

Abstract
RNA editing plays a critical role in the life cycle of hepatitis delta virus (HDV). The host editing enzyme ADAR1 recognizes specific RNA secondary structure features around the amber/W site in the HDV antigenome and deaminates the amber/W adenosine. A previous report suggested that a branched secondary structure is necessary for editing in HDV genotype III. This branched structure, which is distinct from the characteristic unbranched rod structure required for HDV replication, was only partially characterized, and knowledge concerning its formation and stability was limited. Here, we examine the secondary structures, conformational dynamics, and amber/W site editing of HDV genotype III RNA using a miniaturized HDV genotype III RNA in vitro. Computational analysis of this RNA using the MPGAfold algorithm indicated that the RNA has a tendency to form both metastable and stable unbranched secondary structures. Moreover, native polyacrylamide gel electrophoresis demonstrated that this RNA forms both branched and unbranched rod structures when transcribed in vitro. As predicted, the branched structure is a metastable structure that converts readily to the unbranched rod structure. Only branched RNA was edited at the amber/W site by ADAR1 in vitro. The structural heterogeneity of HDV genotype III RNA is significant because not only are both conformations of the RNA functionally important for viral replication, but the ratio of the two forms could modulate editing by determining the amount of substrate RNA available for modification.
Keywords: Hepatitis delta virus, RNA editing, ADAR1, RNA secondary structure
INTRODUCTION
Hepatitis delta virus (HDV) increases the severity of liver disease in patients simultaneously infected with its helper, Hepatitis B Virus (HBV) (Rizzetto 1983). The ca. 1680-nt HDV genome is a single-stranded circular RNA; both the genome and the antigenome, a circular intermediate through which HDV replicates, form a characteristic unbranched rod structure due to base-pairing involving ∼70% of positions over the entire length of the molecule (Wang et al. 1986; Lai 1995). HDV encodes just one protein, hepatitis delta antigen (HDAg). Because of this limited coding capacity, HDV relies heavily on host functions and the structure of its RNA.
HDV uses the site-specific RNA adenosine deaminase activity of the host RNA editing enzyme ADAR1 to produce two essential forms of HDAg from the same open reading frame (Polson et al. 1996; Jayan and Casey 2002b; Wong and Lazinski 2002). The specific site modified is termed the amber/W site because editing at this site changes an amber stop codon to a tryptophan (W) codon. In the current model for editing in the HDV replication cycle (Polson et al. 1996), editing occurs by deamination to inosine of the amber/W adenosine in a fraction of full-length antigenomic RNAs. Genomes synthesized from such edited antigenomes subsequently serve as templates for antigenomic sense mRNAs in which the amber stop codon has been replaced with a tryptophan codon. In mRNAs derived from edited antigenomes, an additional 19 or 20 codons are thereby translated to produce the long form of delta antigen, HDAg-L (Weiner et al. 1988; Xia et al. 1990; Casey et al. 1992; Wang et al. 1992). HDAg-L enables viral particle formation by interacting with the envelope protein of HBV, and inhibits replication (Kuo et al. 1989; Chang et al. 1991; Ryu et al. 1992). In mRNAs derived from antigenomes that are not edited, translation terminates at the amber stop codon, and the short form of the delta antigen protein, HDAg-S, is produced; this form of HDAg is required for RNA replication (Kuo et al. 1989). Because of the opposing roles of HDAg-S and HDAg-L, editing levels must be maintained within a limited range that balances these functions (Jayan and Casey 2002; Sato et al. 2004; Jayan and Casey 2005).
The structural requirements of substrates for site-specific editing by ADAR are not fully understood, but it appears that editing sites must be present in the midst of base-paired structures (for reviews, see Seeburg et al. 1998; Bass 2002). The unbranched rod structure of the HDV genotype I antigenome presents the amber/W adenosine in such a structure, and has been shown by site-directed mutagenesis to be required for editing (Casey et al. 1992; Polson et al. 1996). However, for HDV genotype III, which is the most distantly related of the HDV clades and is associated with the most severe disease (Casey et al. 1993; Manock et al. 2000; Nakano et al. 2001), the base-pairing in the immediate vicinity of the amber/W adenosine is disrupted in the unbranched rod structure. Indirect analysis of the ability of editing to occur at the amber/W site in this structure suggested that it is not the substrate for editing in this genotype (Casey 2002). Based on RNA secondary structure predictions, site-directed mutagenesis, and analysis of editing in mutant RNAs in transfected cells, a model was proposed in which amber/W site editing in HDV genotype III requires a branched RNA secondary structure in which an ∼80 base-pair region of the unbranched rod structure, involving 219 of the 1680 nucleotide antigenome, is rearranged such that two ca. 25 bp stem–loops are formed (Casey 2002); these stem–loops are separated by a ca. 25 base-paired region that includes the amber/W editing site. According to this model, HDV genotype III antigenome RNA can form at least two conformations: the unbranched rod structure, which is a poor substrate for amber/W site editing but which is required for replication, and a branched structure, which can be edited. While the approaches used in previous studies allowed observation of editing during the course of HDV RNA replication, structural information obtained from site-directed mutagenesis is limited, and the energetics and structural dynamics of the RNA were not examined.
Here, we report the use of a miniaturized HDV genotype III RNA to analyze both the secondary structures of the RNA and the ability of different conformations to be edited at the amber/W site in vitro. Our results demonstrate that HDV genotype III RNA forms two secondary structures following transcription in vitro. The most energetically stable structure is the characteristic unbranched rod that is required for HDV RNA replication; the other is a metastable branched structure similar to that previously proposed as the substrate for amber/W site editing. Only the branched conformation is a substrate for editing at the amber/W site by ADAR1 in vitro. We suggest that editing in genotype III is determined, in part, by the ability of nascent antigenomic RNA transcripts to fold into the branched structure rather than the unbranched rod structure.
RESULTS
In order to simplify the analysis of HDV genotype III antigenome RNA secondary structures and to examine the ability of these structures to act as substrates for editing by ADAR1, we created a cDNA clone, pMD-III-2, designed to produce a 320-nt miniaturized RNA derived from the genotype III antigenome (Fig. 1). This cDNA construct will produce an RNA containing sequences (positions 970–1104) that encode the C-terminal region of HDAg, connected by a stem–loop linker to sequences from the noncoding side (486–620) that are base-paired with the first region in the unbranched rod structure of the full length antigenome (Fig. 1).
FIGURE 1.

Schematic diagram of HDV RNA and a miniaturized HDV cDNA construct created for in vitro analyses of RNA structure and editing. (Upper) The elongated circle represents the HDV antigenome RNA in the characteristic unbranched rod structure; vertical lines indicate base pairing between the side encoding HDAg and the noncoding side. The amber stop codon (UAG) is shown, and a six-point star represents the location of the amber/W site. The HDAg-S coding region is indicated by the thick black bar; the W and the open bar to the right denote the additional amino acids added to HDAg-L as a result of RNA editing at the amber/W site. Shaded boxes (sequence numbering refers to the genomic strand) show sequences amplified and cloned to generate pMD-III-2 (lower). The dashed line in the lower panel indicates a linker sequence that includes a BstX I restriction site for cloning, and that forms a terminal stem–loop. The locations of the HindIII site and T7 polymerase promoter are indicated; other regions of the plasmid are not shown. Arrowheads indicate the direction of transcription for both the antigenome (upper) and MD-III-2 (lower). SL1 and SL2 refer to secondary structures that are predicted to form as part of possible branched secondary structures (see Fig. 2).
