase | 18 | 100 | 150 | 320 | |
 |
1 Prion | 30.4 TIM | 3.9 | 40 | 0.4 | 100 |
 |
17 eNOS | 24 nNOS | 7 | 60 | N.D. | N.D. |
 |
3.8 PI3K | 11.0 Integrase | 4 | 100 | N.D. | 220 |
Abbreviations: cDHFR, chicken DHFR; β-gal, β-galactosidase; pDHFR, Pneumocystis carinii DHFR; TS, thymidylate synthase; VEGF, vascular endothelial growth factor receptor tyrosine kinase; IGF-1, insulin-like growth factor receptor tyrosine kinase; TIM, triosephosphate isomerase; eNOS, endothelial nitric oxide synthase; nNOS, neuronal nitric oxide synthase; PI3K, phosphoinositide 3-kinase; N.D., not determined.
Table adapted, with permission, from Ref. [8].
Unpublished observations.
Kd.
Maximal non-effective concentration.
A virtue of this hypothesis was that it was directly testable. Both dynamic light scattering (DLS) and transmission electron microscopy unambiguously detected particles when aqueous mixtures of these inhibitors were examined (Figure 1a and 1b). Both techniques agreed that these particles were huge (≥200 nm in diameter)-up to two orders of magnitude larger than the enzymes that they were inhibiting. Consistent with these particles being colloid-like aggregates of organic small molecules, they were sensitive to ionic strength. On moving to lower ionic strength, particle size decreased but the number of particles appeared to increase and inhibition improved. At high ionic strength, particle size increased and the apparent number of particles diminished, as did inhibition [8].
FIGURE 1.

Aggregating promiscuous inhibitors have characteristic physical features and behaviors that distinguish them from classical inhibitors. Their distinct physical properties lend themselves to detection by direct biophysical measurement of particle size and shape; their presence is often indicated by steep dose-response curves. (a) Particle formation by the promiscuous inhibitor tetraiodophenolphthalein, as visualized by transmission electron microscopy (Bar = 100 nm) and (b) by dynamic light scattering. (c) The dose-response curve of Rottlerin, a promiscuous inhibitor at micromolar concentrations (circles) compared to that of BZB, a pure competitive inhibitor (squares), both acting against the enzyme β-lactamase. Reprinted, with permission, from Refs. [8] and [13].
By 2001, it was clear that there was a chemically disparate class of nuisance compounds that inhibited enzymes not through a classic, single-molecule mechanism, but rather through sequestration. Because these compounds inhibited multiple enzymes, we began to call them ‘promiscuous’ inhibitors, a term probably first coined by Mic Lajines at Pharmacia. What remained unclear was the range of molecules that behaved this way, their relationship with the nuisance hits that had bedeviled HTS campaigns, and their mechanism of action.
What sorts of compounds aggregate?
Many of the compounds that had been shown to aggregate were conjugated dye-like molecules. Indeed, such compounds continue to be discovered as aggregation-based inhibitors. The recent study by Tipton and colleagues [10] on the inhibition of Phosphomannomutase-phosphoglucomutase by the dye disperse blue 56 is a good example of this class of molecules. These dye-like compounds would probably have been detected by the computational filters implemented in pharmaceutical companies to flag nuisance compounds. For example, there is a large overlap between such molecules and those flagged by programs such as REOS [3] and the ‘frequent hitters’ virtual screening program used at Roche [11].We wanted to learn if more drug-like, arguably more pharmaceutically relevant, molecules might also form aggregates.
Initially, thirty compounds from an in-house Pharmacia screening deck were tested [8]. Twenty of these inhibited a panel of unrelated enzymes (β-lactamase, DHFR and chymotrypsin), non-competitively in a time-dependent manner at micromolar or tens-of-micromolar concentrations. The compounds also formed particles in a DLS assay. We concluded that these screening compounds were promiscuous aggregators, at least in biochemical buffers at micromolar concentrations, and that aggregators could be found in real pharmaceutical screening decks.
