

Abstract
Background
Over-expression of IL-6 has been implicated in cholangiocarcinoma growth but the cellular mechanisms involved are unknown. Our aims were to assess the mechanisms by which over-expression of IL-6 promotes transformed cell growth in malignant cholangiocytes.
Methods
Stably transfected cell lines over-expressing IL-6 were derived from malignant human cholangiocytes. Transformed cell growth was assessed by anchorage independent growth in vitro and by xenograft growth in nude mice. Expression of the anti-apoptotic protein Mcl-1 was quantitated by immunoblot analysis and by real-time PCR. Gene silencing was performed using siRNA. Dominant negative upstream kinase activators and isoform-specific constructs were used to evaluate the involvement of p38 MAP kinase signaling pathways.
Results
Over-expression of IL-6 increased xenograft growth, anchorage independent growth and cell survival but did not significantly alter cell proliferation. The basal expression of Mcl-1 was increased in IL-6 over-expressing cells. Selective knockdown of Mcl-1 by siRNA increased gemcitabine-induced cytotoxicity. Moreover, IL-6 increased Mcl-1 mRNA and protein expression via a p38 MAPK dependent mechanism.
Summary
These data demonstrate a major role of survival signaling pathways in mediating the effects of IL-6 over-expression in cholangiocarcinoma growth. Mcl-1 is identified as a mediator of IL-6-induced tumor cell survival and shown to be transcriptionally regulated by IL-6 via a p38 MAPK dependent pathway. We conclude that modulation of IL-6 mediated survival signaling pathways involving the p38 MAPK or downstream targets such as Mcl-1 may prove useful therapeutic strategies for human cholangiocarcinoma.
Keywords: Apoptosis, Biliary tract tumors, Kinases, Tumorigenesis
Abbreviations: IL-6, Interleukin-6; MAPK, mitogen activated protein kinase; Mcl-1, myeloid cell leukemia-1; MKK3, mitogen-activated protein kinase kinase 3; siRNA, small interfering double stranded RNA
Introduction
Primary malignancies arising from the epithelial lining of the biliary tract, or cholangiocarcinoma, are aggressive tumors that are increasing in incidence worldwide (1–4). At present the available treatments for cholangiocarcinoma are of limited efficacy, and prognosis is poor. The molecular mechanisms underlying the formation or progression of cholangiocarcinoma are unknown. However, a better understanding of these mechanisms is likely to result in more specific and effective therapies that may eventually improve survival.
Clinically, these tumors often arise in the setting of chronic biliary tract inflammation (5). Biliary tract inflammation or sepsis is associated with increased concentrations of the inflammatory cytokine, Interleukin-6 (IL-6) in the bile and serum (6–9). IL-6 is produced in the liver by several different cell types, including biliary epithelial cells or cholangiocytes, in response to inflammatory mediators (10–12). IL-6 has multiple functions, and in addition to mediating the acute phase response and immune responses, can influence the growth of normal as well as tumor cells. Dysregulation of IL-6 production has been implicated in several human conditions characterized by excessive tissue growth (13). Within the liver, deregulation of IL-6 can contribute to tumor formation. For example, double transgenic mice expressing IL-6 and IL-6 receptors under the control of liver-specific promoters spontaneously develop hepatocellular hyperplasia and adenomas (14). Several observations suggest an important role for increased IL-6 expression in the pathogenesis or progression of cholangiocarcinoma. IL-6 levels are elevated in the serum of patients with cholangiocarcinoma (15). Moreover, IL-6 expression is up-regulated in malignant cholangiocytes in vitro (16, 17). Normal cholangiocytes can respond to IL-6 by a variety of biological responses, including effects on cell proliferation (10, 16, 18, 19). Additionally, we and others have shown that IL-6 increases proliferation in malignant cholangiocytes, and furthermore can activate cell survival pathways (16, 20–22).
Based on these observations, IL-6 has been implicated as a critical growth factor for cholangiocarcinoma (21, 23). However, the cellular mechanisms by which IL-6 promotes tumor growth are unknown. Tumor growth can be enhanced by either increased proliferation or increased cell survival. Understanding the precise cellular mechanisms by which IL-6 promotes cholangiocarcinoma growth will be essential for the development of effective therapies. Therefore, our aims were to: (1) evaluate the effect of autocrine IL-6 production on the growth of human malignant cholangiocytes; (2) evaluate the relative contributions of proliferative and survival effects of IL-6 on tumor growth, and (3) identify intracellular mechanisms by which autocrine IL-6 production contributes to cholangiocarcinoma progression.
Materials and Methods
Cell lines and culture
KMCH-1, Mz-ChA-1 and TFK-1 human cholangiocarcinoma cells were obtained and cultured as previously described (12, 24, 25). The CC-LP-1 human cholangiocarcinoma cells were a kind gift of Dr. Patricia Whiteside (University of Pittsburgh, PA). These cell lines are derived from human intrahepatic (KMCH-1 or CC-LP-1), extrahepatic (TFK-1), or gallbladder (Mz-ChA-1) tumors. The KMCH-1 and CC-LP-1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum, 1% sodium pyruvate, and 1% antibiotic-antimycotic mix, whereas the Mz-ChA-1 and TFK-1 cells were cultured in CMRL 1066 media containing 10% fetal bovine serum, 1% L-glutamine, and 1% antibiotic-antimycotic mix.
Generation of stably transfected cell lines
KMCH-1 or Mz-ChA-1 cells were stably transfected with the pTarget (Promega, Madison, WI) expression plasmid containing full-length IL-6 under the control of a CMV promoter. These cell lines were designated as KM-IL-6 or Mz-IL-6 whereas empty vector controls cells were designated as KM-1 or Mz-1 respectively. Conditions for transfection were initially optimized using the MT-1 gal vector, and assaying for β-galactosidase expression. Plasmids were purified using the Qiagen Plasmid Midi Kit (Qiagen Inc., Valencia, CA), and linearized by restriction enzyme digestion prior to transfection using Trans-IT (Panvera, Madison, WI). After 48 hrs, the media was replaced, and the cells were grown and passaged for three weeks in media containing G418. Subsequent studies were performed using a mixed population of stable transfectants without isolation of specific clones. Stable transfection was confirmed by reverse transcriptase PCR and using an IL-6 bioassay to verify IL-6 over-expression. For studies of p38 MAPK signaling pathways, KMCH-1 malignant human cholangiocytes cells were stably transfected with pRc/RSV-Flag MKK3(A), (encoding a dominant interfering upstream activator of p38 MAPK with double point mutations in Ser 189 and Thr 193 replaced by Ala) and designated as KM-p38dn, as previously described (24). Compared to control KM-1 cells, KM-p38dn cells have reduced activation of p38 MAPK.
Growth in soft agar
To assess the effect of anchorage independent growth, cells (1,000 cells/35x10 mm plate) were grown in soft agar for 21 days at 37°C using a two layer agar system and the number of colonies quantitated as previously described (24).