Predicted secondary structures of MD-III-2 RNA include unbranched rod and metastable branched conformations
Potential secondary structures of MD-III-2 RNA were calculated using the massively parallel genetic algorithm MPGAfold (Shapiro and Navetta 1994; Shapiro and Wu 1996; Wu and Shapiro 1999; Shapiro et al. 2001a,b). Previous studies have shown that MPGAfold focuses on a few significant RNA conformations and is capable of identifying functional folding intermediates with biological significance (Shapiro et al. 2001a; Kasprzak et al. 2005; Gee et al. 2006; Tortorici et al. 2006). Briefly, in this algorithm, populations of randomly seeded RNA structures (of a given sequence, unchanged during a run) evolve via structural recombination and mutation over many generations until the population variance is minimized. One result obtained is the population consensus structure at the end of the run, but it is also possible to observe structures present as intermediates at earlier generations. Because of the stochastic nature of the algorithm, many runs are performed to obtain overall consensus results. An important parameter that affects the outcome of MPGAfold runs is the size of the population (i.e., number of structures evolving in parallel) analyzed. Runs with smaller populations more frequently converge to lower fitness (higher free energy) conformers as final structures; these structures may appear as intermediate or metastable conformations in runs using larger populations.
The results of MPGAfold runs on the MD-III-2 RNA sequence with population sizes ranging from 4 K to 64 K are shown in Table 1 and Figure 2. The predicted structures fall into two predominant categories: those that are unbranched, or nearly so (Fig. 2, A1–A3), and those with more extensive branching (Fig. 2, B1–B3). Although structure A1 contains a short cruciform, we consider it to be unbranched here because it is mostly unbranched and is much more similar to the unbranched structures A2 and A3 than to the extensively branched structures B1–B3. Not shown in Figure 2 is a structure very similar to B2, except the base-pairing is slightly altered in the stem for which the loop initiates at position 244; in Table 1, this structure is counted with B2. Including this alternative B2 structure, all the conformations shown in Figure 2 account for 89.4% of final structures found in all runs.
TABLE 1.
Percent of MPGAfold runs with different population sizes in which indicated structures were observed as final solutions
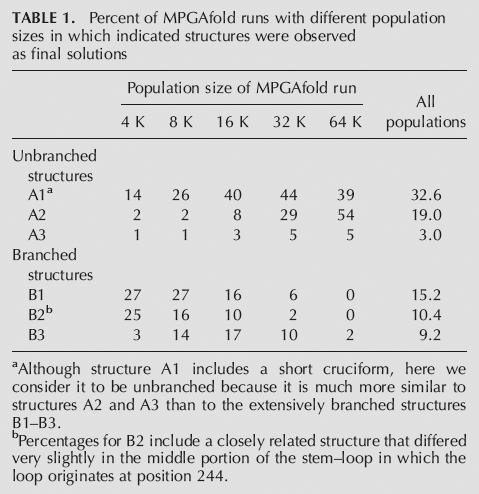
FIGURE 2.

Drawings of the unbranched (A1–A3) and branched (B1–B3) secondary structures predicted to be formed by MD-III-2 RNA. The structures and energies shown were calculated using the massively parallel genetic algorithm MPGAfold (Shapiro and Navetta 1994; Shapiro and Wu 1996, 1997; Wu and Shapiro 1999; Shapiro et al. 2001a,b). Represented here are the dominant structures from the final solutions of MPGAfold runs in all population levels (see Table 1). Numbering refers to sequence positions in the miniaturized cDNA clone pMD-III-2. The amber/W adenosine at position 104 is indicated by a black, five-pointed star. The black bar indicates sequence positions 266–278, which are base-paired around the amber/W site in branched structure B1. SL1 and SL2 indicate stem–loops previously found to be important for editing (Casey 2002). First positions in terminal loops are as follows: 59 in SL1, and 212 in SL2.
An unbranched rod structure is characteristic of HDV RNA, and base-pairs that contribute to this structure are required for replication (Casey 2002; Sato et al. 2004). Thus, it is perhaps not surprising that unbranched rod structures were the most frequently observed solutions (Fig. 2; Table 1). Structures A1–A3 are similar overall and have similar predicted free energies. A2 and A3 are both unbranched rods, but contained slightly different bulges and internal loops. A1 is also nearly identical to A2, except for a short cruciform composed of 4 bp and 6 bp stem–loops. The most energetically stable structure is A2 (Fig. 2), which was also found to be the most energetically stable using the mfold algorithm for RNA secondary structure analysis (Mathews et al. 1999; Zuker 2003).
The predicted extensively branched structures shared some features with each other and, in some cases, with structures A1–A3 as well. Previous analysis of editing using site-directed mutants in Huh-7 cells indicated that editing at the amber/W site requires the distal portions of two stem–loops and base-pairing in the immediate vicinity of the amber/W site (Casey 2002). All three branched structures include one of these stem–loops, a 69-nt stem–loop between positions 28 and 96 denoted SL1 (Fig. 2). In addition to SL1, all three structures contained the second stem–loop, SL2, although the length of the stem at the base of SL2 varies among these three structures. Structures B2 and B3 also shared an additional stem–loop in which the loop initiates at position 244 (Fig. 2). The secondary structure in the immediate vicinity of the amber/W adenosine varied among the three branched structures (Fig. 2). In structures B1 and B3 the amber/W site is in a similar base-paired context involving the same positions, but the base-pairing is more extensive for B1; B2 differs in that the amber/W site occurs in a loop. Elements of the unbranched rod structure were also present in structures B1–B3. The structure of the region from positions 1–24/280–320 in all three structures was the same as that in the unbranched rod structures. At the other end of the folded MD-III-2 RNA, B2 and B3 were identical to the unbranched rods over a longer region (positions 113–198) than was B1 (positions 37–172).
The distribution of structures was not uniform for runs with different population sizes (Table 1). The unbranched structures A1–A3, which are predicted to be more energetically stable than the branched structures, tended to predominate as solutions in larger population runs (Table 1). However, branched conformations were common as final solutions in smaller population runs, and were frequently observed as intermediate structures that persisted for many generations during the course of runs, even those with large populations. An example of a typical run in which branched structures persisted as intermediate structures is shown in the Stem Trace plot in Figure 3. In this 64 K run, structure B2 emerged as the consensus structure at generation 80, then was replaced at generation 138 by structure B1, which persisted for 182 generations before giving way to A1, a near unbranched rod that subsequently yielded to A2, the lowest energy structure. Structure B1 was observed as an intermediate consensus structure in 51% of 64 K runs. The above observations of the distribution of the branched and unbranched predicted structures, peaking in frequency at different population levels, combined with the persistence of the branched structures as intermediates strongly suggest that this RNA sequence has a propensity to form metastable structures.
FIGURE 3.