Next, compounds that we considered to be genuine leads were explored. We picked 15 heavily-studied kinase inhibitors, including quercetin, rottlerin and bisindoylmaleimide, that Cohen and colleagues [12] had shown to be promiscuous among kinase targets. Of these, eight were active against three counter-screen enzymes (β-lactamase, chymotrypsin and MDH) with what were by now tell-tale features: non-competitive, time-dependent inhibition with steep dose-response curves (Figure 1c). All eight formed particles in the hundreds of nanometer size range by DLS (Table 2). [13] We concluded that these eight, highly-studied leads behaved as promiscuous aggregation-based inhibitors in the micromolar and tens-of-micromolar concentration range. Subsequently, investigators have found that other widely used biological reagents can behave this way. An example of these are the flavenoids, which seem particularly common in screening hit-lists, as pointed out by Rishton [14]. Similarly, Zavodszky and Kuhn have found that Fuchsin and Morelloflavone will inhibit thrombin as promiscuous aggregators (Zavodszky, M. and Kuhn, L., pers. commun.). Other classes of reagents that can behave as aggregators include cholesterol-based molecules, some of which will aggregate and inhibit at micromolar concentrations [15].
TABLE 2.
Some kinase inhibitors and drugs form promiscuous aggregatesa
| Kinase Inhibitor | Structures | IC50 versus β-lactamase (μM) | DLS conc. (μM) | Intensity (kcps) | Diameter (nm) |
|---|---|---|---|---|---|
| Rottlerin |  |
1.2 | 15 | 11.8 | 99 |
| Quercetin |  |
4 | 100 | 65.0 | >1000c |
| K-252c |  |
8 | 10 | 13.5 | 780 |
| Bisindolylmaleimide IX |  |
5 | 60 | 25.6 | 578 |
| Bisindolylmaleimide I | 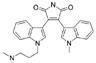 |
60 | 400 | 2.9 | 287 |
| U0126 | 30 | 80 | 53.7 | 432 | |
| Indirubin |  |
20 | 10 | 62.5 | >1000c |
| Indigo | 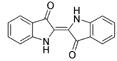 |
30 | 20 | 32.1 | >1000c |
| Drug | |||||
| Clotrimazole |  |
20 | 50 | 35.9 | 323 |
| Benzyl benzoate | 90 | 250 | 30.5 | 893 | |
| Nicardipine | 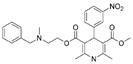 |
20 | 60 | 23.8 | 514 |
| Delavirdine | 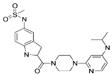 |
90 | 100 | 43.3 | 207 |
If drug-like molecules can behave as aggregation-based inhibitors, what about actual drugs? Our hope was that drugs would not aggregate, affording us a biophysical criterion for drug-like. We tested more than 50 commercially available drugs against a panel of enzymes including MDH, chymotrypsin and β-lactamase [15]. Most did not inhibit these enzymes, even at 400 μM, which was consistent with our expectations. However, seven drugs inhibited all three enzymes in the tens- to hundreds-of-micromolar concentration range (four of these are shown in Table 2). These drugs showed all the characteristics of aggregation-based activity: non-competitive, time-dependent inhibition of multiple enzymes, high sensitivity to enzyme concentration and to detergent, and formation of particles in the hundreds-of-nanometers size range as determined by DLS.
Thus, whereas most drugs were well behaved, some clearly formed aggregates in aqueous buffers. How can this behavior be reconciled with their status as specific agents? One explanation is that the aggregation was observed in biochemical buffers that lacked the adjuvants, serum proteins and bile salts that might disrupt or saturate aggregates in the body. Also, inhibition occurred at concentration ranges three orders of magnitude higher than the IC50 values of the drugs for their targets. Thus, we do not consider the promiscuous inhibition of these drugs to reflect on their behavior at their target in vivo (for another possible in vivo effect of aggregation, see below). Still, drugs are molecules too and are subject to the associations and equilibria that other organic molecules obey. Under screening conditions and at screening concentrations, drugs can aggregate and nonspecifically inhibit just like any other molecule.