Nude mouse xenograft model
Male athymic nu/nu mice, 8 weeks of age, were obtained from Charles River Laboratories (Wilmington, MA), and fed food and water ad libitum. The mice were housed 4 per cage and fluorescent light was controlled to provide alternate light and dark cycles of 12 hours each. The animals received a subcutaneous injection of either Mz-1 or Mz-IL-6 cells (3 X 106 viable cells suspended on 0.5 mL of extracellular matrix gel) on their right flank. Tumor volume was estimated by serial measurements obtained twice a week. The xenografts were excised and sections obtained for analysis of apoptosis or proliferation by quantitating TUNEL positive or PCNA positive cells respectively. TUNEL analysis was performed using a commercially available kit (Wako Chemicals, Tokyo, Japan). Following counterstaining with hematoxylin solution, xenograft sections were examined by light microscopy using an Olympus BX-40 microscope equipped with a CCD camera. Approximately 200 cells per slide were counted in a coded fashion in seven non-overlapping fields. Immunohistochemistry was performed using antibodies to PCNA or Mcl-1. The number of PCNA-positive cholangiocytes was examined under a light microscope (Olympus Optical Co., BX 40, Tokyo, Japan). Approximately 200 cells per slide were counted in a coded fashion in several non-overlapping fields. Animal protocols were approved by the Institutional Animal Care and Use Committee.
Quantitative Real-Time PCR
RNA was isolated using an RNA isolation kit (Bio-Rad, Hercules, CA), and cDNA generated by reverse transcription using 1 μg of total RNA and the reverse transcription kit (Invitrogen, Carlsbad, CA). Quantitative real-time PCR was performed on a MX 3000P™ PCR Instrument (Stratagene, San Diego, CA) and using SYBR Green as the detection fluorophore. Each 20-μl reaction mixture consisted of 2 μl of cDNA (50 ng/μl), 10 μl of 2× Universal SYBR Green PCR Master Mix (Sigma, St.Louis, MO), and 2 μl of 20 nM forward and reverse primers. Optimization was performed for each gene-specific primer prior to the experiment to confirm that primer concentrations and reaction conditions did not produce artefactual amplification signals in control tubes that did not contain any template. Primer sequences were designed using Primer Express Software (PerkinElmer Life Sciences) and sequences used were as follows: Mcl-1 forward: TAA GGA CAA AAC GGG ACT GG, Mcl-1 reverse: 5′-CCT CTT GCC ACT TGC TTT TC-3′; IL-6 forward; 5′-GCA GAA TGA GAT GAG TTG TC; IL-6 reverse; 5′-GCC TTC GGT CCA GTT GCC TT -3′; β-actin forward: 5′-CCA AGG CCA ACC GCG AGA AGA TGA C-3′, and β-actin reverse: 5′-AGG GTA CAT GGT GGT GCC GCC AGA C-3′. PCR parameters were as follows: 10 min at 95 °C, and then 40 cycles of 30 s at 95 °C, 1 min at 65°C, and 1 min at 72°C. The specificity of the produced amplification product was confirmed by melting curve analysis of the reaction products using SYBR Green as well as by visualization on ethidium bromide-stained 1.8% agarose gels to confirm a single band of the expected size. Each sample was tested in triplicate. Threshold values were determined for each sample/primer pair and average and SE values were calculated. The mRNA level of β-actin was used as an internal control, and gene specific mRNA expression was normalized against β-actin expression.
Cell Proliferation
Cells were seeded into 96-well plates (10,000 cells/well), and incubated in a final volume of 200 μl medium. The cell proliferation index was assessed as we have previously described using a commercially available colorimetric assay (CellTiter 96AQueous; Promega Corp., Madison, WI) (21).
Cell cycle analysis
Cell cycle analysis was performed as previously described (26). Cells were collected, and re-suspended in a propidium iodide solution containing 0.1 M propidium iodide, 0.1% v/v Triton X-100, and 20% RNAse A, in PBS. After incubation on ice, in the dark, for 2 hrs, the samples were analyzed by flow cytometry. 10,000 events were recorded and the proportion of cells in various phases of the cell cycle were analyzed using the ModFitLT DNA analysis program (Becton Dickinson, San Jose, CA).
Apoptosis Assay
The cells were seeded into a 96 well plate at a density of 2x104 cells per well and incubated under the experimental conditions indicated in a final volume of 200 μl. Cells with morphological changes indicative of cell death by apoptosis were identified and quantitated either as previously described using fluorescence microscopy and staining with 4′, 6-diamidino-2-phenylindole (DAPI) or using the APOPercentage Apoptosis Assay (Biocolor, Belfast, Northern Ireland) following the manufacturer’s instructions (21).
Immunoblot analysis
Analysis and quantitation was performed as previously described (20). In brief, cell lysates obtained from confluent cells were separated on 4–12% gradient polyacrylamide gels (Novex, San Diego, CA) under reducing conditions and electro-blotted to positively charged 0.45 μM nitrocellulose membrane (Millipore, Bedford, CA). Membranes were incubated overnight at 4°C with the respective anti-human primary antibody, used at a 1:1000 dilution. After washing, the membrane was incubated with the secondary antibody at a 1:2000 dilution for 3–4 hours at 4°C prior to visualization using an enhanced chemiluminescence kit (ECL plus; Amersham Biosciences, Piscataway, NJ).
Bcl-2 family Gene Expression
Macroarray experiments were performed using the GE Array expression kit (SuperArray Inc., Bethesda, MD) using total RNA isolated from cells incubated with or without IL-6 for 24 hours and following the manufacturer’s instructions (27). Expression analysis was performed using the GEArrayAnalyzer™ software (SuperArray Inc., Bethesda, MD). Background subtraction was performed by using plasmid DNA (PUC 18) as negative controls, and gene expression was normalized to GAPDH expression.
RNA interference
SiRNA specific to Mcl-1 and control siRNA were obtained from Qiagen-Xeragon (Germantown, MD). KMCH cells were transfected as previously described (24). Briefly, 0.1 μg siRNA were mixed with 6 μl transfection agent (TransIt TKO, Mirus Corp., Madison, WI) and the mixture was incubated in 1 ml of media at room temperature for 15–20 min prior to adding to cultured cells grown to 50–60% confluency. 48 hours later, efficacy of gene silencing was assessed by immunoblot analysis.
Transient transfections
Cells (3 × 105) were plated in 35-mm dish in culture media for 24 hours. Transient transfections were performed as previously described in cultured cells at 40–60% confluency (25). In brief, the media was then replaced, and plasmids transfected in serum-free media using 2 μg of plasmid DNA per dish and TransIT-LT1 transfection reagent (Mirus Corp, Madison, WI). After 4–6 hr, the medium was replaced with regular media containing 10% serum and cells incubated for 48 hrs prior to study.
Reagents and Plasmids
The FLAG-tagged wild type and dominant negative forms (AF or KM) of human p38α, p38β, p38γ, and p38δ were a kind gift of Dr J Han (Scripps Research Institute, La Jolla, CA) and have been previously described (28–31). The AFs are p38 mutants that cannot be phosphorylated, since the TGY dual phosphorylation site has been changed to AGF, whereas the kinase-dead KM mutants were generated by a mutation of the ATP-binding site Lys to Met (K to M).