Stem Trace plot illustrating maturation of the HDV construct MD-III-2 (320 nt) structure in one full 64 K population MPGAfold run, 581 generations long. Plotted here are the population consensus (population histogram peak) structures. In a Stem Trace plot, individual stems, encoded as triplets (5′start position, 3′stop position, number of base pairs), are depicted along the Y-axis in their order of appearance in the consecutive structures, which are depicted along the X-axis. Thus, one secondary structure corresponds to the set of stems, which would be intersected by a vertical line at a given position x. Since the data plotted here comes from an individual MPGAfold run, the consecutive X-axis positions also correspond to generations of the genetic algorithm run (hence, the two labels on the X axis). Labels above the plot indicate major conformation types, which reflect the histogram peak structures at different stages of the genetic algorithm's run. Structures prior to generation 52 are immature, and do not form any persistent states. Labels A1, A2, B1, and B2 indicate correspondence of the intermediate and final states to the secondary structures shown in Figure 2. Stable conformations tend to form plateaus, such as that associated with the branched editing structure (B1) in the middle of the run (from generation 138 until generation 319), or the final rod structure (A2) (from generation 393 until generation 581). The plot shown here is a black-and-white rendition of an interactive plot normally color-coded to depict the frequency of stems.
MD-III-2 RNA forms at least two secondary structures following transcription in vitro
To analyze the ability of MD-III-2 RNA to form these secondary structures, and to directly assess their roles in amber/W site editing, MD-III-2 RNA was synthesized in vitro with T7 RNA polymerase. Following electrophoresis of MD-III-2 RNA on a nondenaturing polyacrylamide gel, two RNA species with different electrophoretic mobilities were detected (Upper and Lower, Fig. 4A). RNA was purified from these two bands, denatured, and electrophoresed on a denaturing polyacrylamide gel containing 7 M urea; RNA purified from both bands in the native gel migrated with the same mobility in the denaturing gel (Fig. 4B), indicating that the different mobilities in the native gel are due to different RNA secondary structures rather than different sequence lengths.
FIGURE 4.

Conformational heterogeneity of MD-III RNA. (A) RNAs were synthesized in vitro using T7 polymerase, then electrophoresed on a 6% polyacrylamide gel, as described in Materials and Methods. Newly synthesized MD-III-2 RNA transcripts resolve as two bands, denoted Upper and Lower, on a nondenaturing gel. MD-III-2SF RNA, which contains mutations that destabilize the unbranched rod structure, resolves as a single band, commensurate in migration to the Upper band of MD-III-2 RNA. (B) Denaturing gel electrophoresis of MD-III-2 RNA isolated from the Upper and Lower bands of a nondenaturing polyacrylamide gel. Upper and Lower band RNAs were purified from the nondenaturing gel in (A), denatured and electrophoresed on a polyacrylamide gel containing 7 M Urea, as described in Materials and Methods.
To determine how the MD-III-2 RNA species in the Upper and Lower bands in the native gel might be related to the predicted branched and unbranched secondary structures, we created a mutant construct, pMD-III-2SF, in which the bases in the distal portions of the SL1 and SL2 helices were flipped (i.e., C–G pairs were changed to G–C pairs, A–U pairs to U–A pairs). In RNA transcribed from this construct the predicted stability of the unbranched rod structure is severely disrupted, but the predicted stability of branched structures B1–B3 is minimally affected. We observed that upon electrophoresis in a nondenaturing polyacrylamide gel MD-III-2SF RNA migrated as a single species with the same mobility as MD-III-2 RNA in the band marked Upper (Fig. 4A). This result suggests that MD-III-2 RNA in the more slowly migrating Upper band is in a conformation that is unaffected by the stem-flip mutations, possibly one of the branched structures (B1–B3) shown in Figure 2, and that MD-III-2 RNA in the more rapidly migrating Lower band is in the unbranched rod conformation.
The branched conformation of MD-III-2 RNA is less energetically stable than the unbranched form
The branched conformations of MD-III-2 RNA are predicted to be considerably less stable than the unbranched structure, about 6–8 kcal/mol. To determine the relative stabilities of these structures, both Upper and Lower RNAs were isolated from a native polyacrylamide gel, incubated at 40°C for varying lengths of time, then electrophoresed on a second native gel (Fig. 5). At this temperature we observed that RNA in the branched conformation converted almost completely to the unbranched form by 45 min (Fig. 5). We also observed conversion of the branched to the unbranched conformation at 37°C, but the process was much slower (data not shown). The structural transition was not reciprocal; there was no conversion of RNA in the unbranched conformation to the branched form (Fig. 5). These results, which are consistent with the results from MPGAfold, confirm that the branched structure is less energetically stable than the unbranched conformation, and demonstrate that a fraction of HDV genotype III RNA forms a metastable branched structure during or shortly following transcription.
FIGURE 5.

The branched conformation of MD-III-2 RNA is less stable than the unbranched rod conformation. Upper and Lower bands were isolated from a nondenaturing gel. Purified RNA was diluted in water and incubated at 40°C. Aliquots were removed and placed on ice at indicated time points, then electrophoresed on a nondenaturing 6% polyacrylamide gel. Arrows indicate migrations of branched and unbranched RNAs.
Amber/W site editing of MD-III-2 RNA in vitro depends on the RNA conformation
A stem-flip mutant similar to MD-III-2SF was efficiently edited in cells transfected with an HDV genotype III editing reporter construct (Casey 2002). We therefore expected that MD-III-2SF RNA would be edited at the amber/W site by ADAR-1 in vitro. Because MD-III-2 Upper RNA exhibited the same migration as MD-III-2SF RNA on the nondenaturing gel, we expected that RNA in this conformation would be edited similarly. We also expected that MD-III-2 Lower RNA, which is likely to be in an unbranched rod structure, would be edited much less efficiently. To test these hypotheses, we purified MD-III-2SF RNA and both Upper and Lower MD-III-2 RNAs from a native polyacrylamide gel and incubated equal amounts of these RNAs with a nuclear extract from cells transfected with an ADAR1 expression construct. Electrophoresis of the RNAs through a second native gel indicated that the conformation and integrity were not affected during the purification and incubation (data not shown). Editing at the amber/W site was detected by RT-PCR and subsequent StyI digestion of radiolabeled PCR products (Casey and Gerin 1995; Polson et al. 1996; Casey 2002). Consistent with our hypotheses, amber/W site editing was clearly evident in MD-III-2 Upper RNA and in MD-III-2SF RNA (Fig. 6, left panel); no amber/W site editing was detected in MD-III-2 Lower RNA (Fig. 6, right panel). The gel shown in Figure 6 is representative of results from several experiments using independently prepared nuclear extracts and RNAs. In all cases the pattern of editing was the same: Upper RNA and SF RNA were edited at similar levels (range 4.5%–22%), and editing of Lower RNA was either 10-fold lower or undetectable in a given experiment (range 0%–2%). Thus, the low levels of editing shown in Figure 6 do not necessarily indicate that the branched MD-III-2 RNA is a poor substrate for ADAR1.
FIGURE 6.

