

Abstract
Previous data showing that several chloroquine analogs containing an intramolecular hydrogen bonding motif were potent against multidrug-resistant P. falciparum, led to the exploration of the importance of this motif. A series of 116 compounds containing four different alkyl linkers and various aromatic substitutions with hydrogen bond accepting capability was synthesized. The series showed broad potency against the drug-resistant W2 strain of P. falciparum. In particular, a novel series containing variations of the α-aminocresol motif gave 8 compounds with IC50's more potent than 5 nM against the W2 strain. Such simple modifications, significantly altering the pKa and sterics of the basic side chain in chloroquine analogs, may prove to be part of a strategy for overcoming the problem of worldwide resistance to affordable antimalarial drugs.
Keywords: Parallel synthesis, parallel purification, antimalarial, 4-aminoquinoline, drug-resistance
Introduction
Despite over one hundred years of drug development efforts, Malaria remains one of the most devastating infectious diseases in the world.1, 2 The current epidemic is fueled by the development of drug-resistant strains of P. falciparum, the parasite responsible for the most deadly cases of malaria.1 Research over the last two decades has shown that despite worldwide resistance to chloroquine (CQ)3 and emerging resistance to mefloquine,4 there is still significant potential to discover new quinoline antimalarials with activity against even the most drug-resistant strains of P. falciparum.5, 6
Prior work in our laboratory systematically examined modifications to both the quinoline ring and to the basic side chain of 4-aminoquinolines.7,8 While the structure-activity relationships showed reasonable tolerance for both types of modifications, the side chain modifications produced compounds with the greatest increase in potency towards drug-resistant parasite strains. This phenomena has been reported by other workers.5, 9 Our studies revealed that a previously unappreciated structural motif -- the presence of a single aromatic ring containing a hydrogen-bond acceptor attached to the basic nitrogen -- gave the most potent compounds. Interestingly, we noted a similar motif in the class of antimalarial compounds called α-aminocresols, first reported in the mid 1940's.10,11 (Figure 1) Burckhalter et al. discovered that 4-tert-butyl-2-dimethylaminomethyl-phenol (1) was effective against trophozoite-induced avian malaria and made a library of over 100 analogs using the Mannich reaction.10,12 Since only in vivo screening was available at the time, it is difficult to compare the reported activities with those of modern in vitro screening methods. While bioavailability and metabolic stability strongly bias measurements of activity in any in vivo assay, qualitative comparisons amongst similar compounds are reasonable for interpreting a structure-activity relationship (SAR). The early work on α-aminocresols showed the importance of having the hydroxy group ortho to the α-amino group (para- α-aminocresols showed no activity) and the benefit of an additional hydrophobic substituent on the ring. Optimization of these compounds led to the more potent bis-α-aminocresols such as 2.10 (Figure 1) Burckhalter later attached this motif to the 7-chloroquinoline ring found in chloroquine to create the drug amodiaquine (3) which was far more potent than any of the original α-aminocresols.12 Since the development of amodiaquine, little further work has been reported on the α-aminocresols, apart from modifications to amodiaquine.
Figure 1.

Structures of α-aminocresol antimalarial compounds and the drug amodiaquine.
The success of chloroquine diverted attention away from amodiaquine until the emergence of drug-resistance was reported by Moore and Lanier in 1961.13-15 The US Army then renewed its antimalarial screening program and re-discovered amodiaquine along with the quinolinemethanol class of drugs. The quinolinemethanols are derived from quinine and were optimized into the drug mefloquine (4).16 The common structural feature shared by both these compounds is a basic nitrogen within hydrogen-bonding proximity to a hydroxyl group. Since at physiological pH the basic nitrogen will be protonated, it is possible that the intramolecular hydrogen bonding between the protonated amine (H-bond donor) and the hydroxyl (H-bond acceptor) may be an important feature for activity against chloroquine-resistant P. falciparum. (Figure 2)
Figure 2.

Examples of intramolecular hydrogen bonding in antimalarials active against drug-resistant P. falciparum. Amodiaquine, mefloquine and halofantrine are approved drugs used to treat drug-resistant malaria and quinolines 6 and 7 were recently reported to have potent activity against drug-resistant P. falciparum.8
We hypothesized that exploiting this motif might provide a general approach to the discovery of novel and potent compounds and therefore synthesized a larger set of compounds containing an intramolecular hydrogen-bonding motif in the basic side-chain of the 4-aminoquinoline nucleus. These compounds were synthesized as a parallel library of purified discrete compounds which were then subjected to screening, followed by more detailed studies for active compounds.
Chemistry
We elected to fix the scaffold as a 7-chloroquinoline ring since it remains the most generally potent ring system choice and other modifications do little to increase potency against the drug-resistant strains. We designed our compounds to have four different alkyl groups linking the basic nitrogen center to the quinoline ring. It has been shown that the length of this linker region is important for potency against CQ-resistant P. falciparum, with side-chains less than four methylene units or greater than six methylene units having the greatest potency.5, 6 Out of the four alkyl linkers, we chose two that are linear chains of three and four methylene groups and two that are cyclic, containing either a pyrrolidinyl- or piperidinyl- group. For each of these four linkers, a key secondary amine intermediate was synthesized in bulk and then the terminal nitrogen was reductively alkylated by a set of aldehydes. (Scheme 1)
Scheme 1.

Synthesis of side-chain modified 4-aminoquinolines.
The synthesis of the secondary amine intermediates for the compounds with the propyl and butyl linkers was done similarly. First the 4,7-dichloroquinoline ring was reacted with either 1,3-diaminopropane or 1,4-diaminobutane by refluxing in the neat amine. The doubly arylated diamine side-products were easily removed by an acidic wash step. The primary amine products were then reacted with propionic anhydride to rapidly form the amide, which conveniently precipitated out of solution. The amide was reduced using borane-dimethylsulfide in THF to give the key secondary amine intermediate. The two intermediates with the cyclic side chains were synthesized in a single step through direct coupling of 4,7-dichloroquinoline with the necessary diamine. Both of these cyclic diamines have two reactive amine groups, but in each case the main product in the direct reaction of the diamine with the 4,7-dichloroquinoline was the desired product , which could be purified from the side product by flash chromatography.
The diversity enhancing step of our library synthesis was the reaction of each of the four secondary amine intermediates with a set of aldehydes containing an aromatic ring with a hydrogen bond accepting functional group (Figure 3). The reductive amination was done in situ using sodium cyanoborohydride and excess aldehyde to drive the reaction to completion and reduce the amount of unreacted secondary amine byproduct. Each of the final products was then easily worked up using a two-step scavenge and solid-phase extraction (SPE) procedure. First, an equivalent of resin-bound thionyl chloride was added to each well to scavenge unreacted amine starting material. This step was important because separation of the desired products and starting material proved to be difficult. Each compound was then purified using a capture and release SPE strategy using a strong-cation exchange (SCX) resin. The crude reaction mixtures were first acidified with HCl in methanol to completely protonate the amine products and break up any boron complexes formed during the reduction step. This mixture was then added to a short column of SCX-SPE resin and washed with methanol to remove any non-protonated materials. Elution of the final products was done using a solution of 5% triethylamine in methanol. In greater than 90% of the reactions, this procedure alone led to products with > 80% purity. The products with less than 80% purity were individually purified using preparative HPLC. The reactions in which the aldehyde starting materials contained a basic functional group could not be purified by the SCX-SPE method, and thus all of this set were purified by preparative HPLC.
Figure 3.

Aldehyde diversity elements used for side chain nitrogen substitutions.
Results and Discussion
Of the 124 compounds targeted in our library synthesis, we successfully synthesized 116 compounds (93%). The compounds were on average 89% pure following our SCX-SPE procedure. All compounds with purity less than 80% were purified by preparative HPLC, raising the average purity to 93% for the final set of compounds screened. The primary impurity found in the compounds was triethylamine hydrochloride resulting from the incomplete evaporation of the triethylamine used in the purification processes. Each compound was initially synthesized on a 0.2 mmol scale. For compounds not requiring HPLC purification, the average yield for the final reaction and SPE purification procedure was 78%. The products requiring purification by HPLC had much lower overall yields bringing the average yield for the final 112 compounds down to 55%. Since our initial screening library was synthesized on a relatively small scale, each compound was accurately quantitated using a chemiluminescent nitrogen detector (CLND). Equimolar stock solutions of each compound in DMSO were prepared using the data from the CLND analysis, ensuring accurate measurement of our final compound concentrations tested.