Statistical analysis
Data are expressed as the mean ± standard error (SE) from at least three separate experiments performed in triplicate, unless otherwise noted. Differences between groups was analyzed using a double sided Student’s t test when only two groups were present. Statistical significance was considered as p < 0.05. Statistical analyses were performed with the GB-STAT statistical software program (Dynamic Microsystems Inc., Silver Spring, MD).
Materials
Fetal bovine serum and Bradford reagent were obtained from Sigma (St Louis, MO). CMRL 1066 media, DMEM, L-glutamine, and antibiotic-antimycotic mix were from Gibco BRL (Grand Island, New York). Monoclonal antibody to Mcl-1 was from Santa Cruz Biotechnology (Santa Cruz, CA). Recombinant human IL-6 and IL-6 ELISA assay kits were purchased from Biosource International (Camarillo, CA). Actinomycin D and MG132 were purchased from Calbiochem (La Jolla, CA).
Results
Over-expression of IL-6 increases transformed cell behavior of human malignant cholangiocytes
Cell lines designated as KM-IL-6 and Mz-IL-6 were generated from KMCH-1 or Mz-ChA-1 human malignant cholangiocytes following stable transfection with the pCMV-IL-6 expression plasmid encoding full-length human IL-6. Basal secretion of IL-6 was increased in both the KM-IL-6 and the Mz-IL-6 cells compared to their respective controls (Fig 1A). To ascertain the biological effects of IL-6 over-expression on transformed cell behavior, we began by assessing the extent to which over-expression of IL-6 modulated anchorage independent growth in soft agar. Compared to controls, an increase in the number of colonies formed on soft agar was observed with the IL-6 over-expressing KM-IL-6 or Mz-IL-6 cells (Fig 1B). Furthermore, we evaluated the impact of IL-6 over-expression in vivo by assessing the growth rates of Mz-1 and Mz-IL-6 tumor cell xenografts in nude mice. There was no cachexia noted or difference in the mean weights of the mice with Mz-IL-6 xenografts compared to the control cell xenografts. IL-6 mRNA expression was assessed in tumor xenografts by quantitative real-time PCR and was considerably increased in Mz-IL-6 xenografts compared to control cell xenografts (Fig 2A). Consistent with the observations in the soft agar studies, over-expression of IL-6 increased the rate of xenograft growth in nude mice (Fig 2B). These findings provide direct evidence that over-expression of IL-6 promotes transformed cell behavior and tumor growth. They are consistent with the clinical evidence suggesting that IL-6 can promote human cholangiocarcinoma growth.
Figure 1. Over-expression of IL-6 increases transformed cell growth.

KMCH or Mz-ChA-1 cells were stably transfected with expression plasmids containing full length IL-6 and a mixture of stably transfected clones was obtained. IL-6 over-expressing stable transfectants were designated as KM-IL-6 or Mz-IL-6, whereas control cells were designated as KM-1 or Mz-1 respectively. Panel A. IL-6 secretion in culture supernatant was assessed using an ELISA assay. Both KM-IL-6 and Mz-IL-6 cells had increased IL-6 production compared to their respective controls. Panel B. Transformed cell growth in vitro was assessed by determining anchorage independent growth in soft agar. Both KM-IL-6 and Mz-IL-6 cells had increased colony formation in soft agar after 21 days compared to their respective control KM-1 or Mz-1 cells. * p < 0.05 compared to controls.
Figure 2. Over-expression of IL-6 increases tumor growth in vivo.

3 x 106 cells were injected subcutaneously in the flank of athymic balb/c mice. Panel A. RNA was extracted from xenograft tissues and IL-6 mRNA expression assessed by quantitative real-time PCR. Representative amplification plots are shown. IL-6 mRNA expression normalized to β-actin mRNA expression was 1.32 ± 0.54 for Mz-1 xenografts and 7.86 ± 1.12 for Mz-IL-6 xenografts (data from three experiments performed in triplicate). Panel B. Tumor growth was assessed by serial measurements. Data represents the mean of four xenografts for each group. Differences between the two groups were statistically significant (p < 0.05) beyond day 55. Over-expression of IL-6 increased cholangiocarcinoma cell xenograft growth in nude mice.
Over-expression of IL-6 does not increase proliferation in human malignant cholangiocytes
IL-6 is mitogenic for both non-malignant and malignant human cholangiocytes (10, 12, 16, 21). Thus we began by assessing proliferation in malignant cholangiocytes over-expressing IL-6. Surprisingly, proliferation of neither the KM-IL-6 nor the Mz-IL-6 cells was significantly different from their respective controls in short-term assays up to 72 hrs (Fig 3A). We next analyzed the population doubling (PD) rate after serial passaging to determine if a proliferative advantage occurred over prolonged time periods. The proliferative rate for IL-6 over-expressing KM-IL-6 cells was 0.028 ± 0.013 PD/hr, and was not significantly different from that of control cells, which was 0.027 ± 0.004 PD/hr after the ninth passage. Similar results were observed with Mz-IL-6 cells. Next, we assessed the ability of IL-6 over-expressing cells to proliferate during conditions of reduced growth factor availability. However, there was no significant difference between either the KM-IL-6 or the Mz-IL-6 cells and their respective controls in growth under serum-free or reduced serum (1% serum) conditions (Fig 3B). In combination, these observations indicate that over-expression of IL-6 does not confer a proliferative advantage to malignant cholangiocytes.
Figure 3. Over-expression of IL-6 does not increase proliferation.
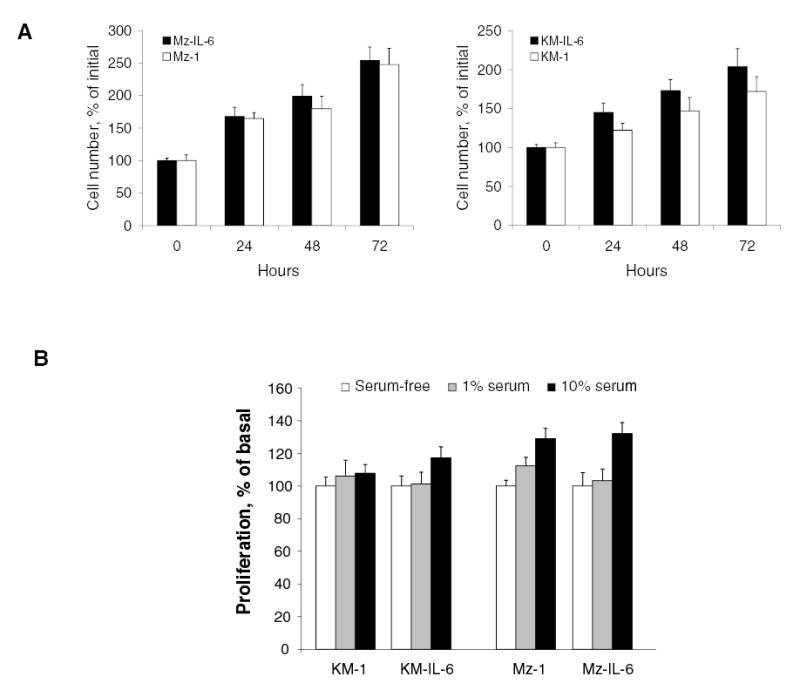
Panel A. Cell proliferation was analyzed at the indicated times using a viable cell assay in IL-6 over-expressing KM-IL-6 or Mz-IL-6 cells, or their respective control KM-1 or Mz-1 cells. The differences between the two groups were not significant at any of the time points (p > 0.05). Panel B. The effect of IL-6 over-expression on proliferation under serum-free or reduced (1%) serum conditions was assessed. Cells over-expressing IL-6 (KM-IL-6 or Mz-IL-6) and their respective controls (KM-1 or Mz-1) were grown in the presence of varying concentrations of serum, and proliferation assessed after 24 hours. The differences between any of the groups was not significant (p > 0.05).