Mechanism of aggregation-based inhibition
Whereas early studies suggested that there was an association between aggregate and inhibition [8], the mechanistic link was unclear. After all, association with a solid support typically leaves an enzyme or a protein uninhibited, so why should association with an aggregate of organic molecules be so detrimental?
The first question was whether the aggregates directly physically interact with enzyme, which was implied but never demonstrated by the initial studies. Two lines of evidence came to support this view. The simplest was co-precipitation of enzyme and aggregates followed by gel electrophoresis. Aggregates were incubated with enzyme and co-precipitated by centrifugation on a bench-top microfuge. The precipitant was run on an agarose gel and silver-stained. In the absence of aggregate, no protein band was observed, nor was one observed when the supernatant was run out on the gel. When the precipitant from the protein-aggregate solution was run, a clear enzyme band was visible, consistent with the idea that the aggregates, on pull-down, co-precipitated enzyme (Figure 2) [16]. Also supporting direct association was the formation of punctate fluorescence on mixing green fluorescent protein (GFP) with an aggregating molecule (Figure 2) [16].
FIGURE 2.
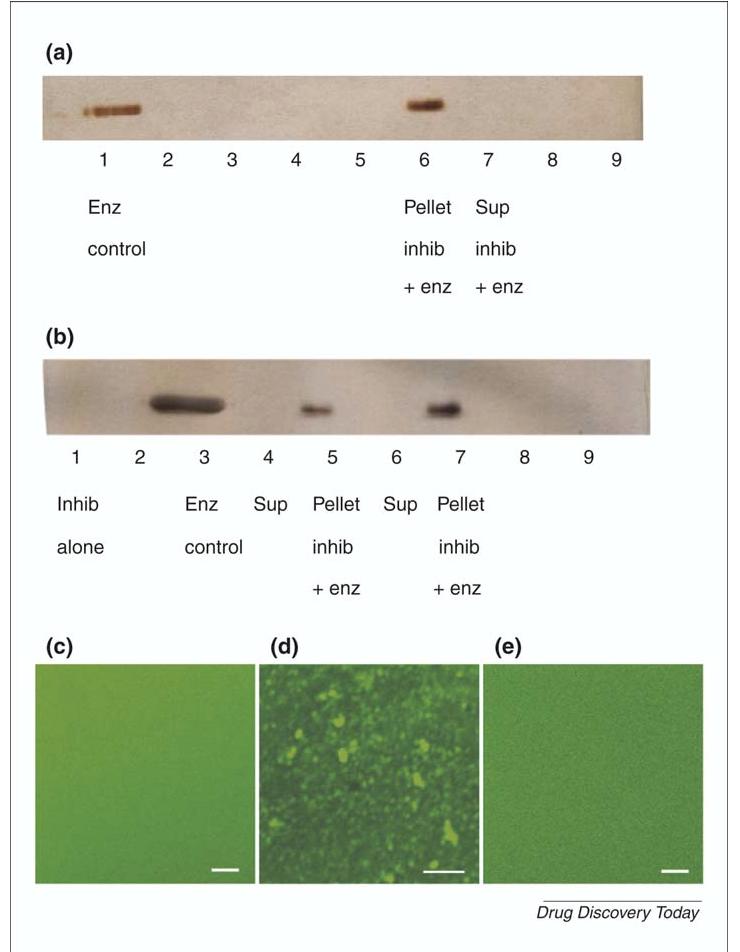
Direct association between enzymes and aggregates. Several lines of evidence suggest that aggregates act by direct association with enzymes, including co-precipitation of enzyme by aggregates and the concentration of green fluorescent protein (GFP) fluorescence on the surface of aggregates. Co-precipitation can be measured by incubating the enzyme with an aggregating inhibitor followed by spinning the aggregate down in a microfuge and running the precipitant via an SDS-PAGE (sodium dodecyl sulphate polyacrylamide gel electrophoresis). Localization of GFP can be observed by the punctuate GFP fluorescence that develops in the presence of an aggregating inhibitor. (a) Co-precipitation of β-lactamase by tetraiodophenolphthalein, a promiscuous aggregator, followed by gel electrophoresis. SDS-PAGE and silver-stain analysis of supernatants and pellets from centrifugation of β-lactamase in the presence or absence of inhibitor, with and without Triton X-100. (b) Same as in (a), except with the promiscuous inhibitor 4-bromophenylazo-(4′)-phenol. (c) Fluorescence of GFP in the absence and (d) in the presence of the aggregator tetrai-odophenophthalein. (e) Same as in (c), but with addition of the detergent Triton-X 100 at 0.01% concentration. Scale bar = 5 μm. Adapted, with permission, from Ref. [16].