All of the compounds were initially assayed for growth inhibition in a cell-based assay against two strains of P. falciparum at two fixed concentrations (30 nM and 200 nM) to give a survey of all the activities (Figure 4). The 3D7 strain is a representative CQ-sensitive strain while the W2 strain is a multi-drug resistant parasite strain. The majority of compounds within this series are active at 30 nM. This indicates that the aminocresol motif , when attached to the distal basic center of quinolines, does lead to potent compounds against both the 3D7 and W2 strains. A comparison of the different linkers between the distal basic center and the aromatic nucleus reveals that the compounds with the propyl and butyl alkyl linkers are more generally potent than those with either of the two cyclic linkers. It was hoped that rigidity from the cyclic groups linking the basic center to the heterocyclic nucleus would introduce a conformational constraint leading to a more potent and/or selective compound. Conformational restraints are known to influence the thermodynamics of ligand/receptor interactions and have been statistically shown to have improved bioavailability over more flexible compounds.17 It is a possibility that the compounds with the conformationally restricted linkers are no longer able to access the optimal conformation for binding to a particular receptor. Such was the case in the development of the drug cromakalim. The class of β-blocking agents cromakalim was derived from were known to have antihypertensive activity side effects, but the conformationally constrained analog cromakalim possessed only the antihypertensive activity and was devoid of any β-blocking activity.18, 19 A broader evaluation of conformationally restricted linkers in aminoquinolines is necessary to further understand the uniform loss of potency in the series of compounds with cyclic linkers between the basic center and aromatic core. Interestingly, only one compound with a structurally rigid linker (e, Figure 2) showed growth inhibition against the drug-resistant W2 strain at the 30 nM dose. When the compounds were screened at 200 nM, several of the compounds with cyclic linkers began to show activity.
Figure 4.

Activity screening data for all compounds against the 3D7 (CQ-sensitive) and W2 (CQ-resistant) parasite strains.
Earlier work on modifications to the side chain of CQ-analogs had shown that a compound with the propyl diaminoalkane side chain was 10-times more potent than that with the butyl diaminoalkane side chain against a CQ-resistant parasite strain.6 Surprisingly, in our study more compounds with the butyl linker were active at 30 nM than those with the propyl linker. The butyl linker has four methylene groups separating the basic center from the aromatic core, similar to CQ, so we expected this set of compounds with greater structural similarity to CQ to be less active against the drug-resistant P. falciparum strain.
Full dose-response curves were done for compounds showing the greatest potency against the multi-drug resistant W2 strain. The data for compounds containing the aminocresol motif are shown in Table 1. Several compounds, with both the propyl and butyl linkers, produced low nanomolar IC50s against both parasite strains. A few of the compounds such as 11v and 14aa had significantly weaker activities against the W2 strain, but all of the other compounds showed activity against W2 within 4-fold of that for 3D7. Interestingly, it appears that electronic factors of the aromatic ring have minimal effect, with both electron-donating and the electron-withdrawing groups giving similar activities. However, both compounds with nitro substitutions (11ad & 11ae) and the difluoro-substituted compound (11ac) had IC50s at least an order of magnitude higher. This indicates that stronger electron withdrawing groups on the ring begin to decrease activity. Several of the compounds with heterocycle aromatic substitutions at the distal basic center, analogous in H-bonding capacity to the aminocresol motif, were selected and their IC50 values are shown in Table 2. The inhibitory activities of these compounds were slightly weaker than the benzyl substituted compounds, but still in the targeted potency range (< 50 nM). The methyl and chloro groups on the 5-position of the furan ring had little effect on the activity of the compounds. Within this series, the compounds with a 4 carbon diaminoalkyl side chains actually showed slightly higher potency than the 3 carbon linker equivalents.
Table 1.
Inhibitory activities for 3D7 and W2 of benzyl substituted compounds.
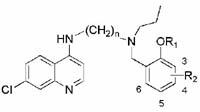 |
| 3D7 IC50 (nM) |
W2 IC50 (nM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| entry |
no. |
n |
R1 |
R2 |
average |
SD |
average |
SD |
| CQ | 17 | 0.48 | 394 | 67 | ||||
| 1 | 11p | 3 | H | H | 0.6 | 0.02 | 1.8 | 0.05 |
| 2 | 11q | 3 | H | 3-F | 0.9 | 0.03 | 2.6 | 0.82 |
| 3 | 11r | 3 | H | 3-OMe | 3.0 | 0.59 | 4.5 | 0.42 |
| 4 | 11s | 3 | H | 5-OMe | 1.0 | 0.02 | 2.0 | 0.03 |
| 5 | 11t | 3 | H | 5-OCF3 | 2.5 | 2.0 | 8.0 | 0.04 |
| 6 | 11v | 3 | H | 5-OH | 2.9 | 0.54 | 22.5 | 3.7 |
| 7 | 11x | 3 | Me | 5-F | 5.2 | 1.6 | 13.3 | 0.77 |
| 8 | 11y | 3 | Me | 6-F | 8.3 | 0.64 | 16 | 1.8 |
| 9 | 11aa | 3 | -OCH2O- | 1.9 | 0.02 | 3.3 | 0.61 | |
| 10 | 14p | 4 | H | H | 7.0 | 0.57 | 20 | 0.86 |
| 11 | 14q | 4 | H | 3-F | 1.4 | 0.39 | 4.6 | 0.19 |
| 12 | 14r | 4 | H | 3-OMe | 1.0 | 0.80 | 4.3 | 0.27 |
| 13 | 14s | 4 | H | 5-OMe | 6.8 | 0.44 | 27 | 1.7 |
| 14 | 14t | 4 | H | 5-OCF3 | 2.4 | 0.04 | 5.9 | 1.7 |
| 15 | 14v | 4 | H | 5-OH | 2.2 | 1.0 | 8.9 | 0.27 |
| 16 | 14x | 4 | Me | 5-F | 1.9 | 0.21 | 4.9 | 0.15 |
| 17 | 14y | 4 | Me | 6-F | 10 | 0.53 | 11 | 1.3 |
| 18 | 14aa | 4 | -OCH2O- | 4.2 | 0.16 | 23 | 5.3 | |
| 19 | 11ac | 3 | H | 3-F, 5-F | 21 | 9.8 | 34 | 4.0 |
| 20 | 11ad | 3 | H | 3-NO2 | 320 | 29 | 427 | 45 |
| 21 | 11ae | 3 | H | 5-NO2 | 38 | 3.4 | 125 | 0.41 |
| 22 | 11af | 3 | H | 5-tBu | 41 | 5.0 | 64 | 0.76 |
Table 2.
Inhibitory activities for 3D7 and W2 of heterocycle substituted compounds.
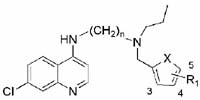 |
| 3D7 IC50 (nM) |
W2 IC50 (nM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| entry | no. | n | X | R2 | average | SD | average | SD |
| 1 | 11a | 3 | O | H | 12 | 2 | 33 | 4 |
| 2 | 11b | 3 | O | 5-Me | 14 | 0.03 | 39 | 4 |
| 3 | 11c | 3 | O | 5-Cl | 7 | 0.3 | 11 | 1 |
| 4 | 11f | 3 | S | H | 32 | 2 | 140 | 6 |
| 6 | 14a | 4 | O | H | 3 | 0.3 | 4 | 0.1 |
| 7 | 14b | 4 | O | 5-Me | 7 | 0.3 | 30 | 0.05 |
| 8 | 14c | 4 | O | 5-Cl | 4 | 1 | 6 | 0.5 |
| 9 | 14f | 4 | S | H | 7 | 0.5 | 22 | 5 |
The parent drug compound CQ is thought to work by interfering with the parasites ability to polymerize toxic free heme into a non-toxic heme polymer called hemozoin.20-22 The mechanism of resistance to CQ is now known to be primarily due to the PfCRT protein which alters the accumulation of drug within the parasite food vacuole, where heme polymerization takes place.23,24 The PfCRT protein shows homology to known drug/metabolite transporters and can confer CQ-resistance with a K76T mutation.25, 26 It has been hypothesized the wild-type K76 residue in PfCRT is positively charged at physiological pH and the electrostatic repulsion with CQ may disfavor the transport of CQ out of the food vacuole, causing the toxic effects that lead to parasite death.27, 28 The K76T mutation eliminates this positive charge and CQ is then transported out of the food vacuole, where it can no longer exert its activity. This mechanistic understanding further demonstrates the importance of the positively charged basic side chain of CQ-analogs in determining the level of activity against CQ-resistant parasite strains. In vitro selection of CQ-resistant P. falciparum mutants resistant against halofantrine 5, which contains an intramolecular hydrogen bond to its basic center, selects a PfCRT mutant S163R that has renewed sensitivity to CQ despite having the K76T mutation.27, 28 The S163R mutation reinstates a positive charge into the PfCRT protein providing further evidence that an electrostatic binding interaction between the drug and the transporter is a key determinant in the rate of drug efflux. This new insight into the mechanism of resistance to CQ and other heme-binding antimalarials shows that cross-resistance among the current battery of heme-binding antimalarials is complex and resistance to one drug can renew sensitivity to another. Also, there must be a structure-activity relationship for binding to PfCRT that is dependent on steric and electrostatic properties of the basic side chains of the heme-binding antimalarials. The addition of substitutions to the basic nitrogen could renew activity against the drug-resistant strains by altering the binding affinity to the PfCRT binding site. The addition of an aromatic group on the basic nitrogen introduces substantial steric bulk to the basic side chain, significantly changing the surface area of the ligand.