Over-expression of IL-6 promotes cell survival in malignant cholangiocytes
Since tumor growth can be enhanced by increased cell survival in addition to alterations in cell proliferation, we next evaluated the effect of IL-6 over-expression on cell survival. We began by exploring the influence of IL-6 over-expression on survival effects in vitro by quantitating apoptosis during incubation with a variety of cytotoxic stimuli. Compared to controls, apoptosis was significantly decreased in IL-6 over-expressing KM-IL-6 cells (Fig 4A). Similar results were observed with Mz-IL-6 cells (Fig 4B). Furthermore, incubation with IL-6 decreased apoptosis induced by the chemotherapeutic agent gemcitabine in several different human cholangiocarcinoma cell lines (Fig 4C). We further compared the effect of IL-6 over-expression on cellular apoptosis and proliferation in vivo, assessed by immunohistochemical techniques, in IL-6 over-expressing Mz-IL-6 or control Mz-1 xenografts in nude mice (Fig 5). While tissue sections from xenografts of IL-6 over-expressing cells had decreased numbers of apoptotic cells compared to control xenografts, the number of proliferating cells was not significantly altered. Therefore, IL-6 activates survival signaling mechanisms in malignant cholangiocytes. In combination, these data provide compelling evidence indicating that autocrine IL-6 production in malignant cholangiocytes promotes transformed cell growth by enhancing cell survival rather than affecting cell proliferation.
Figure 4. Over-expression of IL-6 decreases apoptosis in vitro.

Apoptosis was assessed in KM-IL-6 cells and KM-1 cells (Panel A), or Mz-IL-6 and Mz-1 cells (Panel B) incubated with 30 μM Camptothecin (CPT), 30 μM Gemcitabine (GEM) or 200 nM TRAIL. After 24 hours, the cells were stained with DAPI and the number of cells with morphological features of apoptosis was identified by fluorescence microscopy. There was a significant reduction in apoptosis in the KM-IL-6 cells compared to controls for all agents. * p < 0.05 compared to KM-1 cells. Panel C. Apoptosis was assessed in human malignant cholangiocyte cells pre-treated in the presence or absence of 10 ng/ml IL-6 for 24 hours followed by incubation with 30 μM gemcitabine for 24 hours. Apoptosis was quantitated using the APO Percentage apoptosis assay, and cellular dye uptake analyzed according to the manufacturer’s protocol. Data represents mean and standard error from four separate studies. IL-6 decreased gemcitabine-induced apoptosis in each cell line. * p < 0.05 compared to controls.
Figure 5. Over-expression of IL-6 decreases apoptosis in vivo.

Tissue sections were obtained from xenografts of IL-6 over-expressing (Mz-IL-6) or control (Mz-1) malignant human cholangiocytes in nude mice. Immunohistochemistry for PCNA and TUNEL analysis were performed to identify tumor cell proliferation and apoptosis, respectively. Representative histological sections are shown, with the arrows depicting some positive cells. The number of PCNA positive, or TUNEL positive cells for at least 300 total cells (by nuclear counts) were quantitated for each of 5 separate sections and the mean ± standard error of the percentage of positively staining cells for each group is depicted in the graph. Although the number of TUNEL positive (apoptotic) cells was reduced in Mz-IL-6 xenografts, the number of PCNA positive (proliferating) cells was not different between the two groups. * p < 0.01 compared to Mz-1 controls.
IL-6 increases Mcl-1 expression
The Bcl-2 family of proteins are potent endogenous regulators of cell survival, and can modulate apoptosis induced by different stimuli (32–34). To identify members of the Bcl-2 family that contributed to the survival effects of IL-6, we used a commercial gene hybridization macroarray to analyze the expression of members of this group in KMCH cells stimulated with IL-6 (10 ng/ml, 24 hours). Although the expression of most members of the Bcl-2 family was not altered by IL-6, a greater than two-fold increase in expression of Mcl-1 was noted (Fig 6). Mcl-1 is anti-apoptotic and has been implicated as a survival factor for cholangiocarcinoma (35–37). Moreover, Mcl-1 has been shown to be regulated by IL-6 in a variety of hepatic and other epithelial cancer cell lines such as basal cell carcinoma, gastric cancer and cervical cancer (37–41). We further assessed the effect of IL-6 on Mcl-1 protein expression in human malignant cholangiocytes. Basal expression of Mcl-1 by quantitative immunoblot analysis was increased in IL-6 over-expressing KM-IL-6 or Mz-IL-6 cells. Moreover, Mcl-1 expression was also increased in xenografts of IL-6 over-expressing malignant cholangiocytes compared to control xenografts (Fig 7). We next explored the pathophysiological relevance of the observed differences in basal expression of Mcl-1 in IL-6 over-expressing cells using RNA interference. Cells in which Mcl-1 expression was reduced by siRNA showed reduced resistance to gemcitabine compared to controls (Fig 8). These findings indicate that Mcl-1 contributes to the survival effects of IL-6. Furthermore, these observations suggest that the increased basal levels of Mcl-1 in cells over-expressing IL-6 may contribute to chemoresistance in cholangiocarcinoma.
Figure 6. Interleukin-6 increases Mcl-1 expression.

Total RNA was isolated from cells incubated in the presence or absence of 10 ng/ml IL-6 for 24 hours. Biotinylated cDNA probes were synthesized, and hybridized to nylon membranes containing two spots of each of various Bcl-2 family member genes (Human apoptosis original series GE array, SUpperArray, Bethesda, MD). Gene expression was quantitated by chemiluminescence , and normalized to GAPDH expression. Results represent average of two separate experiments.
Figure 7. Over-expression of IL-6 increases Mcl-1 protein expression in vivo.

Mcl -1 mRNA expression was assessed in IL-6 over-expressing (Mz-IL-6) or control (Mz-1) malignant cholangiocyte xenografts in nude mice by quantitative real-time PCR. The data represents mean and standard error from six experiments. * p < 0.05 compared to Mcl-1 mRNA expression in control Mz-1 xenografts. Immunohistochemistry for Mcl-1 was performed in sections obtained from the Mz-IL-6 or Mz-1 xenografts and representative histological sections are shown. An increase in Mcl-1 expression occurs in vivo in Mz-IL-6 xenografts compared to Mz-1 xenografts in nude mice.
Figure 8. siRNA to Mcl-1 decreases survival effects of IL-6.