In vitro editing of MD-III-2 RNAs by ADAR-1. MD-III-2 RNAs were purified from a native polyacrylamide gel (Fig. 4A) and incubated with nuclear extract from HEK293 cells transfected with a human ADAR-1 expression construct, as described in Materials and Methods. Amber/W site editing was detected by RT-PCR followed by StyI restriction digestion (editing creates a StyI site); this method has been shown to accurately determine the extent of amber/W site editing (Casey et al. 1992; Polson et al. 1996; Jayan and Casey 2002a). RT-PCR products, either uncut (−) or cut (+) with StyI, were analyzed by polyacrylamide gel electrophoresis. Bands produced as a result of StyI digestion are derived from edited RNAs and are indicated as “edited”; uncut RT-PCR products are derived from unedited RNAs and are indicated as “unedited.” Band intensities were quantified by phosphoimager. Percent editing was determined by dividing the sum of edited band intensities by the sum of the intensities of edited and unedited bands. Upper, MD-III-2 Upper RNA; Lower, MD-III-2 Lower RNA; SF, MD-III-2SF RNA. (A) Comparison of in vitro editing of Upper RNA and SF RNA. (B) Comparison of in vitro editing of Upper and Lower RNAs.
Secondary structure analysis of the two conformations of MD-III-2 RNA
We used Ribonuclease (RNase) digestion analysis to determine the identities of the secondary structures formed by MD-III-2 RNA. RNA was purified from the Upper and Lower bands of a nondenaturing gel, labeled at either the 5′ or 3′ end, then digested with either site specific RNase T1 (G specific) or RNase A (U and C specific) to detect single stranded regions of RNA, or with RNase V1 to detect paired nucleotides. A representative gel showing digestion patterns of 5′-end labeled RNA incubated with RNase T1 or RNase A is shown in Figure 7A. It is clear that the overall cleavage patterns are different for Upper and Lower RNA, as expected for RNAs with different secondary structures.
FIGURE 7.

Secondary structure analysis of MD-III-2 Upper and Lower RNAs. (A) Gel purified Upper (U) and Lower (L) RNAs were either untreated (lanes −) or digested with RNase T1 or RNase A, (lanes ++, 1 unit; lanes +, 0.1 units). The gel shown is representative of several independent experiments. The lanes of the gel showing RNAse T1 and RNase A-treated RNAs have been separated to facilitate labeling; thus, the lanes on the left showing untreated RNA also serve as controls for the lanes on the right side of the panel. Black and shaded symbols indicate cleavage sites specific to enzymatic treatment; circles, RNase T1; squares, RNase A. Cleavage efficiency is indicated by darkness of symbols (black, strong cleavage; gray, weak cleavage). Evaluation of cleavage at a few locations was obscured by the presence of bands in lanes containing undigested RNA; these bands could be due to prematurely terminated T7 polymerase transcripts that copurified with the Upper and Lower RNA when isolated from the nondenaturing gel. Numbering refers to base assignments in MD-III-2 RNA. Locations of bands were determined by comparison with bands generated by RNase T1 sequencing or hydroxide treatment (not shown). Because of the length of the RNA analyzed, in some cases bands indicated in the figure were definitively identified on gels electrophoresed for longer times or in RNAs that were labeled at the 3′ end rather than the 5′ end. Vertical lines labeled SL1, SL2, and UB refer to the locations of sequences involved in structure elements depicted in panels B and C. (B,C) Enzyme digestion data for RNase T1 (circles), RNase A (squares), and RNase V1 (gray arrows) with predicted secondary structures consistent with cleavage patterns. Gray letters denote bases for which the sensitivity to RNase could not be fully evaluated due to the presence of bands in undigested RNA lanes. A six-point star indicates the amber/W adenosine. UB indicates a portion of the unbranched rod structure common to structures A1, A2, A3, B2, and B3 (Fig. 2). (B) Summary of digestion data and structure of gel-purified Upper RNA. (C) Summary of digestion data and structure of gel-purified Lower RNA.
The digestion pattern of Upper RNA is most consistent with a branched conformation identical to structure B1 (Fig. 7B). Structures B2 and B3 are ruled out because base pairing in these predicted structures is identical to unbranched structures A1–A3 from position 128–181, and it is clear that the RNase T1 and RNase A digestion patterns of Upper and Lower RNA are different in this region (Fig. 7A). In structure B1 all susceptible nucleotides in the loops of SL1 (G59 and C62) and SL2 (U212, G213, U214, and G215) were cleaved by RNase T1 or RNase A, as were most nucleotides in other predicted bulges and internal loops (Fig. 7B). Overall, RNase T1 cleaved at all nine guanylates predicted to be unpaired in structure B1, whereas digestion was apparent at five (G76, G85, G105, G237, and G278) of the 65 guanylates predicted to be base-paired. The susceptibility of G76, G85, and G278 could be due to the location of these bases at the ends of helices. RNase V1 digestion at G237, which forms a U–G wobble pair with U189, along with RNase A digestion at U236, which forms the adjacent G–U wobble pair, could indicate that base-pairs form in this region, but are not very stable. RNase A cleavages occurred primarily at positions predicted to be unpaired, but cleavage was detected at some positions that were predicted to be at the ends of helices (C24, C31, U38, C58, C91, C101, U103, U135, and C241) or in G–U wobble pairs (U39, U106, U236, and U 239); most of these cleavages were observed to be weak, and some (C24, C31, C91, and U239) were found to be sites of RNase V1 cleavage, an indication that base-pairing is occurring to some degree at these positions. It appears that positions in the proximal portions of SL1 and SL2 that are predicted to be base-paired are more susceptible to cleavage by RNase T1 and RNase A than those in the distal portions of these stems. This increased susceptibility could indicate that the base pairs in these parts of the structure are able to fluctuate. Although base-pairing in the immediate vicinity of the amber/W site is greater in Upper than in Lower RNA, the evidence of weak RNase T1 cleavage at G105 and weak RNase A cleavage at C101, U103, and U106 could indicate that this region of the structure is only modestly stable.
The digestion pattern of Lower RNA was consistent with an unbranched rod conformation similar to structure A2 (Fig. 7C). Structures A1 and A3 are not consistent with the RNase digestion data. In particular, the stem–loop in A1 in which the loop starts at position G59 is also present in structure B1, but the digestion pattern of Lower RNA clearly differs from Upper RNA in this region (Fig. 7A). Structure A3 is identical to A2 except for the base-pairing pattern of positions 62–72/229–246; RNase digestion at C62 and G237, which are unpaired in A2 but not A3, is more consistent with A2. In structure A2 RNase T1 cleavage occurred at 7 of 10 unpaired guanylates, and of the 65 guanylates predicted to be paired, just 3 (G30, G67, and G85) served as cleavage sites. All three of these sites are found at the ends of helices and might thus be more susceptible to cleavage than other base-paired guanylates; also RNase V1 cleavage at G30 is consistent with this position being base-paired, as shown in Figure 7C. Overall, RNase A digestion was also consistent with the structure in Figure 7C. Similar to the pattern of RNase T1 digestion, paired bases found to be susceptible to cleavage (C24, C27, U55, and C81) occurred at the ends of helices. Two digestion patterns of Lower RNA were more difficult to reconcile with the predicted structure. Digestion at C56 and C57, which are predicted to be in the midst of a four base-pair helix, could indicate, along with cleavages at U52–U55, that the entire U52–C57 segment is unpaired. However, it seems more likely that structural heterogeneity occurs in this region such that some RNAs adopt the conformation shown in Figure 7B, while others are in a form in which U52–U55 are paired with G249–A252, leaving C55–57, C247, and U248 to form an asymmetric internal loop. The lack of RNase A cleavage among uridylates and cytidylates in the large internal loop (G132–U136/U173–G177) was unexpected, and could indicate that this region is structured, perhaps involving pyrimidine–pyrimidine base pairs (Lescrinier et al. 2003); RNase V1 cleavage at U174 and U175 is consistent with this interpretation.