What was it about aggregate-protein sequestration that leads to enzyme inhibition? One hypothesis was that the aggregates were denaturing the protein. However, the retention of GFP fluorescence when associated with aggregate was inconsistent with major denaturation (Figure 2). Consistent with this observation was the lack of selectivity of aggregates for destabilized compared with stabilized mutant β-lactamases. If aggregates were working by protein unfolding, one would expect the destabilized β-lactamase to be more susceptible to inhibition than the stabilized enzyme, but this was not the case. Additionally, aggregation-based inhibition was rapidly reversible by addition of detergent even when such detergent was added after the assay had begun [16]. This implies that aggregation-based inhibition is dynamically reversible and is inconsistent with gross structural changes on association between enzyme and inhibitory particle.
Thus, aggregates associate with protein but do not appear to globally denature them. How exactly this association takes place, what drives it, and why it leads to inhibition remain open questions. Preliminary electron microscopy studies suggest that the aggregates adsorb to the surface of the aggregate particles, but these studies do not rule out absorption by diffusion into the aggregates (not shown). Why either should lead to inhibition is unclear. Possible mechanisms include local denaturation of the protein, freezing out of conformational modes necessary for catalysis, or the existence of high local concentrations of organic molecule on the aggregate surface with which the protein is associated by nonspecific interactions. The nature of the interactions driving aggregation of the organic molecules and, subsequently, sequestration of the protein, also remains open: it is tempting to posit hydrophobic effects, given their well-known lack of specificity, but other terms might also be important, including liquid crystal-like stacking.
Failing a detailed understanding of mechanism, it is tempting to infer chemical patterns that predict aggregation-based inhibition. Based on over 1100 molecules tested for aggregation-based inhibition, Tom Doman [15,17] at Eli Lilly and Anang Shelat [17] and Kip Guy at UCSF have developed computational models to predict aggregation. Using a Random Forest algorithm to classify molecules into aggregators or non-aggregators, Doman correctly predicted the aggregator or non-aggregator status of 89% of the 298 random molecules that the method had not previously seen [17]; using a naïve Bayesian classifier, Shelat and Guy predicted the status of 80% of the same molecules [17]. Given the diversity of the molecules tested, this level of success is encouraging, and one can imagine using these methods for an early alert of colloidal aggregation, for example, when evaluating hits from a screening campaign. Whether these prediction rates are high enough for reliable classification of an entire library is unclear. When one is in doubt as to whether or not an inhibitor is acting through aggregation, experiment must be the final arbiter (Box 1).
BOX 1 Evaluating candidate hits for aggregation-based inhibition:
- Is inhibition significantly attenuated by small amounts of nonionic detergent?
- If so, the compound is very likely acting through aggregation.
- In cases where you cannot use detergent, e.g. cell-based assays, it may be possible to use high (1 mg/ml) concentrations of serum albumin instead.
- Is inhibition significantly attenuated by increasing enzyme concentration?
- If so, the compound is very likely an aggregator. Except when the enzyme concentration to Ki ratio is high, increasing enzyme concentration should not affect percent inhibition.
Is inhibition competitive? If so, the compound is unlikely to be an aggregator.
Does the inhibitor retain activity after spinning for several minutes in a microfuge? If not, particle formation is likely (see point 5).
Can you directly observe particles in the 50 to 1000 nm size range? We have typically used dynamic light scattering for this. Formation of particles does not guarantee promiscuous inhibition, but it is a worrying sign.