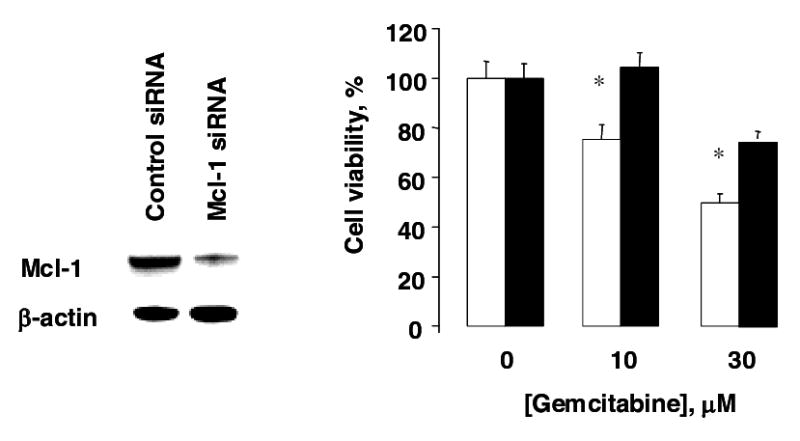
KM-IL-6 cells were incubated with siRNA to Mcl-1 to decrease cellular expression (white bars), or control siRNA (black bars). An immunoblot was performed after 48 hours (right panel). Cells were treated with the chemotherapeutic drug gemcitabine at the indicated concentrations and cell viability was assessed after 24 hours (left panel). Incubation with siRNA to Mcl-1 increased the cytotoxic effects of gemcitabine in IL-6 over-expressing cells compared to incubation with control siRNA. * p < 0.05 compared to control siRNA.
Mcl-1 expression is transcriptionally regulated by IL-6
To elucidate the mechanisms by which IL-6 regulates Mcl-1 expression, we began by assessing Mcl-1 mRNA production during incubation with IL-6 using quantitative real-time PCR. Stimulation of KMCH malignant cholangiocytes with IL-6 (10 ng/ml) increased Mcl-1 mRNA expression by 456 ± 100% (Fig 9). Mcl-1 has a short-half life, and can be rapidly degraded by proteasomal hydrolysis. Incubation of KMCH cells with the proteasomal inhibitor MG132 (50 nM) increased Mcl-1 protein expression to 140 ± 12% of controls after 24 hours. These data indicate that cellular Mcl-1 protein expression in malignant cholangiocytes under basal conditions is sensitive to proteasomal hydrolysis. Thus, Mcl-1 protein expression can be modulated by effects on protein degradation as well as by increased transcription. Nevertheless, basal Mcl-1 protein expression was almost completely abolished by pre-incubation with Actinomycin D, but was not significantly altered by pre-incubation with MG132 in IL-6 over-expressing cells. Similar results were observed in KMCH malignant cholangiocytes treated with IL-6. Overall, these data indicate that IL-6 can increase cellular Mcl-1 protein expression by several mechanisms including an increase in mRNA production.
Figure 9. Transcriptional Regulation of Mcl-1 by IL-6.

KMCH cells were treated with 10 ng/ml IL-6 for the indicated times. RNA was isolated and cDNA generated by reverse transcription using MMLV reverse transcriptase. Mcl-1 mRNA expression was quantitated by real-time PCR using SYBR Green as the fluorophore, and expressed relative to β-actin mRNA expression assessed concurrently in the same samples. IL-6 stimulation increased Mcl-1 mRNA expression in a time-dependent manner. A representative amplification plot is shown along with data summarizing four separate determinations done in triplicate. The PCR products were verified by melting curve analysis as well as by 1.8% agarose gel electrophoresis of the PCR product (not shown).
Up-regulation of Mcl-1 by IL-6 involves p38 MAPK signaling
We have previously shown that the p38 MAPK pathway is aberrantly activated in malignant, but not in non-malignant cholangiocytes by IL-6 (12, 21, 26). Moreover, inhibition of p38 MAPK activation decreases xenograft growth and anchorage independent growth of malignant cholangiocytes (24). These observations suggest that downstream targets regulated by p38 MAPK activation may contribute to promotion of tumor growth by IL-6. Therefore, we first explored the role of p38 MAPK signaling in Mcl-1 expression by IL-6. IL-6 stimulation of KMCH malignant cholangiocytes increased Mcl-1 protein expression (Fig 10A). Similar results were observed in Mz-ChA-1, TFK-1 and CC-LP malignant human cholangiocytes after IL-6 treatment. Next, we assessed Mcl-1 expression using KM-p38dn cells, derived from KMCH malignant cholangiocytes stably transfected with a mutated MKK3 (A) that have reduced p38 MAPK activation following stimulation (24). In contrast to parental KMCH cells, IL-6 stimulation of KM-p38dn cells did not alter Mcl-1 expression.
Figure 10. IL-6 increases Mcl-1 expression via a p38 MAPK pathway.
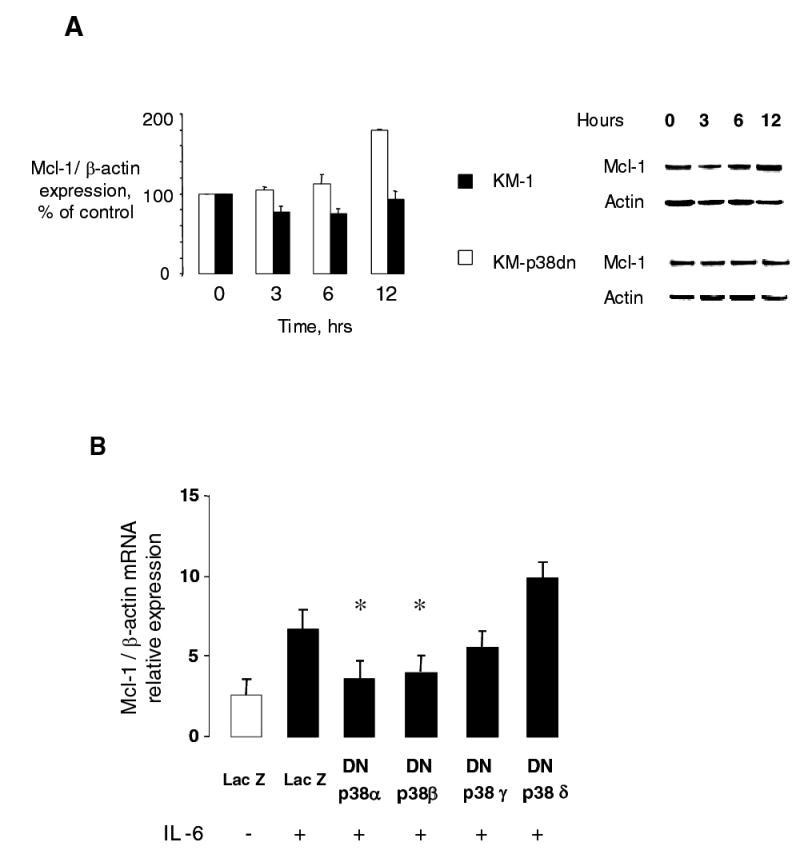
Panel A. KMCH cells stably transfected with a dominant negative MKK3 (KM-p38dn), or control cells (KM-1) were serum starved for 12 hours prior to stimulation with 10 ng/ml IL-6. Immunoblot analyses of cell lysates were done using monoclonal mouse anti-Mcl-1 and polyclonal β-Actin antibody (loading control). IL-6 increased Mcl-1 expression in a time dependent manner in the controls but did not increase Mcl-1 in the KM-p38dn cells. Thus, regulation of Mcl-1 expression involves p38 MAPK activation. Representative immunoblots and quantitative data from 3 separate experiments are shown for KM-1 (white bars) and KM-p38dn (black bars) cells. Panel B. KMCH cells were transfected with Flag tagged dominant negative p38 MAPK α, β, γ or δ isoforms or LacZ expressing control plasmids. After 48 hours, cells were stimulated with 10 ng/ml IL-6 for 40 minutes. Mcl-1 mRNA expression was quantitated by real-time PCR, and expressed relative to that of β-actin mRNA concurrently assessed in the same samples. The increase in Mcl-1 mRNA expression by IL-6 was significantly inhibited by dominant negative p38α or p38β isoforms. * p < 0.05 compared to Lac Z transfected IL-6 treated controls.