The addition of a hydrogen-bond acceptor in proximity to the protonated basic nitrogen center raises the pKa of the nitrogen, significantly changing the electrostatic character of the basic side chain at physiological pH. This hypothesis is consistent with our data showing that electron-donating substitutions on the ring tend to increase activity, since electron donating groups should further increase the pKa of the basic center. This would also be consistent with the sharp decrease in activity for the compounds with the strong electron withdrawing substitutions. Given that our primary screen is a cell-based assay, it is difficult to correlate small shifts in pKa with observed activity, but the general trends fit the hypothesis that changing the pKa of the basic center is important for activity.
Conclusions
It remains unknown what the possible mode of action was for the original α-aminocresols since more attention was focused on the quinoline hybrids such as amodiaquine. It is possible that the α-aminocresols have a different mode of action from chloroquine and hybrid 4-aminoquinoline-α-aminocresol hybrid compounds such as amodiaquine exert their activity through interaction with multiple targets. This possibility would partially explain their continued activity against strains of P. falciparum that have grown resistant to chloroquine. It is also possible that the steric and electrostatic changes to the side chain of the CQ-analogs simply reduce binding to the PfCRT efflux protein that infers CQ-resistance. Further experiments to address this hypothesis are warranted by the strong activity of this class of quinolines.
Experimental Methods
General
All reagents and starting materials were purchased from commercial sources and used without further purification. Dichloromethane, tetrahydrofuran, and methanol were dried using the solvent purification system manufactured by Glass Contour Inc. (Laguna Beach, CA). 1H and 13C NMR were recorded on the Varian Utility 400 MHz spectrometer, in CDCl3, CD3OD, or d-DMSO solvent. Chemical shifts were reported as parts per million (ppm) downfield from an internal tetramethylsilane (TMS) standard (δ = 0.0 for 1H NMR) or from solvent reference. Coupling constants (J values) were measured in hertz (Hz). Electrospray mass spectra (ES-MS) were collected on a Waters ZQ 4000 mass spectrometer. Nitrogen composition was measured using the Antek 6000 online chemiluminescent nitrogen detector (CLND). Crude products were purified by flash chromatography using 230-400 mesh silica gel (SiliCycle). All parallel synthesis steps were carried out in polypropylene fritted Bohdan 48-well MiniBlocks™ Purity was assessed using two orthogonal HPLC methods. HPLC method A: using an Xterra reversed phase C18 column (4.6 × 20 mm, 3 μm) running a binary gradient with water (w/ 0.05% TFA) and methanol (w/ 0.05% TFA). Purity was measured at 220 nM on a 10 minute gradient running from 0 - 100% methanol/TFA at 1 mL/min on a Waters Alliance HPLC. HPLC method B: using an Xterra reversed phase phenyl column (4.6 × 20 mm, 3 μm) running a binary gradient with water (w/ 0.05% TFA) and methanol (w/ 0.05% TFA). Purity was measured at 220 nM on a 10 minute gradient running from 0 - 100% methanol/TFA at 0.5 mL/min on a Waters Alliance HPLC.
N-(7-Chloro-quinolin-4-yl)-N′-propyl-propane-1,3-diamine (10)
To a stirring solution of 9 (prepared as described by De et al.9) (10 g, 42 mmol) in chloroform (200 mL) and pyridine (3.83 mL, 47 mmol), propionic anhydride (6.0 mL, 47 mmol) was added dropwise by a syringe at 0 °C under an inert atmosphere. The reaction was allowed to warm until room temperature and then stirring was continued for 1 hour. Excess anhydride was then quenched by addition of several drops of water. The crude reaction mixture was then dried down under vacuum, redissolved in ethyl acetate (300 mL) and washed with brine (5 × 200 mL). Solvent was removed under vacuum and the resulting residue placed on a high-vacuum for 18 hours to remove the trace amounts of pyridine remaining. The remaining 11.14 g (90%) of fluffy white solid amide was then immediately dissolved in dry THF for the reduction. The flask was cooled to 0 °C and Borane-methyl sulfide (15.4 mL, 4 eq.) was added slowly while stirring under an inert atmosphere. The reaction was then heated to reflux for 1 hour, before cooling to room temperature and careful quenching with water. After the addition of water no longer evolved bubbles, 25 mL of 37% HCl was carefully added to the reaction and the mixture heated to reflux for 1 hour to break up any boron complexes formed with the product. After cooling this mixture, the the pH was adjusted to basic with solid K3PO4 (pH > 10), and extracted into chloroform (200 mL). The organic solvent was removed under vacuum and the crude product was purified using silica gel with CH2Cl2/MeOH/Et3N (8.9 : 1 : 0.1) as an eluent to give 10 (7.5 g, 63%) as a pure white solid, 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C15H20ClN3 [M + H]+ 278.1. Found: 278.4. 1H NMR (400 MHz, CDCl3): δ 8.48 (d, J = 5.6, 1H), 7.91 (d, J = 2.0, 1H), 7.88 (br-s, 1H), 7.71 (d, J = 8.9, 1H), 7.30 (dd, J = 8.9, 2.0, 1H), 6.29 (d, J = 5.6, 1H), 3.48 (s, 1H), 3.78 (q, J = 5.6, 2H), 2.91 (t, J = 5.2, 2H), 2.64 (t, J = 6.8, 2H), 1.90 (m, 2H), 1.60 (m, 2H), 0.98 (t, J = 7.2, 3H). 13C NMR (400 MHz, CDCl3): 151.9, 150.5, 148.8, 134.5, 128.2, 124.8, 122.3, 117.5, 98.2, 51.9, 49.3, 43.8, 27.1, 23.2, 11.8.
N-(7-Chloro-quinolin-4-yl)-N'-propyl-butane-1,4-diamine (13)
To a stirring solution of 12 (prepared as described by De et al.9) (10 g, 40 mmol) in chloroform (200 mL) and pyridine (3.83 mL, 47 mmol), propionic anhydride (6.0 mL, 47 mmol) was added dropwise by a syringe at 0 °C under an inert atmosphere. The reaction was allowed to warm until room temperature and then stirring was continued for 1 hour. Excess anhydride was then quenched by addition of several drops of water. The crude reaction mixture was then dried down under vacuum, redissolved in ethyl acetate (300 mL) and washed with brine (5 × 200 mL). Solvent was removed under vacuum and the resulting residue placed on a high-vacuum for 18 hours to remove the trace amounts of pyridine remaining. 11.26 g (92%) of the amide was then immediately dissolved in dry THF for the reduction. The flask was cooled to 0 °C and Borane-methyl sulfide (15.4 mL, 4 eq.) was added slowly while stirring under an inert atmosphere. The reaction was then heated to reflux for 1 hour, before cooling to room temperature and careful quenching with water. After the addition of water no longer evolved bubbles, 25 mL of 37% HCl was carefully added to the reaction and the resulting mixture heated to reflux for 1 hour to break up any boron complexes formed with the product. After cooling this mixture, the pH was adjusted to basic with solid K3PO4 (pH > 10), and extracted into chloroform (200 mL). The organic solvent was removed under vacuum and the crude product was purified using silica gel with CH2Cl2/MeOH/Et3N (8.9 : 1 : 0.1) as an eluent to give 13 (76.2 g, 58 %) as an off-white solid, 100% pure by HPLC Method A; 93% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C16H22ClN3 [M + H]+ 292.2. Found: 292.4. 1H NMR (400 MHz, CDCl3): δ 8.51 (d, J = 5.2, 1H), 7.94 (d, J = 2.0, 1H), 7.71 (d, J = 8.9, 1H), 7.32 (dd, J = 8.9, 2.0, 1H), 6.36 (d, J = 5.2, 1H), 6.10 (s, 1H), 3.48 (s, 1H), 3.29 (q, J = 4.4, 2H), 2.71 (t, J = 6.4, 2H), 2.60 (t, J = 7.2, 2H), 1.86 (quin, J = 6.4, 2H), 1.69 (m, 2H), 1.55 (m, 4H), 0.94 (t, J = 7.2, 3H). 13C NMR (400 MHz, CDCl3): δ 152.0, 150.3, 149.2, 134.8, 128.5, 125.0, 121.9, 117.5, 98.9, 52.0, 49.2, 43.3, 27.9, 26.4, 23.2, 11.9.