DISCUSSION
Our previous studies indicated that RNA editing in HDV genotype III requires a branched secondary structure that differs substantially from the unbranched rod, the structure which is characteristic of HDV RNA (Casey 2002; Cheng et al. 2003). Although the branched nature of the RNA editing substrate was strongly supported by these earlier studies, it was not possible, based on site-directed mutagenesis alone, to precisely identify the secondary structure nor to determine how this structure is formed. Here, we report the use of a truncated 320nt RNA, MD-III-2, derived from an HDV genotype III isolate to analyze the secondary structure, conformational dynamics, and amber/W site editing of HDV genotype III RNA in vitro. Our results show that MD-III-2 RNA forms a metastable branched secondary structure that is efficiently edited by the host RNA adenosine deaminase ADAR1 in vitro. RNase digestion analysis of the secondary structure of this branched RNA was most consistent with structure B1 (Fig. 2), which differs by about 80 bp from the unbranched rod, and is nearly identical to that previously proposed (Casey 2002).
In order to create MD-III-2 RNA, we used the sequence of an HDV genotype III isolate that is epidemiologically unrelated to the HDV genotype III prototype isolate obtained from Peru (Casey et al. 1993). Previous analysis of replication and editing of the prototype led to the model describing the use of a branched structure for genotype III editing (Casey 2002). Similar to MD-III-2 RNA, a miniaturized RNA derived from the prototype isolate formed both branched and unbranched structures, and only the branched structure could be edited in vitro (data not shown). Furthermore, secondary structure analysis (Mathews et al. 1999; Zuker 2003) of five additional genotype III sequences that diverge by an average of 7% indicates that these RNAs are also able to form branched structures essentially identical to structure B1 (Casey 2002; data not shown). Thus, the use of the branched structure is likely typical for amber/W site editing in HDV genotype III.
ADAR1 deaminates specific adenosines in RNA based on structural requirements that are not fully understood. The context of the amber/W site in structure B1 is consistent with proposed structures of other known targets of site-specific RNA editing by ADAR1; that is, the amber/W site is in a short base-paired segment that occurs toward one end of a nearly 30-bp region that is disrupted by asymmetric internal loops and mismatches. However, there are a few differences between the structure around the amber/W site in MD-III-2 RNA in structure B1 compared with the structure around the HDV genotype I editing site. First, in structure B1 the amber/W site is in an A–U pair, whereas in genotype I the amber/W adenosine occurs as an A–C mismatch, the mutation of which to an A–U pair decreases editing substantially (Casey et al. 1992; Wong et al. 2001). Second, while MD-III-2 RNA B1 contains an A–A mismatch pair (in an internal loop) two bases away from the amber/W site, mutational and genetic analysis of the type I site has shown that activity is highest when these positions form a G–C or an A–U pair (Casey et al. 1992; Wong et al. 2001). Finally, while MD-III-2 RNA B1 exhibits significant base-pairing in the region 3′ of the amber/W site, the presence of a 6nt asymmetric internal loop 7 nt 3′ of the site is curious; in a double-stranded RNA substrate a 6-nt symmetric internal loop prevented editing within a 20-nt region (Lehmann and Bass 1999). There are two asymmetric internal loops downstream of the HDV genotype I editing site, but they are smaller (4 nt), and are positioned differently with respect to the target adenosine. Understanding the significance of the differences between the secondary structures around the genotype I and genotype III editing sites will likely have implications for determining what constitutes a suitable site for ADAR1 editing, a highly selective process for HDV (Polson et al. 1998), as well as host mRNA substrates (for review, see Bass 2002).
Metastable RNA structures have been shown to play important functional roles in several biological systems (Nagel and Pleij 2002), including the ribozymes of HDV (Diegelman-Parente and Bevilacqua 2002). The branched structure B1 that is a substrate for HDV genotype III editing is another such metastable structure. The most stable structures for MD-III-2 RNA are unbranched rods such as A1–A3 (Fig. 2), but the amber/W site is not edited when the RNA is in the unbranched configuration. The MPGAfold analysis suggested that MD-III-2 RNA is likely to form both types of secondary structure. Structure B1 occurred as an intermediate consensus structure in 51% of the 64K runs, and if we count branched conformers similar to it, this number increases to 71%. That about 30% of runs did not involve such intermediate branched structures could indicate the existence of two distinct folding pathways: one leading to the metastable branched structure B1 (in which the amber/W site can be edited), the other bypassing this branched structure and leading to an unbranched rod conformation, possibly via other nonediting-competent branched conformers shown in Figure 2.
The in vitro transcription results presented here indicate that MD-III-2 RNA is indeed structurally heterogeneous, consistent with the MPGAfold analysis. Both editing-competent branched (i.e., B1) and editing-incompetent unbranched structures were formed following transcription in vitro with T7 polymerase, and these structures were confirmed by the RNase digestion analysis. These results indicate the predictive power of this algorithm, particularly for identifying functionally important metastable structures. Although it is possible that cellular or viral factors (e.g., HDAg) could influence the efficiency of branched RNA formation in infected cells, we conclude that all of the information necessary for MD-III-2 to fold into the metastable structure B1 resides in the RNA sequence itself; no cellular or HDV factors (except the RNA) were present in the transcription reaction.
Cotranscriptional folding has been shown to be important for the formation of functionally significant RNA structures (for review, see Pan and Sosnick 2006), including the HDV ribozyme (Diegelman-Parente and Bevilacqua 2002). In infected cells, the distribution of HDV RNA between the unbranched rod and structure B1 could well be affected by cotranscriptional folding or by the propensity of nascently transcribed RNA to proceed along different folding pathways leading to these structures. A circular permutant of MD-III-2 RNA that starts and ends at positions 155 and 154, respectively, formed the branched structure when transcribed in vitro just as readily as MD-III-2 RNA (data not shown); this result could indicate that cotranscriptional folding has a limited role in the formation of the branched structure. Nevertheless, folding may yet occur during or shortly following transcription in cells; transcription of HDV RNA in infected cells is likely to occur much more slowly than with T7 polymerase in vitro, and a slower transcription rate might be expected to favor the formation of branched structures such as B1. For example, in vivo, folding of a Tetrahymena group I pre-RNA differed considerably depending on the polymerase involved (Koduvayur and Woodson 2004). Further investigation, both in vitro and in cells, will be necessary to determine the extent to which cotranscriptional folding contributes to the formation of the branched and unbranched structures of HDV genotype III RNA. Such studies will likely be augmented by folding algorithms, including MPGAfold (Shapiro et al. 2001a; Kasprzak et al. 2005; Gee et al. 2006), that can incorporate the transcription process into the analysis (Meyer and Miklos 2004; Xayaphoummine et al. 2005).