There are several p38 MAPK isoforms, and inhibition of the upstream MKK3 globally decreases activation of the downstream isoforms. However, p38 isoforms can have divergent transcriptional responses (28, 42). To clarify the involvement of specific p38 MAPK isoforms in regulation of Mcl-1 expression by IL-6, KMCH cells were transiently transfected with p38 α, β, γ and δ isoform specific dominant negative constructs, and the expression of Mcl-1 was assessed by quantitative real-time PCR (Fig 10B). Inhibition of the p38 α or β MAPK isoforms abolished the effect of IL-6 on Mcl-1 mRNA expression. These observations demonstrate a role for Mcl-1 as one of the transcriptionally regulated downstream mediators of the effects of aberrant p38 MAPK signaling in promoting cholangiocarcinoma growth.
Discussion
The novel findings of this study relate to the identification of survival signaling by IL-6 as a contributor to transformed cell growth in human cholangiocarcinoma. Mcl-1 is identified as a downstream effector of the survival effects of IL-6 and as a key mediator of chemoresistance in cholangiocarcinoma. These data identify a primary involvement of survival signaling by IL-6 during tumor growth in malignant cholangiocytes and show that enforced expression of IL-6 (a) increases cell survival but not proliferation in malignant cholangiocytes; (b) increases tumor cell growth in xenografts in nude mice as well as anchorage independent growth in vitro; (c) decreases chemotherapeutic drug induced apoptosis by up-regulation of the anti-apoptotic protein Mcl-1 and (d) increases constitutive expression of Mcl-1 via a p38 MAPK dependent mechanism. These observations are of biological and clinical significance given the role of IL-6 as an autocrine factor involved in human cholangiocarcinoma as well as the involvement of p38 MAPK signaling in mediating growth of these tumors.
Survival effects have been reported for IL-6 in a variety of malignancies such as basal cell carcinoma, prostate cancer, esophageal carcinoma and multiple myeloma (38, 43–45). Tumor growth can be promoted by cell survival resulting from modulation of apoptosis by cytokines. Although autocrine promotion of tumor growth has been proposed, the present study is the first to systematically evaluate the role of mitogenic or survival signaling in the growth of human cholangiocarcinoma cells in vitro and in vivo. Despite the well-characterized effect of IL-6 as a mitogen for biliary epithelia, autocrine IL-6 production does not provide a proliferative advantage to malignant cholangiocytes. In contrast there is ample evidence of increased survival signaling. Furthermore, activation of survival signaling may potentially contribute to resistance to therapeutic modalities such as chemotherapy and radiation therapy. Thus, manipulation of IL-6 survival signaling is likely to be a powerful strategy for the treatment of cholangiocarcinoma.
Mcl-1 was originally identified as a result of its up-regulation in a human myeloblastic leukemia cell line that was differentiated along the monocyte lineage (46). The mcl-1 knockout mouse is embryonic lethal but without any obvious effects on embryogenesis. However, Mcl-1 can act as a survival factor in hematopoeitic cells in cell culture as well as in transgenic mice (47). Furthermore, Mcl-1 may play an essential role in cytokine mediated survival of hematopoeitic cells and function as an early response gene when stimulated with granulocyte/macrophage colony stimulating factor, (48–50). Our findings are consistent with these observations and suggest that Mcl-1 can act as an IL-6 early response gene in mediating survival signaling in malignant cholangiocytes. Although our data do not directly implicate Mcl-1 in transformed cell growth, we have previously reported that inhibition of p38 MAPK activation in malignant cholangiocytes decreases xenograft growth in vivo and anchorage independent growth in vitro (24). Further detailed studies to investigate the involvement of Mcl-1 in promoting transformed cell behavior, in addition to the documented effect on chemo-sensitivity, are warranted based on these reports and the results of our current studies showing that Mcl-1 is a downstream effector of the p38 MAPK pathway.
IL-6 can activate multiple intracellular signaling pathways including the p38 MAPK, the JAK-STAT, the ERK1,2 and the PI3-kinase pathways. Of these, inhibitor studies have identified the PI3-kinase and the p38 MAPK pathway as being involved in cell survival signaling in cholangiocytes. IL-6 induced PI 3 kinase signaling resulting in activation of Mcl-1 have been reported in several different settings (39, 41, 49, 51). In contrast, the role of p38 MAPK in mediating Mcl-1 expression has not previously been reported to the best of our knowledge. Our findings reported suggest that p38 MAPK α and β isoforms are critically involved in mediating Mcl-1 expression by IL-6. Although our study has focused on the role of p38 MAPK given the importance of this pathway in mediating transformed cell growth in malignant cholangiocytes, the findings are not inconsistent with concomitant activation of other signaling pathways such as the PI 3-kinase or JAK-STAT pathways. Indeed, given the importance of survival signaling, there is likely to be redundancy in IL-6 signaling pathways, or cross-talk between different signaling pathways.
In this study, we have identified cellular mechanisms by which over-expression of IL-6, an autocrine factor involved in cholangiocarcinoma growth, can promote cell survival and enhance tumor growth. We propose that persistant stimulation of biliary tract epithelia by inflammatory stimuli results in deregulated IL-6 production. Over-expression of IL-6 subsequently promotes cell survival pathways that contribute to the growth of transformed cells as well as resistance to therapy (Fig 11). Based on this model, targeted efforts at ameliorating the survival signaling by IL-6 such as by disruption of autocrine IL-6 production, or by manipulating mediators of intracellular signaling such as the p38 MAPK or Mcl-1 may be appropriate strategies to decrease growth or increase responses to conventional treatments for cholangiocarcinoma.
Figure 11. Aberrant IL-6 production and cellular responses in the pathogenesis of cholangiocarcinoma.