(7-Chloro-quinolin-4-yl)-pyrrolidin-3-yl-amine (15)
A solution of 4,7-dichloroquinoline (5 g, 25 mmol) and 3-aminopyrrolidine dihydrochloride (7.95 g, 50 mmol) in diisopropylethylamine (100 mL) was heated at reflux for 4 h. After concentration under vacuum, the crude reaction mixture was purified on silica gel with CH2Cl2/MeOH/Et3N (8.9 : 1 : 0.1) as an eluent to give 15 (4.3 g, 70%). 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C13H14ClN3 [M + H]+ 248.1. Found: 248.3. 1H NMR (400 MHz, CDCl3): δ 8.69 (d, J = 4.8, 1H), 8.02 (d, J = 2.0, 1H), 7.90 (d, J = 8.8, 1H), 7.42 (dd, J = 8.8, 2.0; 1H), 6.82 (d, J = 4.8, 1H), , 2H), 2.60 (t, J = 7.2, 2H), 1.86 (quin, J = 6.4, 2H), 1.69 (m, 2H), 1.55 (m, 4H), 0.94 (t, J = 7.2, 3H). 13C NMR (400 MHz, CDCl3): δ 152.2, 150.4, 149.7, 134.2, 128.0, 126.3, 123.7, 119.1, 102.8, 60.3, 51.1, 50.2, 34.5.
(7-Chloro-quinolin-4-yl)-piperidin-4-yl-amine (17)
A solution of 4,7-dichloroquinoline (10 g, 50 mmol) and 4-aminopiperidine (8.4 mL, 250 mmol) in diisopropylethylamine (200 mL) was heated at 100 °C for 20 hours. After partial concentration under vacuum, the crude reaction mixture was purified on silica gel with CH2Cl2/MeOH/Et3N (8.9 : 1 : 0.1) as an eluent to give 17 (8.6 g, 65%). 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C14H16ClN3 [M + H]+ 262.1. Found: 262.3. 1H NMR (400 MHz, CDCl3): δ 8.62 (d, J = 5.2, 1H), 7.96 (d, J = 2.0, 1H), 7.85 (d, J = 8.9, 1H), 7.35 (dd, J = 8.9, 2.0, 1H), 6.75 (d, J = 5.2, 1H), 5.241 (s, 1H), 3.48 (d, J = 12.0, 2H), 2.89 (m, 1H), 2.82 (t, J = 11.6, 2H), 1.97 (d, J = 12.0, 2H), 1.61 (m, 4H). 13C NMR (400 MHz, CDCl3): δ 157.2, 151.6, 150.0, 134.5, 128.4, 125.6, 125.3, 121.9, 108.8, 51.1, 48.4, 35.5.
General Procedure for Reductive Alkylation Diversity Step
A stock solution (36 mM) of the appropriate secondary amine intermediate (10, 13, 16 or 18) was made in dry MeOH and aliquoted to the wells of a 48-well reaction block (2 mL/well; 0.07 mmol/well). To each well was then added an aldehyde (Figure 3) (0.22 mmol, 4 eq.) with care taken to reduce exposure to moisture in the air. The reaction block was sealed with a rubber gasket and 220 μL (0.22 mmol) of a dry solution of NaBH3CN in THF (1 M) was added by syringe to each well through the gasket and the block was shaken at room temperature for 18 hours. Each reaction was then quenched with 0.4 mL of a saturated solution of HCl in MeOH. CAUTION: EXPLOSION HAZARD. After bubbling had ceased, 100 mg of polymer-bound sulfonyl chloride (Aldrich, 100-200 mesh, 1.5 mmol/g) was added to each well and the resulting mixture was shaken for 4 hours. The solution remaining in each well was transferred into another 48-position reaction block containing cartridges loaded with SCX-SPE medium. Each well was washed with 1% TFA in MeOH (2 × 1 mL), MeOH (2 × 2 mL) and eluted with 5% TEA in MeOH (2 × 2 mL). The eluted products were collected into glass test tubes and solvent was removed on a GeneVac HT-4 (10 mbar, 4.0 h, 35 °C). The partially purified products were then dissolved in saturated HCl/MeOH (1 mL) and dried again to form the HCl salts of each compound. The dry HCl salts were then each dissolved in 1mL d6-DMSO and subjected to LC/MS/CLND for analysis of purity, identity and yield. A 10 mM stock solution of each compound was made in d6-DMSO based on the CLND quantitation data and selected compounds were analyzed by 1H-NMR.
General Procedure for purification of library members
Compounds with purity < 80% by analytical HPLC were dissolved in a total of 1 mL DMSO and purified with a semi-preparative reversed-phase Xterra column (19 × 50 mm, particle size 5 μm) running a 10 - 100% gradient of 0.5%TEA-H2O/methanol with a 20 mL/min flow rate on a Parallex Flex HPLC system. Fraction collection was automatically triggered by UV absorbance above 0.1 AU at 254nm. Sample and fraction data were then transferred to the Waters OpenLynx operating software, which coordinated the injection, mass spectrometric analysis, and data processing for each fraction. A Gilson 215 liquid handler and a Gilson 208 injection module were used to inject samples into a Waters ZQ 4000 mass spectrometer rigged for flow injection with an electrospray probe and single quadrapole detector operating in positive ion mode. All fraction plates were then dried down using a GeneVac Mega 980 solvent evaporator. Fractions that exhibited a peak in the mass spectra of the correct molecular weight were then dissolved in 1 mL of freshly prepared saturated HCl in methanol, and like fractions were combined. Combined fractions were then dissolved in 0.5 mL of d6-DMSO and were analyzed by LC/MS/CLND for purity, identity and yield. 10 mM stock solutions in d6-DMSO were prepared based on the CLND quantitation data.
N′-(7-Chloro-quinolin-4-yl)-N-furan-2-ylmethyl-N-propyl-propane-1,3-diamine (11a) 89% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C20H24ClN3O [M + H]+ 358.2. Found: 358.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.93 (br, 0.5 H), 9.75 (t, J = 5.2, 1H), 8.70 (d, J = 9.2, 1H), 8.56 (d, J = 7.2, 1H), 8.05 (d, J = 1.6, 1H), 7.74 (dd, J = 9.2, 1.6, 1H), 7.70 (d, J = 3.2, 1H), 6.88 (d, J = 7.2, 1H), 6.72 (d, J = 3.2, 1H), 6.44 (d, J = 3.2, 1H), 5.27 (d, J = 2.2, 1H), 4.36 (br-s, 2H), 3.59 (m, 2H), 3.32 (H20 peak), 3.04 (m, 2H), 2.87 (m, 2H), 2.13 (dd, J = 6.4, 6.4, 2H), 1.70 (m, 2H), 0.83 (t, J = 7.6, 3H).
N′-(7-Chloro-quinolin-4-yl)-N-(5-methyl-furan-2-ylmethyl)-N-propyl-propane-1,3-diamine (11b) 59% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C21H26ClN3O [M + H]+ 372.2. Found: 372.5. 1H NMR (d6-DMSO, 400 MHz): δ 9.96 (br, 1H), 9.88 (t, J = 5.2, 1H), 8.83 (d, J = 9.2, 1H), 8.60 (d, J = 7.2, 1H), 8.12 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 6.93 (m, 1H), 6.61 (d, J = 3.2, 1H), 6.08 (d, J = 3.2, 1.2, 1H), 5.32 (d, J = 1.6, 1H), 4.43 (d, J = 4.8, 2H), 3.61 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.74 (m, 2H), 2.09 (s, 3H), 1.65 (m, 4H), 0.87 (t, J = 7.2, 3H).
