

Today I will be discussing where some of the new heart failure therapies may be headed as we move beyond the guidelines. These predictions are difficult in cardiology, especially so in heart failure, because these days the shelf-life for any “new” idea seems to be only about 6 months.
Heart failure is generally thought to begin after an index event that produces a decline in the pumping capacity of the heart. This event can be sustained pressure or volume overload, an acute myocardial infarction, a genetic abnormality that disrupts contraction—it could be any one of a number of things. In many ways, the exact cause of the decline in pumping capacity may be somewhat irrelevant. What is important is that, in response to this decline, a variety of compensatory mechanisms are activated that are designed to maintain cardiovascular homeostasis for periods of months to years; during that period, patients tend to remain asymptomatic.
The prevailing wisdom is that when these compensatory mechanisms are excessively activated, they themselves can cause secondary damage to the heart and circulation (Fig. 1). It is this secondary damage that drives the disease process of heart failure forward, largely through the mechanism of cardiac remodeling. As the heart remodels, it not only gets bigger, but the cardiac walls get thinner and the pumping capacity of the heart declines. With the transition from a small heart to a big heart, patients generally go from asymptomatic to symptomatic heart failure.
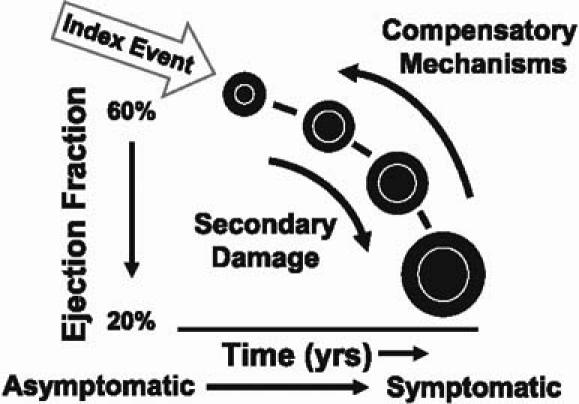
Fig. 1 When compensatory mechanisms are excessively activated, they themselves can cause secondary damage to the heart and circulation.
From: Mann DL. Mechanism and models in heart failure: a combinatorial approach. Circulation 1999;100:999–1008. Reproduced by permission of the American Heart Association.
Our current therapies in heart failure fall into 3 general categories: cardiorenal agents (diuretics), cardiocirculatory agents (inotropics and vasodilators), and neurohormonal agents (angiotensin-converting enzyme [ACE] inhibitors/angiotensin receptor [ATR] blockers, β-blockers, aldosterone antagonists, and hydralazines/nitrates). We use diuretics largely for symptomatic relief in both acute and chronic heart failure. We use inotropic agents and vasodilators for symptomatic relief primarily in acute heart failure. And we use neurohormonal therapy to target the various compensatory mechanisms that can be activated, namely the renin–angiotensin system and the adrenergic system.
What is coming next in heart failure? How many pills can people actually take? What other treatments should we think about for the future? As we've focused our attention on cardiac remodeling, we've come to appreciate that it is not so much a manifestation of chronic disease as a direct cause of disease progression. As the heart gets bigger and the walls get thinner, wall stress increases, which in turn increases the amount of work that the heart does—the afterload. This has the undesired consequence of increasing oxygen consumption, which leads to episodic subendocardial ischemia (Fig. 2). We now recognize that a lot of our congestive heart failure patients (even patients with dilated as opposed to ischemic cardiomyopathy) can have high troponin T levels, probably as a consequence of episodic subendocardial ischemia. When you stretch the myocardium, you activate a variety of maladaptive genes, which can cause further disease progression. Finally, as the heart dilates, there can be progressive functional mitral regurgitation that leads to sustained volume overload over a period of months to years, further worsening heart failure. So the process of cardiac remodeling, in and of itself, can lead to disease progression, independent of the neurohormonal status of the patient.
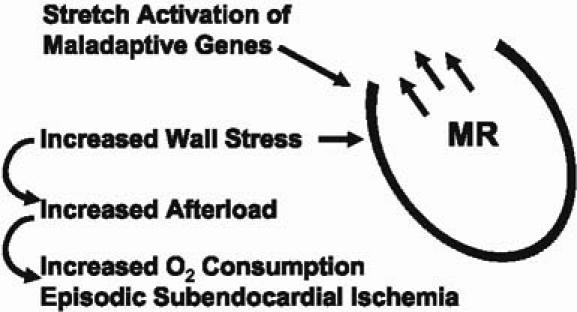
Fig. 2 Adverse effects of left ventricular remodeling.