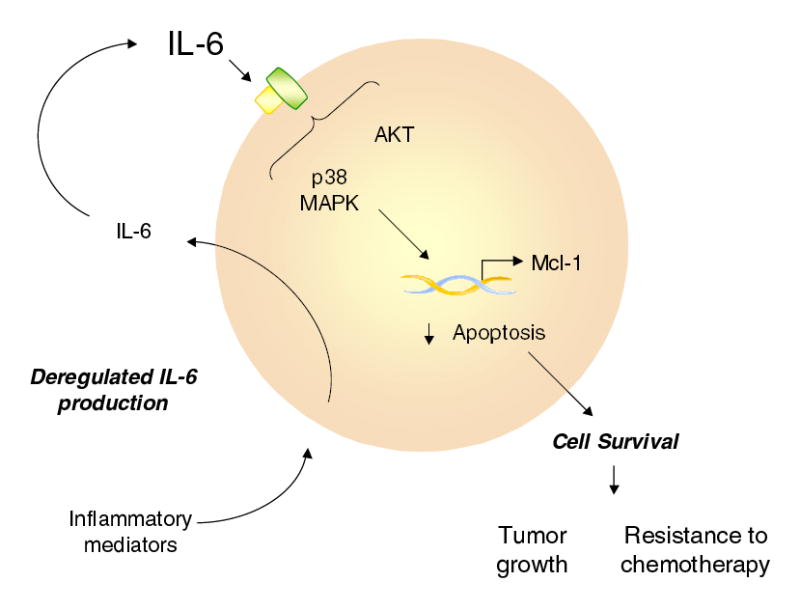
Chronic biliary tract inflammation predisposes to cholangiocarcinoma. We postulate that persistent stimulation by inflammatory mediators results in deregulated production of IL-6 in cholangiocytes. IL-6 acts in an autocrine or paracrine manner in malignant cholangiocytes to activate intracellular kinase signaling pathways involved in cell survival. Downstream effectors of survival signaling may involve anti-apoptotic proteins such as Mcl-1, which can influence tumor cell responses to chemotherapy as well as tumor growth. Therefore, interventions targeted towards upstream kinases activated by IL-6 such as p38 MAPK and AKT, or manipulation of effectors such as Mcl-1 may be appropriate therapeutic targets for human cholangiocarcinoma.
Acknowledgments
The technical assistance provided by Laura Tadlock, Valorie Chiasson and Linda Brooks are gratefully acknowledged. This work was supported by the Scott, Sherwood and Brindley Foundation, and Grants DK069370 from the National Institutes of Health.
References
- 1.Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001 Jun;33(6):1353–1357. doi: 10.1053/jhep.2001.25087. [DOI] [PubMed] [Google Scholar]
- 2.Patel T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer. 2002 May 3;2(1):10. doi: 10.1186/1471-2407-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan SA, Taylor-Robinson SD, Toledano MB, Beck A, Elliott P, Thomas HC. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J Hepatol. 2002 Dec;37(6):806–813. doi: 10.1016/s0168-8278(02)00297-0. [DOI] [PubMed] [Google Scholar]
- 4.Taylor-Robinson SD, Toledano MB, Arora S, Keegan TJ, Hargreaves S, Beck A, et al. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968–1998. Gut. 2001 Jun;48(6):816–820. doi: 10.1136/gut.48.6.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gores GJ. Cholangiocarcinoma: current concepts and insights. Hepatology. 2003 May;37(5):961–969. doi: 10.1053/jhep.2003.50200. [DOI] [PubMed] [Google Scholar]
- 6.Rosen HR, Winkle PJ, Kendall BJ, Diehl DL. Biliary interleukin-6 and tumor necrosis factor-alpha in patients undergoing endoscopic retrograde cholangiopancreatography. Dig Dis Sci. 1997 Jun;42(6):1290–1294. doi: 10.1023/a:1018822628096. [DOI] [PubMed] [Google Scholar]
- 7.Akiyama T, Hasegawa T, Sejima T, Sahara H, Seto K, Saito H, et al. Serum and bile interleukin 6 after percutaneous transhepatic cholangio-drainage. Hepatogastroenterology. 1998 May;45(21):665–671. [PubMed] [Google Scholar]
- 8.Kimura F, Miyazaki M, Suwa T, Sugiura T, Shinoda T, Itoh H, et al. Serum interleukin-6 levels in patients with biliary obstruction. Hepatogastroenterology. 1999 May;46(27):1613–1617. [PubMed] [Google Scholar]
- 9.Scotte M, Daveau M, Hiron M, Delers F, Lemeland JF, Teniere P, et al. Interleukin-6 (IL-6) and acute-phase proteins in rats with biliary sepsis. Eur Cytokine Netw. 1991 May;2(3):177–182. [PubMed] [Google Scholar]
- 10.Matsumoto K, Fujii H, Michalopoulos G, Fung JJ, Demetris AJ. Human biliary epithelial cells secrete and respond to cytokines and hepatocyte growth factors in vitro: interleukin-6, hepatocyte growth factor and epidermal growth factor promote DNA synthesis in vitro. Hepatology. 1994 Aug;20(2):376–382. [PubMed] [Google Scholar]
- 11.Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16(3–4):249–284. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 12.Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999 Apr;29(4):1037–1043. doi: 10.1002/hep.510290423. [DOI] [PubMed] [Google Scholar]
- 13.Kallen KJ. The role of transsignalling via the agonistic soluble IL-6 receptor in human diseases. Biochim Biophys Acta. 2002 Nov 11;1592(3):323–343. doi: 10.1016/s0167-4889(02)00325-7. [DOI] [PubMed] [Google Scholar]
- 14.Maione D, Di Carlo E, Li W, Musiani P, Modesti A, Peters M, et al. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. EMBO J. 1998 Oct 1;17(19):5588–5597. doi: 10.1093/emboj/17.19.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goydos JS, Brumfield AM, Frezza E, Booth A, Lotze MT, Carty SE. Marked elevation of serum interleukin-6 in patients with cholangiocarcinoma: validation of utility as a clinical marker. Ann Surg. 1998 Mar;227(3):398–404. doi: 10.1097/00000658-199803000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokomuro S, Tsuji H, Lunz JG, III, Sakamoto T, Ezure T, Murase N, et al. Growth control of human biliary epithelial cells by interleukin 6, hepatocyte growth factor, transforming growth factor beta1, and activin A: comparison of a cholangiocarcinoma cell line with primary cultures of non-neoplastic biliary epithelial cells. Hepatology. 2000 Jul;32(1):26–35. doi: 10.1053/jhep.2000.8535. [DOI] [PubMed] [Google Scholar]
- 17.Sugawara H, Yasoshima M, Katayanagi K, Kono N, Watanabe Y, Harada K, et al. Relationship between interleukin-6 and proliferation and differentiation in cholangiocarcinoma. Histopathology. 1998 Aug;33(2):145–153. doi: 10.1046/j.1365-2559.1998.00445.x. [DOI] [PubMed] [Google Scholar]
- 18.Yokomuro S, Lunz JG, III, Sakamoto T, Ezure T, Murase N, Demetris AJ. The effect of interleukin-6 (IL-6)/gp130 signalling on biliary epithelial cell growth, in vitro. Cytokine. 2000 Jun;12(6):727–730. doi: 10.1006/cyto.1999.0612. [DOI] [PubMed] [Google Scholar]
- 19.Sakamoto T, Liu Z, Murase N, Ezure T, Yokomuro S, Poli V, et al. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology. 1999 Feb;29(2):403–411. doi: 10.1002/hep.510290244. [DOI] [PubMed] [Google Scholar]
- 20.Yamagiwa Y, Marienfeld C, Meng F, Holcik M, Patel T. Translational regulation of x-linked inhibitor of apoptosis protein by interleukin-6: a novel mechanism of tumor cell survival. Cancer Res. 2004 Feb 15;64(4):1293–1298. doi: 10.1158/0008-5472.can-03-2517. [DOI] [PubMed] [Google Scholar]
- 21.Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999 Nov;30(5):1128–1133. doi: 10.1002/hep.510300522. [DOI] [PubMed] [Google Scholar]
- 22.Patel T. Dysregulation of cholangiocyte apoptosis by cytokines. In: G Paumgartner, editor. Diseases of the Liver and Bile Ducts. New aspects and clinical implications. Lancaster, England: Kluwer Academic Publishers; 1998.