N-(5-Chloro-furan-2-ylmethyl)-N′-(7-chloro-quinolin-4-yl)-N-propyl-propane-1,3-diamine (11c) 76% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C20H23Cl2N3O 391.1218. Found: 391.1214. 1H NMR (d6-DMSO, 400 MHz): δ 9.96 (br, 1H), 9.88 (t, J = 5.2, 1H), 8.83 (d, J = 9.2, 1H), 8.60 (d, J = 7.2, 1H), 8.12 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 6.93 (m, 1H), 6.61 (d, J = 3.2, 1H), 6.08 (d, J = 3.2, 1.2, 1H), 5.32 (d, J = 1.6, 1H), 4.43 (d, J = 4.8, 2H), 3.61 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.74 (m, 2H), 2.09 (s, 3H), 1.65 (m, 4H), 0.87 (t, J = 7.2, 3H).
N′-(7-Chloro-quinolin-4-yl)-N-propyl-N-thiophen-2-ylmethyl-propane-1,3-diamine (11f) 90% yield; 82% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C20H24ClN3S [M + H]+ 374.1. Found: 374.4. 1H NMR (d6-DMSO, 400 MHz): δ 11.16 (br, 0.5 H), 9.83 (t, J = 5.2, 1H), 8.75 (d, J = 9.2, 1H), 8.59 (d, J = 7.2, 1H), 8.13 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 7.61 (d, J = 5.2, 1H), 7.41 (d, J = 3.2, 1H), 7.03 (dd, J = 3.2, 3.2; 1H), 6.93 (d, J = 7.2, 1H), 4.56 (d, J = 2.2, 1H), 3.63 (m, 2H), 3.32 (H20 peak), 3.14 (m, 2H), 2.93 (m, 2H), 2.18 (m, 2H), 1.76 (m, 2H), 0.87 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-phenol (11p) 78% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C22H26ClN3O [M + H]+ 384.2. Found: 384.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.34 (br, 0.5 H), 10.06 (br, 1H), 9.77 (t, J = 5.2, 1H), 8.73 (d, J = 9.2, 1H), 8.59 (d, J = 7.2, 1H), 8.11 (d, J = 1.6, 1H), 7.77 (dd, J = 9.2, 1.6, 1H), 7.46 (d, J = 7.2, 1H), 7.17 (d, J = 7.2, 1H), 6.91 (m, 2H), 6.75 (d, J = 7.2, 1H), 5.32 (d, J = 2.4, 1H), 4.23 (br-s, 2H), 3.61 (m, 2H), 3.33 (H20 peak), 3.15 (m, 2H), 2.96 (m, 2H), 2.18 (m, 2H), 1.77 (m, 2H), 0.86 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-6-fluoro-phenol (11q) 30% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C22H25ClFN3O [M + H]+ 402.2. Found: 402.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.38 (br-s, 1H), 9.75 (t, J = 5.2, 1H), 8.70 (d, J = 9.2, 1H), 8.58 (d, J = 7.2, 1H), 8.10 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 7.37 (d, J = 7.6, 1H), 7.17 (t, J = 9.2, 1H), 6.91 (d, J = 7.2, 1H), 6.87 (d, J = 7.2, 1H), 6.80 (m, 1H), 4.29 (s, 2H), 3.61 (m, 2H), 3.35 (H20 peak), 3.15 (m, 2H), 2.97 (m, 2H), 2.16 (m, 2H) 1.76 (m, 2H), 0.87 (t, J = 7.2, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-6-methoxy-phenol (11r) 77% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C23H28ClN3O2 413.1870. Found: 413.1871. 1H NMR (d6-DMSO, 400 MHz): δ 10.13 (br, 1H), 9.75 (t, J = 5.2, 1H), 9.39 (br, 1H), 8.70 (d, J = 9.2, 1H), 8.57 (d, J = 7.2, 1H), 8.11 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 7.06 (d, J = 8.4, 1H), 6.95 (d, J = 8.4, 1H), 6.90 (d, J = 7.2, 1H), 6.75 (t, J = 8.4, 1H), 4.24 (br-s, 2H), 3.61 (m, 2H), 3.33 (H20 peak), 3.14 (m, 2H), 2.95 (m, 2H), 2.17 (m, 2H), 1.76 (m, 2H), 0.86 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-4-methoxy-phenol (11s) 67% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C23H28ClN3O2 413.1870. Found: 413.1882. 1H NMR (d6-DMSO, 400 MHz): δ 10.20 (br-s, 1H), 9.76 (br-s, 1H), 9.68 (t, J = 5.2, 1H), 8.69 (d, J = 9.2, 1H), 8.59 (d, J = 6.8, 1H), 8.08 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 7.15 (d, J = 2.9, 1H), 6.90 (d, J = 7.2, 1H), 6.75 (m, 2H), 4.20 (br-s, 2H), 3.65 (s, 3H), 3.60 (m, 2H), 3.33 (H20 peak), 3.15 (m, 2H), 2.97 (m, 2H), 2.16 (m, 2H), 1.77 (m, 2H), 0.87 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-4-trifluoromethoxy-phenol (11t) 70% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C23H25ClF3N3O2 467.1587. Found: 467.1599. 1H NMR (d6-DMSO, 400 MHz): δ 10.77 (br-s, 1H), 10.03 (br-s, 1H), 9.57 (t, J = 5.2, 1H), 8.61 (d, J = 9.2, 1H), 8.03 (d, J = 1.6, 1H), 7.80 (dd, J = 9.2, 1.6, 1H), 7.55 (d, J = 2.9, 1H), 7.20 (m, 1H), 6.97 (d, J = 8.9, 1H), 6.92 (d, J = 7.2, 1H), 4.26 (br-s, 1H), 3.62 (m, 2H), 3.33 (H20 peak), 3.17 (m, 2H), 2.99 (m, 2H), 2.15 (m, 2H), 1.75 (m, 2H), 0.87 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-benzene-1,4-diol (11v) 84% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C22H26ClN3O2 [M + H]+ 400.2. Found: 400.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.12 (br-s, 1H), 9.95 (br-s, 1H), 9.79 (t, J = 5.2, 1H), 9.56 (br-s, 1H), 8.98 (m, 1H), 8.74 (d, J = 9.2, 1H), 8.59 (d, J = 7.2, 1H), 8.11 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 6.91 (d, J = 7.2, 1H), 6.86 (d, J = 2.9, 1H), 6.71 (d, J = 8.6, 1H), 6.64 (dd, J = 8.6, 2.9; 1H), 4.15 (br-s, 2H), 3.60 (m, 2H), 3.33 (H20 peak), 3.15 (m, 2H), 2.97 (m, 2H), 2.16 (m, 2H), 1.74 (m, 2H), 0.86 (t, J = 7.6, 3H).
N′-(7-Chloro-quinolin-4-yl)-N-(5-fluoro-2-methoxy-benzyl)-N-propyl-propane-1,3-diamine (11x) 94% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C23H27ClFN3O 415.1826. Found: 415.1839. 1H NMR (d6-DMSO, 400 MHz): δ 10.40 (br-s, 1H), 9.81 (t, J = 5.2, 1H), 8.76 (d, J = 9.2, 1H), 8.59 (d, J = 6.8, 1H), 8.11 (d, J = 1.6, 1H), 7.79 (dd, J = 9.2, 1.6, 1H), 7.56 (dd, J = 9.1, 3.1; 1H), 7.21 (dt, J = 3.1, 8.2, 1H), 7.05 (m; 1H), 6.95 (d, J = 7.2, 1H), 4.26 (brs, 2H), 3.79 (s, 2H), 3.61 (m, 2H), 3.33 (H20 peak), 3.17 (m, 2H), 2.95 (m, 2H), 2.17 (m, 2H), 1.77 (m, 2H), 0.87 (t, J = 7.6, 3H).
N′-(7-Chloro-quinolin-4-yl)-N-(2-fluoro-6-methoxy-benzyl)-N-propyl-propane-1,3-diamine (11y) 32% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C23H27ClFN3O 415.1826. Found: 415.1829. 1H NMR (d6-DMSO, 400 MHz): δ 9.89 (m, 2H), 8.79 (d, J = 9.2, 1H), 8.60 (d, J = 7.2, 1H), 8.12 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 7.47 (dd, J = 7.5, 8.2; 1H), 6.95 (m, 2H), 6.84 (m, 1H), 4.26 (br-s, 2H), 3.86 (s, 3H), 3.65 (m, 2H), 3.33 (H20 peak), 3.20 (m, 2H), 2.99 (m, 2H), 2.20 (m, 2H), 1.79 (m, 2H), 0.89 (t, J = 7.6, 3H).