Is the dose-response curve unusually steep? There are classical reasons for curves, but they too are a worrying sign.
Few if any of the experiments listed in Box 1 are definitive by themselves, although the detergent test is fairly reliable. When several of these tests are combined, they are strong indicators of aggregation or non-aggregation based mechanisms of action.
A rapid counter-screen for aggregation-based inhibitors
Two of the characteristics of aggregation-based inhibition listed in the last section lend themselves to rapid detection: their high sensitivity to non-ionic detergents [16,18] and their proclivity to forming large particles. We attempted to exploit these characteristics in a high-throughput manner to explore how prevalent promiscuous aggregators might be in screening decks. We targeted 1030 Rule-of-Five-compliant [1] molecules from a widely-used supplier of screening compounds. These molecules were tested in 96-well plates for inhibition of β-lactamase in the presence and absence of Triton-X 100, and in a second plate-based assay for light scattering by DLS. Some of the molecules tested were predicted to be either aggregators or non-aggregators, based on computational models (above) [15]. Among the most interesting results came from analyzing the 298 drug-like compounds that were selected at random. At 30 mM, 19% of these inhibited β-lactamase in the absence of detergent but not in its presence [17], consistent with aggregation-based inhibition. At 5 mM, 1.4% of these molecules inhibited in the absence but not the presence of detergent (Figure 3). Intriguingly, an even higher percentage of molecules formed particles in a plate-based DLS assay [17]. Even at 5 mM, the high percentage of drug-like screening-relevant molecules that form promiscuous aggregates is sufficient to cloud the results of many screens that do not control for them.
FIGURE 3.

Many ‘drug-like’ molecules form promiscuous aggregates at micromolar concentrations. More than 1000 such compounds were evaluated for detergent-dependent inhibition and dynamic light-scattering, using 96-well plate based assays. Molecules were tested in different sets: two sets of predicted aggregators and two sets of predicted non-aggregators, derived using different computational models, were tested, as was a fifth category chosen at random from a ‘drug-like’ subset of the ChemDiv collection (see text). Reprinted, with permission, from Ref. [17].
Biological implications?
Until now, the relevance of aggregation to biochemical screening has been emphasized; is it conceivable that aggregation affects compound behavior in cell-based assays or in whole animals? At present, there is little evidence to support or falsify activity in cell-based assays. Weighing against such activity, the aggregate might have to cross the cellular membrane for effects to be observed; this is possible, owing to dynamic equilbria between aggregated and non-aggregated compound, but the plausibility of this is unknown. Aggregates could also affect cellular behavior indirectly, either through extracellular receptors or by membrane disruption. Consistent with the latter, some aggregates are hemolytic (Parker, C. pers. commun.).
Recently, Arnold and colleagues [19] have mooted a role for aggregation in whole body pharmacology, through effects on absorption. Several HIV non-nucleoside reverse transcriptase inhibitors (NNRTIs) were found to aggregate into particles in the 90 nm size range under pH and buffer conditions similar to that experienced in the gut. These NNRTIs have activities in patients below (better than) their IC50 values against isolated reverse transcriptase. Arnold and colleagues propose that, after an oral dose, these inhibitors would be present in the gut as aggregates, where they would be preferentially absorbed into specialized, particle-absorbing cells in the GI tract that feed into the lymphatic system. This would deliver them at unusually high concentrations to immune T cells, the preferential target of HIV, explaining their unusual IC50 to effective dose ratios. Indeed, aggregation in the stomach and gut could explain the relatively high bioavailability of several oral drugs that might otherwise be predicted to have low bioavailability. This intriguing hypothesis remains to be directly tested by full animal pharmacology.