The secondary structure dynamics of the RNA are important not only for the formation of the metastable branched structure, but also for the subsequent conversion of RNA in this structure to the unbranched rod. RNA editing occurs on the HDV antigenome, which is a replication intermediate. In order to produce HDAg-L, edited antigenomes must first serve as templates for transcription of genome RNA, which then acts as template for synthesis of mRNAs encoding HDAg-L. Because replication requires the unbranched rod structure of HDV RNA, it seems probable that RNA in the metastable branched structure B1 must first convert to the unbranched rod for transcription to occur. This conversion may have an energy barrier because it involves the rearrangement of nearly 80 base pairs. Indeed, it could be that this energy barrier is the basis of the stability of the metastable structure B1. Cellular and viral factors could play a role in the conversion of this branched structure to the unbranched rod, but they are not required; although purified MD-III-2 RNA was stable in structure B1 at room temperature, it converted readily to the unbranched rod at 40°C (Fig. 5).
Finally, our results suggest that the structural heterogeneity of HDV genotype III RNA could be an important mechanism for controlling editing. Modulation of editing is particularly important for the HDV replication cycle because editing levels determine the balance between the amounts of HDAg-S and HDAg-L produced. MPGAfold analysis of the secondary structure of HDV genotype I RNA indicates that this RNA is not capable of forming an extensive branched structure similar to the genotype III structure B1 (data not shown). Rather, for this RNA, editing occurs on the characteristic stable unbranched rod structure (Polson et al. 1996), and suppression of editing by HDAg-S (Polson et al. 1998; Sato et al. 2004) is likely an important mechanism for preventing excessive editing. However, this mechanism is not employed by genotype III, because genotype III HDAg-S is not an effective inhibitor of amber/W site editing (Cheng et al. 2003). We have shown that HDAg-L can inhibit genotype III editing, and might function in a negative feedback process (Cheng et al. 2003). Because only RNA that adopts the branched conformation B1 can be edited, the distribution of the RNA between different conformations is also a potential determinant of editing levels. This distribution will be affected by both the folding dynamics of the RNA and by the stability of the metastable branched structure. Further studies will be needed to test this hypothesis and reveal the details of the folding dynamics.
MATERIALS AND METHODS
Plasmid construction
Plasmid pMD-III-2, was generated by reverse transcription-polymerase chain reaction (RT-PCR) of an HDV RNA isolate (Manock et al. 2000). Sequences 970–1104 were amplified with primers MD1 and MD2 (Table 2) and sequences 486–620 were amplified with primers MD3 and MD4 (Table 2; nucleotide numbering according to Casey et al. (1993). Amplified cDNA fragments were digested with EcoRI and BstXI and cloned into the EcoRI restriction site of pGEM3Zf (+) (Promega) to yield plasmid pMD-III-2. Digestion of pMD-III-2 with HindIII, followed by transcription with T7 RNA polymerase, produces a 320-nt RNA, MD-III-2. The sequence of the insert can be accessed using GenBank accession number DQ445664.
TABLE 2.
Primers used in this study

Double stem-flip mutant pMD-III-2SF was constructed by double PCR amplification of pMD-III-2 using overlapping mutant primers SF1a and SF1b or SF2a and SF2b (Table 2), as described previously (Casey 2002). In this construct, transversion mutations were made in the most distal paired nucleotides of stem–loops SL1 and SL2 (between the terminal loop and the most distal internal loop) in the predicted branched secondary structures shown in Figure 2; that is, G–C pairs were changed to C–G pairs, A–U pairs to U–A pairs, and U–G pairs to A–U pairs, etc.
Computational analysis of RNA secondary structure
RNA secondary structure analysis was performed on MD-III-2 RNA using the massively parallel genetic algorithm MPGAfold, (Shapiro and Navetta 1994; Shapiro and Wu 1996, 1997; Wu and Shapiro, 1999; Shapiro et al. 2001a,b). First, a pool of all fully complementary stems for a given nucleic acid sequence is generated. Next, a population of thousands of potential RNA structures is randomly seeded from that stem pool. The secondary structures evolve via recombination of maturing structures and mutation of structural motifs (i.e., deletion of stems or addition of new ones from the stem pool). This process takes place in parallel in a two-dimensional array of population elements. At each generation the free energy of the RNA structures is used as a fitness criterion, including efn2 calculations that account for stem stacking across multibranch loops and free ends (Mathews et al. 1999). Evolution of the population of structures continues for a number of generations until the population variance is minimized; i.e., the population converges on a final structure. This structure is not necessarily the lowest energy structure, and the stochastic nature of MPGAfold requires multiple runs to obtain overall consensus results. We performed MPGAfold runs on MD-III-2 RNA with population sizes ranging from 4 K to 64 K structures (1 K = 1024 elements); 100 runs were performed for each population size. The results of MPGAfold runs with all population sizes were combined into one Stem Trace plot (Shapiro and Kasprzak 1996; Kasprzak and Shapiro 1999) in order to visualize the key predicted structural conformations and collect statistics. Stem Trace plots all unique stems, defined as lists of triplets (5′ start position, 3′ stop position, number of base pairs), found in the structures from a solution space on the Y-axis. Consecutive X-axis positions correspond to full structures from a solution space of a folding program. In this study we have plotted both final solutions of MPGAfold runs from multiple population level runs as well as individual runs, such as the one shown in Figure 3, in which the dominant (histogram peak) structures from every generation are shown. Stem Trace plots produce views orthogonal to the widely used stem histogram plot (a dot plot), and they depict structural motifs in their original full structure context. What is also very important, and what distinguishes them from dot plots, is that they allow one to identify significant clusters of full structures with a frequency of occurrence lower than 50%.
RNA preparation and structure isolation
Plasmids were linearized with HindIII and transcribed in vitro with T7 RNA polymerase for 1 h at 37°C. Transcripts were either uniformly labeled with [α]-32P-CTP (3000 Ci/mmol), labeled at the 5′ end using [γ]-33P-ATP and T4 Kinase (NEB), or labeled at the 3′ end using 32P-pCp and RNA ligase (Ambion). Radiolabeled nucleotides were purchased from MP Biomedicals. For all subsequent manipulations, RNA was kept at or below 4°C unless otherwise indicated.
Nondenaturing polyacrylamide gel electrophoresis was performed for 4 h at 30 W on 6% polyacrylamide gels in 0.5× TBE buffer (1× TBE = 89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.3). RNAs were detected with a phosphorimager or with Kodak Biomax MS film, excised, and purified using Ultrafree-MC and Microcon YM-30 columns (Milllipore Corp.) according to the manufacturer's recommendations. RNA recovery rates were calculated based on scintillation counts.
Kinetic analysis of RNA structures
Uniformly labeled RNA transcripts purified from a nondenaturing gel were incubated at 40°C in a circulating water bath. The bath temperature was monitored with a Scientific Precision Thermometer (Fisher); fluctuation was <0.3°C. At indicated time points, aliquots were taken and immediately chilled on ice. Samples were electrophoresed on a second nondenaturing gel at 4°C for 4 h. The gel was exposed to a phosphor screen overnight, which was then scanned with a Molecular Dynamics Storm 475 phosphorimager.