- 23.Okada K, Shimizu Y, Nambu S, Higuchi K, Watanabe A. Interleukin-6 functions as an autocrine growth factor in a cholangiocarcinoma cell line. J Gastroenterol Hepatol. 1994 Sep;9(5):462–467. doi: 10.1111/j.1440-1746.1994.tb01275.x. [DOI] [PubMed] [Google Scholar]
- 24.Yamagiwa Y, Marienfeld C, Tadlock L, Patel T. Translational regulation by p38 mitogen-activated protein kinase signaling during human cholangiocarcinoma growth. Hepatology. 2003 Jul;38(1):158–166. doi: 10.1053/jhep.2003.50257. [DOI] [PubMed] [Google Scholar]
- 25.Tadlock L, Yamagiwa Y, Hawker J, Marienfeld C, Patel T. Transforming growth factor-{beta} inhibition of proteasomal activity. A potential mechanism of growth arrest. Am J Physiol Cell Physiol 2003 Mar 19. [DOI] [PubMed]
- 26.Tadlock L, Patel T. Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology. 2001 Jan;33(1):43–51. doi: 10.1053/jhep.2001.20676. [DOI] [PubMed] [Google Scholar]
- 27.Tadlock L, Yamagiwa Y, Marienfeld C, Patel T. Double-stranded RNA activates a p38 MAPK-dependent cell survival program in biliary epithelia. Am J Physiol Gastrointest Liver Physiol. 2003 Jun;284(6):G924–G932. doi: 10.1152/ajpgi.00355.2002. [DOI] [PubMed] [Google Scholar]
- 28.Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, Han J, et al. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 2003 Feb 14;278(7):4831–4839. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, et al. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997 Nov 28;272(48):30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 30.Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996 Feb 9;271(6):2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 31.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, et al. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta) J Biol Chem. 1996 Jul 26;271(30):17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 32.Kirkin V, Joos S, Zornig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta. 2004 Mar 1;1644(2–3):229–249. doi: 10.1016/j.bbamcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 33.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003 Nov 24;22(53):8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 34.Korsmeyer SJ. Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood. 1992 Aug 15;80(4):879–886. [PubMed] [Google Scholar]
- 35.Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004 May 15;64(10):3517–3524. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 36.Yoon JH, Werneburg NW, Higuchi H, Canbay AE, Kaufmann SH, Akgul C, et al. Bile acids inhibit Mcl-1 protein turnover via an epidermal growth factor receptor/Raf-1-dependent mechanism. Cancer Res. 2002 Nov 15;62(22):6500–6505. [PubMed] [Google Scholar]
- 37.Kobayashi S, Werneburg NW, Bronk SF, Kaufmann SH, Gores GJ. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005 Jun;128(7):2054–2065. doi: 10.1053/j.gastro.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Jourdan M, Veyrune JL, Vos JD, Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003 May 15;22(19):2950–2959. doi: 10.1038/sj.onc.1206423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei LH, Kuo ML, Chen CA, Chou CH, Cheng WF, Chang MC, et al. The anti-apoptotic role of interleukin-6 in human cervical cancer is mediated by up-regulation of Mcl-1 through a PI 3-K/Akt pathway. Oncogene. 2001 Sep 13;20(41):5799–5809. doi: 10.1038/sj.onc.1204733. [DOI] [PubMed] [Google Scholar]
- 40.Lin MT, Juan CY, Chang KJ, Chen WJ, Kuo ML. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human gastric cancer AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis. 2001 Dec;22(12):1947–1953. doi: 10.1093/carcin/22.12.1947. [DOI] [PubMed] [Google Scholar]
- 41.Jee SH, Chiu HC, Tsai TF, Tsai WL, Liao YH, Chu CY, et al. The phosphotidyl inositol 3-kinase/Akt signal pathway is involved in interleukin-6-mediated Mcl-1 upregulation and anti-apoptosis activity in basal cell carcinoma cells. J Invest Dermatol. 2002 Nov;119(5):1121–1127. doi: 10.1046/j.1523-1747.2002.19503.x. [DOI] [PubMed] [Google Scholar]
- 42.Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem. 2003 May 16;278(20):17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 43.Jee SH, Shen SC, Chiu HC, Tsai WL, Kuo ML. Overexpression of interleukin-6 in human basal cell carcinoma cell lines increases anti-apoptotic activity and tumorigenic potency. Oncogene. 2001 Jan 11;20(2):198–208. doi: 10.1038/sj.onc.1204076. [DOI] [PubMed] [Google Scholar]
- 44.Steiner H, Godoy-Tundidor S, Rogatsch H, Berger AP, Fuchs D, Comuzzi B, et al. Accelerated in vivo growth of prostate tumors that up-regulate interleukin-6 is associated with reduced retinoblastoma protein expression and activation of the mitogen-activated protein kinase pathway. Am J Pathol. 2003 Feb;162(2):655–663. doi: 10.1016/S0002-9440(10)63859-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leu CM, Wong FH, Chang C, Huang SF, Hu CP. Interleukin-6 acts as an antiapoptotic factor in human esophageal carcinoma cells through the activation of both STAT3 and mitogen-activated protein kinase pathways. Oncogene. 2003 Oct 30;22(49):7809–7818. doi: 10.1038/sj.onc.1207084. [DOI] [PubMed] [Google Scholar]
- 46.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol. 1996 Mar;166(3):523–536. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 47.Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB, et al. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood. 1998 Nov 1;92(9):3226–3239. [PubMed] [Google Scholar]
- 48.Chao JR, Wang JM, Lee SF, Peng HW, Lin YH, Chou CH, et al. mcl-1 is an immediate-early gene activated by the granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling pathway and is one component of the GM-CSF viability response. Mol Cell Biol. 1998 Aug;18(8):4883–4898. doi: 10.1128/mcb.18.8.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999 Sep;19(9):6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schubert KM, Duronio V. Distinct roles for extracellular-signal-regulated protein kinase (ERK) mitogen-activated protein kinases and phosphatidylinositol 3-kinase in the regulation of Mcl-1 synthesis. Biochem J. 2001 Jun 1;356(Pt 2):473–480. doi: 10.1042/0264-6021:3560473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuo ML, Chuang SE, Lin MT, Yang SY. The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene. 2001 Feb 8;20(6):677–685. doi: 10.1038/sj.onc.1204140. [DOI] [PubMed] [Google Scholar]