N-Benzo[1,3]dioxol-4-ylmethyl-N'-(7-chloro-quinolin-4-yl)-N-propyl-propane-1,3-diamine (11aa) 31% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C23H26ClN3O2 411.1713. Found: 411.1718. 1H NMR (d6-DMSO, 400 MHz): δ 10.95 (br-s, 1H), 9.80 (t, J = 5.2, 1H), 8.73 (d, J = 9.2, 1H), 8.59 (d, J = 6.8, 1H), 8.10 (d, J = 1.6, 1H), 7.78 (dd, J = 9.2, 1.6, 1H), 7.13 (d, J = 7.6; 1H), 6.91 (m, 2H), 6.83 (t, J = 7.6, 1H), 6.05 (s, 2H), 4.23 (br-s, 2H), 3.62 (m, 2H), 3.33 (H20 peak), 3.15 (m, 2H), 2.97 (m, 2H), 2.18 (m, 2H), 1.78 (m, 2H), 0.88 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-4,6-difluoro-phenol (11ac) 65% yield; 92% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C22H24ClF2N3O 419.1564. Found: 419.1562. 1H NMR (d6-DMSO, 400 MHz): δ 8.38 (d, J = 5.3, 1H), 8.21 (d, J = 8.9, 1H), 7.78 (d, J = 1.6, 1H), 7.42 (dd, J = 8.9, 1.6, 1H), 7.28 (m, 1H), 7.01 (m, 1H), 6.83 (m, 1H), 6.44 (d, J = 5.3, 1H), 3.77 (m, 2H), 3.35 (m, 2H), 3.15 (m, 2H), 2.60 (q, J = 6.0, 2H), 1.47 (m, 2H), 0.82 (t, J = 7.6, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-6-nitro-phenol (11ad) 57% yield; 90% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C22H25ClN4O3 [M + H]+ 429.2. Found: 429.5. 1H NMR (d6-DMSO, 400 MHz): δ 8.39 (d, J = 5.3, 1H), 8.21 (d, J = 8.9, 1H), 7.78 (d, J = 1.6, 1H), 7.70 (d, J = 8.4, 1H), 7.44 (dd, J = 8.9, 1.6, 1H), 7.33 (t, J = 5.3, 1H), 7.29 (d, J = 7.2, 1H), 6.61 (m, 1H), 6.48 (d, J = 5.3, 1H), 4.01 (s, 2H), 3.29 (m, 2H), 2.79 (m, 2H), 2.63 (m, 2H), 1.96 (m, 2H), 1.55 (m, 2H), 0.84 (t, J = 7.2, 3H).
2-({[3-(7-Chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-4-nitro-phenol (11ae) 54% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C22H25ClN4O3 [M + H]+ 429.2. Found: 429.5. 1H NMR (d6-DMSO, 400 MHz):: δ 8.37 (d, J = 5.2, 1H), 8.19 (d, J = 9.2, 1H), 8.09 (d, J = 1.6, 1H), 7.95 (dd, J = 9.2, 1.6, 1H), 7.77 (d, J = 1.5, 1H), 7.41 (dd, J = 8.9, 1.5; 1H), 7.31 (m, 1H), 6.74 (t, J = 8.9, 1H), 6.45 (d, J = 5.2, 1H), 3.85 (s, 2H), 3.37 (m, 2H), 3.17 (m, 2H), 2.70 (t, J = 5.8; 2H), 1.91 (dd, J = 5.8, 5.8; 2H), 1.51 (dd, J = 7.2, 7.2; 2H), 0.84 (t, J = 7.2, 3H).
4-tert-Butyl-2-({[3-(7-chloro-quinolin-4-ylamino)-propyl]-propyl-amino}-methyl)-phenol (11af) 74% yield; 85% pure by HPLC Method A; 84% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C26H34ClN3O [M + H]+ 440.2. Found: 440.5. 1H NMR (d6-DMSO, 400 MHz): δ 8.37 (d, J = 5.2, 1H), 8.19 (d, J = 9.2, 1H), 7.77 (d, J = 1.6, 1H), 7.41 (dd, J = 9.2, 1.6; 1H), 7.31 (m, 1H), 7.09 (d, J = 1.6, 1H), 7.05 (dd, J = 8.9, 1.6; 1H), 6.60 (d, J = 8.9, 1H), 6.43 (d, J = 5.2, 1H), 3.70 (s, 2H), 3.26 (m, w/H20 overlap), 2.60 (t, J = 5.8; 2H), 1.88 (m; 2H), 1.49 (dd, J = 7.2, 7.2; 2H), 1.19 (s, 9H), 0.81 (t, J = 7.2, 3H).
N'-(7-Chloro-quinolin-4-yl)-N-furan-2-ylmethyl-N-propyl-butane-1,4-diamine (14a) 64% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C21H26ClN3O [M + H]+ 372.2. Found: 372.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.54 (br, 0.5 H), 9.72 (t, J = 5.2, 1H), 8.78 (d, J = 9.2, 1H), 8.55 (m, 1H), 8.08 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 6.89 (d, J = 3.2, 1H), 6.88 (d, J = 7.2, 1H), 6.63 (d, J = 3.2, 1H), 6.10 (d, J = 3.2, 1H), 4.31 (d, J = 4.8, 2H), 3.56 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.90(m, 2H), 1.86 (m, 2H), 1.73 (m, 4H), 0.87 (t, J = 7.2, 3H).
N'-(7-Chloro-quinolin-4-yl)-N-(5-methyl-furan-2-ylmethyl)-N-propyl-butane-1,4-diamine (14b) 59% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C22H28ClN3O [M + H]+ 386.2. Found: 386.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.54 (br, 1H), 9.74 (t, J = 5.2, 1H), 8.79 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.08 (d, J = 1.6, 1H), 7.77 (dd, J = 9.2, 1.6, 1H), 6.88 (d, J = 3.4, 1H), 6.64 (d, J = 3.2, 1H), 6.13 (d, J = 3.2, 1.2, 1H), 4.32 (d, J = 4.8, 2H), 3.57 (m, 2H), 3.16 (solvent), 3.04 (m, 2H), 2.90 (m, 2H), 2.24 (s, 3H), 1.86 (dt, J = 7.5, 7.5, 2H), 1.72 (m, 4H), 0.87 (t, J = 7.2, 3H).
N-(5-Chloro-furan-2-ylmethyl)-N'-(7-chloro-quinolin-4-yl)-N-propyl-butane-1,4-diamine (14c) 13% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C21H25Cl2N3O [M + H]+ 406.1. Found: 406.4. 1H NMR (d6-DMSO, 400 MHz): δ 10.97 (br, 0.5 H), 9.83 (t, J = 5.2, 1H), 8.85 (d, J = 9.2, 1H), 8.53 (m, 1H), 8.12 (d, J = 1.6, 1H), 7.74 (dd, J = 9.2, 1.6, 1H), 6.89 (d, J = 3.4, 1H), 6.87 (d, J = 7.2, 1H), 6.56 (d, J = 3.2, 1H), 4.38 (d, J = 4.8, 2H), 3.58 (m, 2H), 3.16 (solvent), 3.04 (m, 2H), 2.92 (m, 2H), 1.86 (dt, J = 7.5, 7.5, 2H), 1.73 (m, 4H), 0.87 (t, J = 7.2, 3H).
N'-(7-Chloro-quinolin-4-yl)-N-propyl-N-thiophen-2-ylmethyl-butane-1,4-diamine (14f) 44% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C21H26ClN3S [M + H]+ 388.2. Found: 388.4. 1H NMR (d6-DMSO, 400 MHz): δ 10.79 (br-s, 0.5 H), 9.74 (t, J = 5.2, 1H), 8.78 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.08 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 7.68 (d, J = 5.2, 1H), 7.42 (d, J = 3.2, 1H), 7.10 (dd, J = 3.2, 3.2; 1H), 6.89 (d, J = 7.2, 1H), 4.54 (d, J = 4.8, 2H), 3.54 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.90(m, 2H), 1.85 (m, 2H), 1.71 (m, 4H), 0.86 (t, J = 7.2, 3H).