Nuclear extraction of ADAR 1
HEK 293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1 mM glutamine. Cells were transfected with ADAR-1 expression construct pDL701 (Wong et al. 2003) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications. The pDL701 expression plasmid produces the p110 form of ADAR-1. Nuclear extracts were harvested 48 h post-transfection as previously described (Schreiber et al. 1989). Briefly, cells were washed in 1× phosphate-buffered saline, pelleted, resuspended in cold buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF), and incubated on ice for 15 min. Twenty-five microliters 10% Nonidet NP40 were added and the mixture was vortexed and centrifuged briefly. Pellets were resuspended in ice cold buffer C (20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF), and incubated at 4°C for 15 min. After 5-min centrifugation at 13,000g the supernatant was removed and stored at −80°C.
In vitro editing analysis
For editing analysis, 1 fmol of RNA was incubated at 37°C overnight with 2-μL nuclear extract in 80-μL buffer (0.02 M HEPES pH 7.0, 0.1 M NaCl, 10% glycerol, 5 mM EDTA, 250 units/mL RNasin RNase inhibitor (Invitrogen), 1 mM dithiothreitol, 25 μg BSA, 0.1 mM Phenylmethylsulfonyl fluoride (PMSF) (Cho et al. 2003)). After overnight incubation, RNA was purified by extraction with phenol-chloroform and ethanol precipitation. RNA editing at the amber/W site was analyzed by StyI digestion of RT-PCR products as previously described (Casey and Gerin 1995; Polson et al. 1996; Casey 2002), except that primers E1 and E2 were used for PCR (Table 2), and the annealing temperature was increased to 57°C.
Enzymatic analysis of RNA secondary structure
Three femtomoles of 5′- or 3′-end labeled RNA transcript was incubated in structure buffer containing 10 mM Tris pH 7.0, 100 mM KCl, 10 mM MgCl2, 4 μg yeast RNA, and nuclease-free water. All buffers for RNA structural analysis were obtained from Ambion and used according to the manufacturer's recommendations. RNase digestions were performed at room temperature for 15 min with 1 unit of RNase T1 (Ambion), or either 1 or 0.1 units RNase A (Ambion), as indicated. RNA was precipitated, resuspended in gel loading buffer (95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% Xylene Cyanol, 0.025% Bromophenol Blue) and loaded onto a 6% polyacrylamide/7M urea sequencing gel. RNA sequencing was performed by digestion of end-labeled RNA with RNase T1 for 15 min at 55°C in 1× sequencing buffer (20 mM sodium citrate pH 5, 1 mM EDTA, 7 M urea). To produce an alkaline hydrolysis ladder, RNA was incubated at 95°C for 4, 6, and 8 min in Hydrolysis Buffer (50 mM sodium carbonate pH 9.2, 1 mM EDTA). The gel was fixed in 6% acetic acid/10% ethanol, dried, then exposed to a phosphor screen overnight. Digestion patterns were visualized by radioanalytic imaging (Molecular Dynamics Storm 475).
ACKNOWLEDGMENTS
We thank D. Lazinski (Tufts University) for providing plasmid pDL701. We also thank Kevin Graf for his assistance with the initial computational predictions. This work was supported by Grant R01-AI42324 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. This publication has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.89306.
REFERENCES
- Bass B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey J.L. RNA editing in hepatitis delta virus genotype III requires a branched double-hairpin RNA structure. J. Virol. 2002;76:7385–7397. doi: 10.1128/JVI.76.15.7385-7397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey J.L., Gerin J.L. Hepatitis D virus RNA editing: Specific modification of adenosine in the antigenomic RNA. J. Virol. 1995;69:7593–7600. doi: 10.1128/jvi.69.12.7593-7600.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey J.L., Bergmann K.F., Brown T.L., Gerin J.L. Structural requirements for RNA editing in hepatitis delta virus: Evidence for a uridine-to-cytidine editing mechanism. Proc. Natl. Acad. Sci. 1992;89:7149–7153. doi: 10.1073/pnas.89.15.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey J.L., Brown T.L., Colan E.J., Wignall F.S., Gerin J.L. A genotype of hepatitis D virus that occurs in northern South America. Proc. Natl. Acad. Sci. 1993;90:9016–9020. doi: 10.1073/pnas.90.19.9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang F.L., Chen P.J., Tu S.J., Wang C.J., Chen D.S. The large form of hepatitis delta antigen is crucial for assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. 1991;88:8490–8494. doi: 10.1073/pnas.88.19.8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q., Jayan G.C., Casey J.L. Differential inhibition of RNA editing in hepatitis delta virus genotype III by the short and long forms of hepatitis delta antigen. J. Virol. 2003;77:7786–7795. doi: 10.1128/JVI.77.14.7786-7795.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D.S., Yang W., Lee J.T., Shiekhattar R., Murray J.M., Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J. Biol. Chem. 2003;278:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- Diegelman-Parente A., Bevilacqua P.C. A mechanistic framework for co-transcriptional folding of the HDV genomic ribozyme in the presence of downstream sequence. J. Mol. Biol. 2002;324:1–16. doi: 10.1016/s0022-2836(02)01027-6. [DOI] [PubMed] [Google Scholar]
- Gee A.H., Kasprzak W., Shapiro B.A. Structural differentiation of the HIV-1 polyA signals. J. Biomol. Struct. Dyn. 2006;23:417–428. doi: 10.1080/07391102.2006.10531236. [DOI] [PubMed] [Google Scholar]
- Jayan G.C., Casey J.L. Increased RNA editing and inhibition of hepatitis delta virus replication by high-level expression of ADAR1 and ADAR2. J. Virol. 2002a;76:3819–3827. doi: 10.1128/JVI.76.8.3819-3827.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayan G.C., Casey J.L. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of ADAR1 but not ADAR2 expression. J. Virol. 2002b;76:12399–12404. doi: 10.1128/JVI.76.23.12399-12404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayan G.C., Casey J.L. Effects of conserved RNA secondary structures on hepatitis delta virus genotype I RNA editing, replication, and virus production. J. Virol. 2005;79:11187–11193. doi: 10.1128/JVI.79.17.11187-11193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzak W., Shapiro B. Stem Trace: An interactive visual tool for comparative RNA structure analysis. Bioinformatics. 1999;15:16–31. doi: 10.1093/bioinformatics/15.1.16. [DOI] [PubMed] [Google Scholar]
- Kasprzak W., Bindewald E., Shapiro B.A. Structural polymorphism of the HIV-1 leader region explored by computational methods. Nucleic Acids Res. 2005;33:7151–7163. doi: 10.1093/nar/gki1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koduvayur S.P., Woodson S.A. Intracellular folding of the Tetrahymena group I intron depends on exon sequence and promoter choice. RNA. 2004;10:1526–1532. doi: 10.1261/rna.7880404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo M.Y., Chao M., Taylor J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989;63:1945–1950. doi: 10.1128/jvi.63.5.1945-1950.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M. The molecular biology of hepatitis delta virus. Annu. Rev. Biochem. 1995;64:259–286. doi: 10.1146/annurev.bi.64.070195.001355. [DOI] [PubMed] [Google Scholar]