MR = mitral regurgitation
Mike Bristow and I wrote a recent update on heart failure insights and coined the term “the biomechanical model.” This model suggests that heart failure develops and progresses, at least in part, as a result of the end-organ changes that occur in the remodeled heart; and that the progression of heart failure may occur independently of the neurohormonal status. The unproven implication is that neurohormonal antagonists will not be effective, particularly for people with advanced heart failure. Thus, the cardiac remodeling process itself becomes the therapeutic target.
How might it be possible to deal directly with cardiac remodeling? Last year at the AHA meetings, I presented the results of the Acorn trial (Fig. 3). This was a trial of a fabric mesh weave that was surgically implanted around the ventricle in CHF patients, to limit chamber enlargement and to functionally unload the heart. The study tested the hypothesis that preventing LV remodeling would have a favorable impact on the natural history of the disease. The answer was, “Yes, it did.”
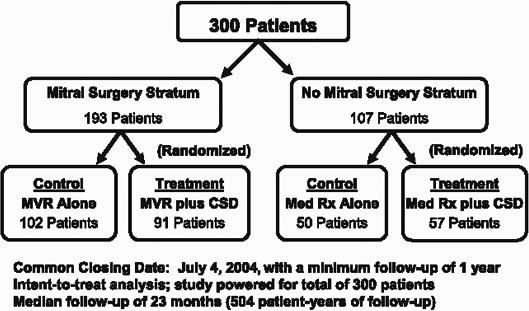
Fig. 3 Design of Acorn randomized trial.
CSD = cardiac support device (fabric mesh); MVR = mitral valve repair; Rx = prescription
There were 2 arms in the study. The 1st arm involved patients who were undergoing mitral valve surgery for functional mitral regurgitation associated with dilated cardiomyopathy. If they needed surgery (about two thirds of the patients did), they were randomized to either mitral surgery alone or mitral surgery plus the cardiac support device (the CSD, or surgically implanted mesh). If the patients didn't need mitral surgery, they were randomized in the 2nd arm to either best medical therapy (β-blockers, ACE inhibitors, and, if needed, spironolactone), or medical therapy plus the CSD.
The primary endpoint was a composite of adverse events: death, a major cardiac procedure indicative of CHF progression (left ventricular assist device, cardiac transplantation, coronary artery bypass grafting, biventricular pacing, or repeat mitral surgery), or a worsening of New York Heart Association functional class. Patients were judged to be improved if they had an improved NYHA class, with no major cardiac procedures. If patients were neither worse nor improved, they were considered to be unchanged. At a median follow-up of 23 months, the primary clinical composite was significantly improved with the CSD (OR, 1.73 [95% CI 1.07–2.79]; P=0.024). Substantially better freedom from major cardiac procedures was the individual component that really drove the primary composite. By limiting cardiac remodeling, we were able to decrease the number of cardiac transplants by 50%, decrease the number of LVADs by 50%, significantly reduce the need for biventricular pacing, and significantly reduce the need for repeat mitral surgery. Interestingly, we actually observed progressive reverse remodeling. In experimental studies, hearts that got smaller also showed evidence of reversal of the expression of a variety of maladaptive genes (Fig. 4). In the Acorn study, patients also tended to feel better with CSD therapy, at least as measured by the Minnesota Living with Heart Failure Questionnaire.
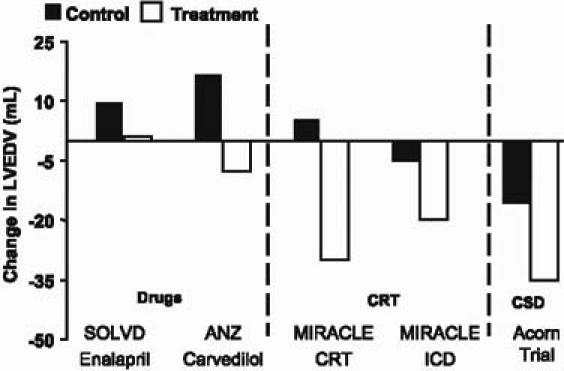
Fig. 4 Comparative changes in left ventricular size.
ANZ = Australia/New Zealand Heart Failure Collaborative Group study (Doughty RN, et al. JACC 1997;29:1066); CRT = cardiac resynchronization therapy; ICD = implantable cardioverter defibrillator; LVEDV = left ventricular end-diastolic volume; MIRACLE = Multicenter InSync Randomized Clinical Evaluation (Medtronic SSE P010015; and Young JB, et al. JAMA 2003;289:2685–94); SOLVD = Studies of Left Ventricular Dysfunction (Greenberg et al. Circulation 1995;91:2573–81) Reproduced by permission of the American Heart Association.