2-({[4-(7-Chloro-quinolin-4-ylamino)-butyl]-propyl-amino}-methyl)-phenol (14p) 83% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C23H28ClN3O [M + H]+ 398.2. Found: 398.5. 1H NMR (d6-DMSO, 400 MHz): δ 9.72 (t, J = 5.2, 1H), 8.77 (d, J = 9.2, 1H), 8.55 (d, J = 6.8, 1H), 8.07 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 7.46 (d, J = 7.6, 1H), 7.24 (t, J = 7.6, 1H), 6.97 (d, J = 7.6, 1H), 6.88 (d, J = 7.2, 1H), 6.82 (t, J = 7.6, 1H), 4.22 (d, J = 4.8, 2H), 3.54 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.90(m, 2H), 1.85 (m, 2H), 1.71 (m, 4H), 0.86 (t, J = 7.2, 3H).
2-({[4-(7-Chloro-quinolin-4-ylamino)-butyl]-propyl-amino}-methyl)-6-fluoro-phenol (14q) 39% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C23H27ClFN3O [M + H]+ 416.2. Found: 416.5. 1H NMR (d6-DMSO, 400 MHz): δ 9.72 (t, J = 5.2, 1H), 8.77 (d, J = 9.2, 1H), 8.55 (d, J = 7.2, 1H), 8.07 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 7.46 (d, J = 7.6, 1H), 7.24 (t, J = 7.6, 1H), 6.96 (d, J = 7.6, 1H), 6.87 (d, J = 7.2, 1H), 6.82 (t, J = 7.6, 1H), 4.23 (d, J = 4.8, 2H), 3.56 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.94(m, 2H), 1.86 (dt, J = 6.4, 6.4, 2H), 1.70 (m, 4H), 0.86 (t, J = 7.2, 3H).
2-({[4-(7-Chloro-quinolin-4-ylamino)-butyl]-propyl-amino}-methyl)-6-methoxy-phenol (14r) 50% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C24H30ClN3O2 427.2026. Found: 427.2036. 1H NMR (d6-DMSO, 400 MHz): δ 9.92 (br-s, 1H), 9.76 (t, J = 5.2, 1H), 8.80 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.10 (d, J = 1.6, 1H), 7.75 (dd, J = 9.2, 1.6, 1H), 7.09 (dd, J = 7.8, 1.2, 1H), 7.02 (dd, J = 7.8, 1.2, 1H), 6.87 (d, J = 7.2, 1H), 6.81 (dd, J = 7.8, 7.8; 1H) 4.24 (d, J = 4.8, 2H), 3.81 (s, 3H), 3.55 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.93 (m, 2H), 1.85 (dt, J = 7.6, 7.6, 2H), 1.72 (m, 4H), 0.85 (t, J = 7.2, 3H).
2-({[4-(7-Chloro-quinolin-4-ylamino)-butyl]-propyl-amino}-methyl)-4-methoxy-phenol (14s) 55% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C24H30ClN3O2 [M + H]+ 428.2. Found: 428.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.01(br-s, 1H), 9.72 (t, J = 5.2, 1H), 8.78 (d, J = 9.2, 1H), 8.5 (m, 1H), 8.08 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 7.16 (d, J = 2.9, 1H), 6.89 (d, J = 4.8, 1H), 6.87 (d, J = 2.9, 1H), 6.81 (m, 1H) 4.21 (d, J = 4.8, 2H), 3.67 (s, 3H), 3.55 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.95 (m, 2H), 1.86 (dt, J = 7.6, 7.6, 2H), 1.72 (m, 4H), 0.85 (t, J = 7.2, 3H).
2-({[4-(7-Chloro-quinolin-4-ylamino)-butyl]-propyl-amino}-methyl)-4-trifluoromethoxy-phenol (14t) 33% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C24H27ClF3N3O2 [M + H]+ 482.2. Found: 482.4. 1H NMR (d6-DMSO, 400 MHz): δ 10.07(brs, 1H), 9.73 (t, J = 5.2, 1H), 8.78 (d, J = 9.2, 1H), 8.5 (m, 1H), 8.08 (d, J = 1.6, 1H), 7.77 (dd, J = 9.2, 1.6, 1H), 7.61 (d, J = 2.9, 1H), 7.26 (dd, J = 8.9, 2.9, 1H), 6.08 (d, J = 8.9, 1H), 6.87 (d, J = 7.2, 1H), 4.26 (d, J = 4.8, 2H), 3.55 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.95 (m, 2H), 1.85 (dt, J = 7.6, 7.6, 2H), 1.72 (m, 4H), 0.85 (t, J = 7.2, 3H).
2-({[4-(7-Chloro-quinolin-4-ylamino)-butyl]-propyl-amino}-methyl)-benzene-1,4-diol (14v) 52% yield; 87% pure by HPLC Method A; 83% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C23H28ClN3O2 [M + H]+ 414.2. Found: 414.5. 1H NMR (d6-DMSO, 400 MHz): δ 9.76 (t, J = 5.2, 1H), 9.67 (br-s, 1H), 8.79 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.09 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 6.88 (d, J = 7.2, 1H), 6.86 (d, J = 2.9, 1H), 6.78 (d, J = 8.6, 1H), 6.69 (dd, J = 8.6, 2.9; 1H), 4.15 (d, J = 4.8, 2H), 3.535 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.95 (m, 2H), 1.85 (dt, J = 7.5, 7.5, 2H), 1.72 (m, 4H), 0.86 (t, J = 7.2, 3H).
N′-(7-Chloro-quinolin-4-yl)-N-(5-fluoro-2-methoxy-benzyl)-N-propyl-butane-1,4-diamine (14x) 39% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; LCMS (ESI) m/z Calcd for C24H29ClFN3O [M + H]+ 430.2. Found: 430.5. 1H NMR (d6-DMSO, 400 MHz): δ 10.13 (br-s, 1H), 9.80 (t, J = 5.2, 1H), 8.82 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.09 (d, J = 1.6, 1H), 7.76 (dd, J = 9.2, 1.6, 1H), 7.56 (dd, J = 9.1, 3.1; 1H), 7.26 (dt, J = 3.1, 8.2, 1H), 7.10 (dd, J = 4.4, 9.1; 1H), 6.88 (d, J = 7.2, 1H), 4.25 (d, J = 4.8, 2H), 3.81 (s, 3H), 3.55 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.95 (m, 2H), 1.85 (dt, J = 7.6, 7.6, 2H), 1.71 (m, 4H), 0.86 (t, J = 7.2, 3H).
N′-(7-Chloro-quinolin-4-yl)-N-(2-fluoro-6-methoxy-benzyl)-N-propyl-butane-1,4-diamine (14y) 22% yield; 100% pure by HPLC Method A; 87% pure by HPLC Method B; HRMS (EI) m/z Calcd for C24H29ClFN3O 429.1983. Found: 429.1972. 1H NMR (d6-DMSO, 400 MHz): δ 9.90 (t, J = 5.2, 1H), 9.75 (br-s, 1H), 8.85 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.12 (d, J = 1.6, 1H), 7.75 (dd, J = 9.2, 1.6, 1H), 7.49 (dd, J = 7.5, 8.2; 1H), 6.97 (d, J = 8.6, 1H), 6.89 (m, 2H), 4.26 (d, J = 4.8, 2H), 3.88 (s, 3H), 3.55 (m, 2H), 3.16 (solvent), 3.05 (m, 2H), 2.95 (m, 2H), 1.88 (m, 2H), 1.74 (m, 4H), 0.86 (t, J = 7.2, 3H).
N-Benzo[1,3]dioxol-4-ylmethyl-N′-(7-chloro-quinolin-4-yl)-N-propyl-butane-1,4-diamine (14aa) 47% yield; 100% pure by HPLC Method A; 100% pure by HPLC Method B; HRMS (EI) m/z Calcd for C24H28ClN3O2 425.1870. Found: 425.1857. 1H NMR (d6-DMSO, 400 MHz): δ 10.68 (br-s, 1H), 9.79 (t, J = 5.2, 1H), 8.80 (d, J = 9.2, 1H), 8.54 (m, 1H), 8.10 (d, J = 1.6, 1H), 7.75 (dd, J = 9.2, 1.6, 1H), 7.12 (d, J = 7.6; 1H), 6.97 (d, J = 7.6, 1H), 6.88 (m, 2H), 6.06 (s, 2H), 4.24 (d, J = 4.8, 2H), 3.53 (m, 2H), 3.16 (solvent), 3.07 (m, 2H), 2.95 (m, 2H), 1.90 (dt, J = 7.5, 7.5, 2H), 1.74 (m, 4H), 0.86 (t, J = 7.2, 3H).