The spirit haunted world of screening
HTS is typically the first- and certainly the most automated-step in the drug discovery pipeline. It is thus unnerving that its results can be among the most ambiguous. This is no trivial fault of instrumentation, assays or analysis, but reflects the breathtaking ambition to rapidly screen libraries composed of 105 to 106 individual organic molecules. Each of these molecules has idiosyncratic physical and chemical properties affecting solubility, reactivity and stability; that many will behave poorly in any given assay is unsurprising. Multiple mechanisms contribute to such misbehavior. Some, like precipitation [7] or instability, contribute to false-negatives in screening, others, such as chemical reactivity [14] optical opacity [3] and oxidation [5], contribute to false-positives. It is the false-positives that are the most costly in terms of time spent, if not opportunity. If their identification continues to be researched it is because molecules that behave badly under one condition might behave well under another. Knowledge-based tools to identify nuisance compounds must thus contend with a context-dependence that confers on the molecules a maddening inconsistency.
Few screening phenomena are more prone to erratic behavior than colloidal aggregation, the observation of which depends on the physical properties of the assays, the presence or absence of adjuvants, such as detergents or serum proteins, and the concentration of the target. It is thus unsettling that so many drug-like organic molecules have this property at micromolar concentrations. However, one might take encouragement from the physical basis of colloid-like particle formation, whose study long-predates HTS. In this sense, one of the more shadowy pathologies of organic molecules in HTS is also among the easiest to detect and control for. Doing so early in the discovery process will save investigators much effort and diminish the number of reports that are based on artifact.
Acknowledgements
I am grateful to my colleagues who undertook the aggregation studies discussed here, especially Susan McGovern, Brian Feng and Kristin Coan, and also James Seidler, Brian Helfand, Anang Shelat and Tom Doman. I thank BF and KC for reading this manuscript. I thank Chris Lipinski for thoughtful conversations. I thank Proterion for use of the plate reading DLS instrument and GM71630.
References
- 1.Lipinski CA, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 2.Rishton GM. Reactive compounds and in vitro false positives in HTS. Drug Discov. Today. 1997;2:382–384. [Google Scholar]
- 3.Walters W, Namchuk M. Designing screens: how to make your hits a hit. Nat. Rev. Drug Discov. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- 4.Veber DF, et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 5.Hajduk PJ, et al. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 6.Martin YC. A bioavailability score. J. Med. Chem. 2005;48:3164–3170. doi: 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- 7.Oldenburg K, et al. High throughput sonication: evaluation for compound solubilization. Comb. Chem. High Throughput Screen. 2005;8:499–512. doi: 10.2174/1386207054867364. [DOI] [PubMed] [Google Scholar]
- 8.McGovern SL, et al. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 9.Straus OH, Goldstein A. Zone behavior of enzymes: illustrated by the effect of dissociation constant and dilution on the system cholinesterase-physostigmine. J. Gen. Physiol. 1943;26:559–585. doi: 10.1085/jgp.26.6.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu HY, et al. Detailed kinetic studies of an aggregating inhibitor; inhibition of phosphomannomutase/phosphoglucomutase by disperse blue 56. Biochemistry. 2004;43:8662–8669. doi: 10.1021/bi0491907. [DOI] [PubMed] [Google Scholar]
- 11.Roche O, et al. Development of a virtual screening method for identification of “frequent hitters” in compound libraries. J. Med. Chem. 2002;45:137–142. doi: 10.1021/jm010934d. [DOI] [PubMed] [Google Scholar]
- 12.Davies SP, et al. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGovern SL, Shoichet BK. Kinase inhibitors: not just for kinases anymore. J. Med. Chem. 2003;46:1478–1483. doi: 10.1021/jm020427b. [DOI] [PubMed] [Google Scholar]
- 14.Rishton GM. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discov. Today. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 15.Seidler J, et al. Indentification and prediction of promiscuous drugs. J. Med. Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 16.McGovern SL, et al. A specific mechanism for non-specific inhibition. J. Med. Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 17.Feng BY, et al. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 18.Ryan AJ, et al. Effect of detergent on “promiscuous” inhibitors. J. Med. Chem. 2003;46:3448–3451. doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- 19.Frenkel YV, et al. Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J. Med. Chem. 2005;48:1974–1983. doi: 10.1021/jm049439i. [DOI] [PubMed] [Google Scholar]