How does the CSD compare with other existing therapies? Angiotensin-converting enzyme inhibitors stabilize cardiac remodeling but don't reverse it. Drugs, such as β-blockers, cause a modest reduction in reverse remodeling. Cardiac resynchronization therapy (CRT) is probably one of the more effective tools we have to achieve favorable remodeling. The CSD device appears to have an effect comparable to that of CRT, and appears to be additive to CRT if you put the two together.
Moving away from the Acorn device, I'd like to talk about 2 other novel therapies for the treatment of mitral regurgitation. As the heart begins to fail, the papillary muscles are pulled apart, the annulus becomes dilated, and you begin to get progressive mitral regurgitation. As I indicated earlier, this sustained volume overload, over a period of months to years, can lead to worsening left ventricular dilatation. Mitral regurgitation begets more mitral regurgitation.
One new device is the Coapsys implant device (Fig. 5), currently in clinical trials, which consists of 2 epicardial pads that are placed on either side of the ventricle. They're connected by a subvalvular cord that actually goes through the ventricle. The anterior pad pushes the annulus together. The posterior pad brings the left ventricular walls together; in the operating room, the surgeon can just tighten the cord linking the 2 pads to adjust the degree of mitral regurgitation. The nice thing about this is that it can all be done off-pump. The RESTOR-MV trial (Randomized Evaluation of a Surgical Treatment for Off-Pump Repair of the Mitral Valve) is a multicenter, prospective, randomized trial that evaluates the safety and efficacy of the Coapsys system. A recently published preliminary series of 19 patients suggested that the device was safe and effective; it reduced intraoperative MR from 2.4 (out of 4) down to 0.4.
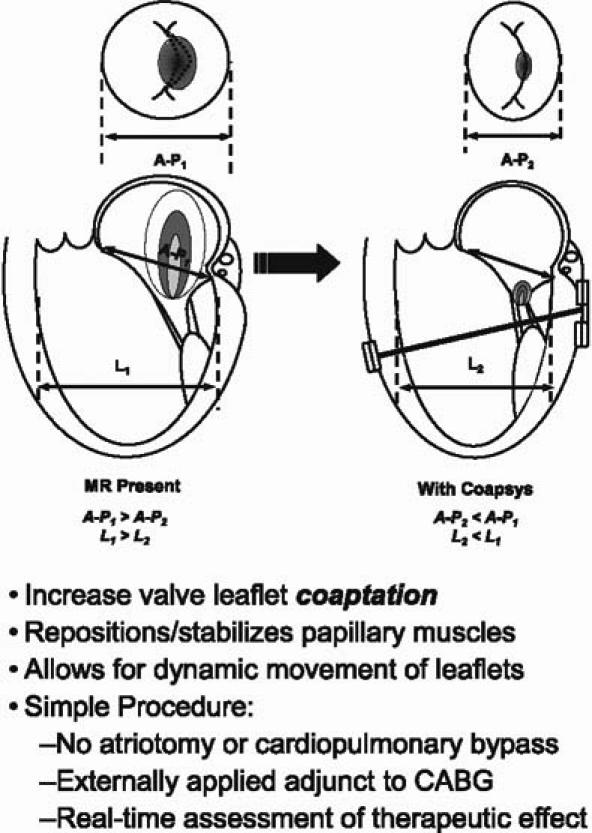
Fig. 5 The Myocor® Coapsys™ System.
A = anterior; L = lateral; MR = mitral regurgitation; P = posterior
Another novel device, developed by Cardiac Dimensions, is a completely percutaneous mitral annular constraint delivered via the coronary sinus. This device is used to “cinch up” a dilated mitral annulus to reduce regurgitation.
Yet another interesting piece of technology is the neurostimulation system developed by BioControl. It's an implantable device that electronically stimulates the vagus nerve to decrease heart rate; vagal nerve stimulation may also have substantial anti-inflammatory effects. This particular device has undergone preclinical testing and is being tested in patients in Europe; it will probably make its way to North America in the next couple of years.
The immortal 20th-century philosopher Yogi Berra said, “The future isn't what it used to be.” Given how rapidly the field of heart failure is moving forward, Yogi might now add, “… and the future is devices, devices, and devices.”
Footnotes
Address for reprints: Douglas L. Mann, MD, Section of Cardiology, Baylor College of Medicine and the Texas Heart Institute, 1709 Dryden Road, 9th FL, BCM620 – FC 9.83, Houston, TX 77030. E-mail: dmann@bcm.tmc.edu
Presented at the Texas Heart Institute's symposium “Evolving Standards in Cardiovascular Care: What Have We Learned? Where Are We Going?” held at the Adam's Mark Hotel; 12 November 2005; Dallas