Measurement of in vitro antimalarial activity
The effects of experimental compounds on the growth of Plasmodium falciparum cultures in vitro were measured using flow cytometry.29 Synchronous cultures of ring stage parasites (200 μL, 0.8% parasitemia, 0.5% hematocrit) were grown in 48-well tissue culture plates (Falcon) with 30 nm and 200 nm concentrations of experimental compounds. Cultures were grown in atmospherically regulated (5 % CO2, 5 %O2) incubators (Sanyo) at 37 °C. Aliquots of 50 μL were removed from each well at 72 hours post drug treatment and resuspended in an equal amount of 2 % paraformaldehyde for approximately 1 hour. Fixed cells were stained with 50 nM YOYO-1 (Molecular Probes) in PBS. Each sample was then incubated for 24 h in the dark at 4 °C before being analyzed on a Becton-Dickenson LSRII flow cytometer to measure the percent of parasitized red blood cells (RBCs). Percent of parasitized RBCs was directly read and subsequently growth inhibition values were calculated as the fraction of parasitized RBCs relative to cultures without drug. All screening was done in triplicate with a no drug negative control and a positive control, chloroquine. Dose response studies followed the same general methodology with the exception that each drug was used at doses ranging from 0.1 to 200 nM in triplicate. Fifty percent inhibitory concentrations (IC50's) were calculated by fitting the data to a variable slope sigmoidal dose response curve using SigmaPlot graphing software.
Acknowledgment
We thank the NIH/NIAID (AI53862) for financial support (J.L.D., R.K.G.), as well as the Burroughs Wellcome Quantitative Biology Fellowship (P.B.M.), and the Sandler Foundation.
References
- 1.Baird JK. Effectiveness of antimalarial drugs. N Engl J Med. 2005;352(15):1565–77. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 2.Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415(6872):680–5. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 3.Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298(5591):210–3. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price RN, Uhlemann A-C, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. The Lancet. 364(9432):438. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridley RG, Hofheinz W, Matile H, Jaquet C, Dorn A, Masciadri R, Jolidon S, Richter WF, Guenzi A, Girometta MA, Urwyler H, Huber W, Thaithong S, Peters W. 4-aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrob Agents Chemother. 1996;40(8):1846–54. doi: 10.1128/aac.40.8.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De D, Krogstad FM, Cogswell FB, Krogstad DJ. Aminoquinolines that circumvent resistance in Plasmodium falciparum in vitro. Am J Trop Med Hyg. 1996;55(6):579–83. doi: 10.4269/ajtmh.1996.55.579. [DOI] [PubMed] [Google Scholar]
- 7.Madrid PB, Sherrill J, Liou AP, Weisman JL, Derisi JL, Guy RK. Synthesis of ring-substituted 4-aminoquinolines and evaluation of their antimalarial activities. Bioorg Med Chem Lett. 2005;15(4):1015–8. doi: 10.1016/j.bmcl.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 8.Madrid PB, Wilson NT, DeRisi JL, Guy RK. Parallel synthesis and antimalarial screening of a 4-aminoquinoline library. J Comb Chem. 2004;6(3):437–42. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De D, Byers LD, Krogstad DJ. Antimalarials: synthesis of 4-aminoquinolines that circumvent drug resistance in malaria parasites. Journal of Heterocyclic Chemistry. 1997;34(1):315–320. [Google Scholar]
- 10.Burckhalter JH, Tendick FH, Jones EM, Holcomb WF, Rawlins AL. Aminoalkylphenols as Antimalarials. I. Simply Substituted a-Aminocresols. J. Am. Chem. Soc. 1946;68:1894–1901. doi: 10.1021/ja01214a008. [DOI] [PubMed] [Google Scholar]
- 11.Wiselogle FY, Council NR. A survey of antimalarial drugs, 1941-1945. Edwards; Ann Arbor, Mich.: 1946. p 2 v. in 3. illus. 28 cm. [Google Scholar]
- 12.Burckhalter JH, Tendick FH, Jones EM, Jones PA, Holcomb WF, Rawlins AL. Aminoalkylphenols as Antimalarials. II. (Heterocyclic-amino)-a-amino-o-cresols. The Synthesis of Camoquin. J. Med. Chem. 1948;70:1363–1373. doi: 10.1021/ja01184a023. [DOI] [PubMed] [Google Scholar]
- 13.Hunsicker LG. The pharmacology of the antimalarials. A rational approach to the therapy of resistant falciparum malaria. Arch Intern Med. 1969;123(6):645–9. doi: 10.1001/archinte.123.6.645. [DOI] [PubMed] [Google Scholar]
- 14.Jeffery GM, Collins WE, Skinner JC. Antimalarial Drug Trials On A Multiresistant Strain Of Plasmodium Falciparum. Am J Trop Med Hyg. 1963;12:844–50. doi: 10.4269/ajtmh.1963.12.844. [DOI] [PubMed] [Google Scholar]
- 15.Moore DV, Lanier JE. Observations on two Plasmodium falciparum infections with an abnormal response to chloroquine. Am J Trop Med Hyg. 1961;10:5–9. doi: 10.4269/ajtmh.1961.10.5. [DOI] [PubMed] [Google Scholar]
- 16.Rieckmann KH, Trenholme GM, Williams RL, Carson PE, Frischer H, Desjardins RE. Prophylactic activity of mefloquine hydrochloride (WR 142490) in drug-resistant malaria. Bull World Health Organ. 1974;51(4):375–7. [PMC free article] [PubMed] [Google Scholar]
- 17.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–23. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 18.Evans JM, Fake CS, Hamilton TC, Poyser RH, Showell GA. Synthesis and antihypertensive activity of 6,7-disubstituted trans-4-amino-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-3-ols. J Med Chem. 1984;27(9):1127–31. doi: 10.1021/jm00375a007. [DOI] [PubMed] [Google Scholar]
- 19.Evans JM, Fake CS, Hamilton TC, Poyser RH, Watts EA. Synthesis and antihypertensive activity of substituted trans-4-amino-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-3-ols. J Med Chem. 1983;26(11):1582–9. doi: 10.1021/jm00365a007. [DOI] [PubMed] [Google Scholar]
- 20.Mungthin M, Bray PG, Ridley RG, Ward SA. Central role of hemoglobin degradation in mechanisms of action of 4-aminoquinolines, quinoline methanols, and phenanthrene methanols. Antimicrob Agents Chemother. 1998;42(11):2973–7. doi: 10.1128/aac.42.11.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Neill PM, Bray PG, Hawley SR, Ward SA, Park BK. 4-Aminoquinolines--past, present, and future: a chemical perspective. Pharmacol Ther. 1998;77(1):29–58. doi: 10.1016/s0163-7258(97)00084-3. [DOI] [PubMed] [Google Scholar]
- 22.Foley M, Tilley L. Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther. 1998;79(1):55–87. doi: 10.1016/s0163-7258(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 23.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6(4):861–71. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper RA, Hartwig CL, Ferdig MT. pfcrt is more than the Plasmodium falciparum chloroquine resistance gene: a functional and evolutionary perspective. Acta Trop. 2005;94(3):170–80. doi: 10.1016/j.actatropica.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 25.Tran CV, Saier MH., Jr. The principal chloroquine resistance protein of Plasmodium falciparum is a member of the drug/metabolite transporter superfamily. Microbiology. 2004;150(Pt 1):1–3. doi: 10.1099/mic.0.26818-0. [DOI] [PubMed] [Google Scholar]
- 26.Martin RE, Kirk K. The malaria parasite's chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol Biol Evol. 2004;21(10):1938–49. doi: 10.1093/molbev/msh205. [DOI] [PubMed] [Google Scholar]
- 27.Wellems TE. Transporter of a malaria catastrophe. Nat Med. 2004;10(11):1169–71. doi: 10.1038/nm1104-1169. [DOI] [PubMed] [Google Scholar]
- 28.Johnson DJ, Fidock DA, Mungthin M, Lakshmanan V, Sidhu AB, Bray PG, Ward SA. Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents. Mol Cell. 2004;15(6):867–77. doi: 10.1016/j.molcel.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barkan D, Ginsburg H, Golenser J. Optimisation of flow cytometric measurement of parasitaemia in plasmodium-infected mice. Int J Parasitol. 2000;30(5):649–53. doi: 10.1016/s0020-7519(00)00035-7. [DOI] [PubMed] [Google Scholar]