- Lehmann K.A., Bass B.L. The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 1999;291:1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- Lescrinier E.M., Tessari M., van Kuppeveld F.J., Melchers W.J., Hilbers C.W., Heus H.A. Structure of the pyrimidine-rich internal loop in the poliovirus 3′-UTR: The importance of maintaining pseudo-2-fold symmetry in RNA helices containing two adjacent non-canonical base-pairs. J. Mol. Biol. 2003;331:759–769. doi: 10.1016/s0022-2836(03)00787-3. [DOI] [PubMed] [Google Scholar]
- Manock S.R., Kelley P.M., Hyams K.C., Douce R., Smalligan R.D., Watts D.M., Sharp T.W., Casey J.L., Gerin J.L., Engle R., et al. An outbreak of fulminant hepatitis delta in the Waorani, an indigenous people of the Amazon basin of Ecuador. Am. J. Trop. Med. Hyg. 2000;63:209–213. doi: 10.4269/ajtmh.2000.63.209. [DOI] [PubMed] [Google Scholar]
- Mathews D.H., Sabina J., Zuker M., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Meyer I.M., Miklos I. Co-transcriptional folding is encoded within RNA genes. BMC Mol. Biol. 2004;5 doi: 10.1186/1471-2199-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel J.H., Pleij C.W. Self-induced structural switches in RNA. Biochimie. 2002;84:913–923. doi: 10.1016/s0300-9084(02)01448-7. [DOI] [PubMed] [Google Scholar]
- Nakano T., Shapiro C.N., Hadler S.C., Casey J.L., Mizokami M., Orito E., Robertson B.H. Characterization of hepatitis D virus genotype III among Yucpa Indians in Venezuela. J. Gen. Virol. 2001;82:2183–2189. doi: 10.1099/0022-1317-82-9-2183. [DOI] [PubMed] [Google Scholar]
- Pan T., Sosnick T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct. 2006;35:161–175. doi: 10.1146/annurev.biophys.35.040405.102053. [DOI] [PubMed] [Google Scholar]
- Polson A.G., Bass B.L., Casey J.L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature. 1996;380:454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- Polson A.G., Ley H.L., 3rd, Bass B.L., Casey J.L. Hepatitis delta virus RNA editing is highly specific for the amber/W site and is suppressed by hepatitis delta antigen. Mol. Cell. Biol. 1998;18:1919–1926. doi: 10.1128/mcb.18.4.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzetto M. The delta agent. Hepatology. 1983;3:729–737. doi: 10.1002/hep.1840030518. [DOI] [PubMed] [Google Scholar]
- Ryu W.S., Bayer M., Taylor J. Assembly of hepatitis delta virus particles. J. Virol. 1992;66:2310–2315. doi: 10.1128/jvi.66.4.2310-2315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S., Cornillez-Ty C., Lazinski D.W. By inhibiting replication, the large hepatitis delta antigen can indirectly regulate amber/W editing and its own expression. J. Virol. 2004;78:8120–8134. doi: 10.1128/JVI.78.15.8120-8134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E., Matthias P., Muller M.M., Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 1989;17 doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg P.H., Higuchi M., Sprengel R. RNA editing of brain glutamate receptor channels: Mechanism and physiology. Brain Res. Brain Res. Rev. 1998;26:217–229. doi: 10.1016/s0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- Shapiro B.A., Kasprzak W. STRUCTURELAB: A heterogeneous bioinformatics system for RNA structure analysis. J. Mol. Graph. 1996;14:194–205. 222–194. doi: 10.1016/s0263-7855(96)00063-x. [DOI] [PubMed] [Google Scholar]
- Shapiro B.A., Navetta J. A massively parallel genetic algorithm for RNA secondary structure prediction. J. Supercomput. 1994;8:195–207. [Google Scholar]
- Shapiro B.A., Wu J.C. An annealing mutation operator in the genetic algorithms for RNA folding. Comput. Appl. Biosci. 1996;12:171–180. doi: 10.1093/bioinformatics/12.3.171. [DOI] [PubMed] [Google Scholar]
- Shapiro B.A., Wu J.C. Predicting RNA H-type pseudoknots with the massively parallel genetic algorithm. Comput. Appl. Biosci. 1997;13:459–471. doi: 10.1093/bioinformatics/13.4.459. [DOI] [PubMed] [Google Scholar]
- Shapiro B.A., Bengali D., Kasprzak W., Wu J.C. RNA folding pathway functional intermediates: Their prediction and analysis. J. Mol. Biol. 2001a;312:27–44. doi: 10.1006/jmbi.2001.4931. [DOI] [PubMed] [Google Scholar]
- Shapiro B.A., Wu J.C., Bengali D., Potts M.J. The massively parallel genetic algorithm for RNA folding: MIMD implementation and population variation. Bioinformatics. 2001b;17:137–148. doi: 10.1093/bioinformatics/17.2.137. [DOI] [PubMed] [Google Scholar]
- Tortorici M.A., Shapiro B.A., Patton J.T. A base-specific recognition signal in the 5′ consensus sequence of rotavirus plus-strand RNAs promotes replication of the double-stranded RNA genome segments. RNA. 2006;12:133–146. doi: 10.1261/rna.2122606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.G., Cullen J., Lemon S.M. Immunoblot analysis demonstrates that the large and small forms of hepatitis delta virus antigen have different C-terminal amino acid sequences. J. Gen. Virol. 1992;73:183–188. doi: 10.1099/0022-1317-73-1-183. [DOI] [PubMed] [Google Scholar]
- Wang K.S., Choo Q.L., Weiner A.J., Ou J.H., Najarian R.C., Thayer R.M., Mullenbach G.T., Denniston K.J., Gerin J.L., Houghton M. Structure, sequence and expression of the hepatitis delta viral genome. Nature. 1986;323:508–514. doi: 10.1038/323508a0. [DOI] [PubMed] [Google Scholar]
- Weiner A.J., Choo Q.L., Wang K.S., Govindarajan S., Redeker A.G., Gerin J.L., Houghton M. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24 delta and p27 delta. J. Virol. 1988;62:594–599. doi: 10.1128/jvi.62.2.594-599.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.K., Lazinski D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. 2002;99:15118–15123. doi: 10.1073/pnas.232416799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.K., Sato S., Lazinski D.W. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–858. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.K., Sato S., Lazinski D.W. Elevated activity of the large form of ADAR1 in vivo: Very efficient RNA editing occurs in the cytoplasm. RNA. 2003;9:586–598. doi: 10.1261/rna.5160403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.C., Shapiro B.A. A Boltzmann filter improves the prediction of RNA folding pathways in a massively parallel genetic algorithm. J. Biomol. Struct. Dyn. 1999;17:581–595. doi: 10.1080/07391102.1999.10508388. [DOI] [PubMed] [Google Scholar]
- Xayaphoummine A., Bucher T., Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005;33:W605–W610. doi: 10.1093/nar/gki447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.P., Chang M.F., Wei D., Govindarajan S., Lai M.M. Heterogeneity of hepatitis delta antigen. Virology. 1990;178:331–336. doi: 10.1016/0042-6822(90)90415-n. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]