

Abstract
Although helix P4 in the catalytic domain of the RNase P ribozyme is known to coordinate magnesium ions important for activity, distinguishing between direct and indirect roles in catalysis has been difficult. Here, we provide evidence for an indirect role in catalysis by showing that while the universally conserved bulge of helix P4 is positioned 5 nt downstream of the cleavage site, changes in its structure can still purturb active site metal binding. Because changes in helix P4 also appear to alter its position relative to the pre-tRNA cleavage site, these data suggest that P4 contributes to catalytic metal ion binding through substrate positioning.
Keywords: RNase P, catalysis, metal ion, substrate recognition
INTRODUCTION
Like other large ribozymes, RNase P is a metalloenzyme that requires the binding of divalent metal ions for both folding and catalysis (Harris and Christian 2003; Fedor and Williamson 2005). However, relatively little is known about the number or position of catalytic metal ions in RNase P because of the fundamental difficulty of distinguishing catalytic metals from those contributing indirectly to catalysis through structural stabilization. Current biochemical evidence indicates that the universally conserved structural element helix P4 binds metal ions and that perturbation of metal binding to helix P4 causes large (104-fold) catalytic defects (Christian et al. 2002; Crary et al. 2002). The long-standing interpretation of these observations has been that the major groove surrounding the universally conserved bulged uridine in helix P4 is the site of catalytic metal ion binding (Frank and Pace 1997; Christian et al. 2002; Crary et al. 2002; Kaye et al. 2002). However, there is no direct evidence that P4 functional groups interact with the same metal ions as those coordinated to the scissile phosphate. In fact, models of the enzyme–substrate complex derived from recent crystal structures of RNase P RNA indicate a structural rather than a catalytic role for the P4 helix (Kazantsev et al. 2005; Torres-Larios et al. 2005). To resolve this important issue, we have used short-range cross-linking and quantitative analysis of thiophic metal ion rescue to distinguish between a structural and catalytic role for metal ion interactions with helix P4.
RESULTS AND DISCUSSION
Direct involvement of helix P4 in catalysis should require its close proximity to the scissile phosphate. We, therefore, examined the location of the P4 helix relative to pre-tRNA by intermolecular crosslinking (Fig. 1). Escherichia coli RNase P RNA containing 4-thiouridine (4sU) at the universally conserved bulged uridine in helix P4 (4sU69 P RNA, Fig. 1A) yields a specific intermolecular cross-link (XL69) to pre-tRNA in the presence of calcium (Ca2+) (Fig. 1B). Ca2+ allows RNase P RNA folding and substrate binding while suppressing enzymatic cleavage, which permits analysis of the ground state enzyme–substrate (E-S) complex (Smith et al. 1992). Incubation of purified XL69 RNA in magnesium (Mg2+) yields a fragment that comigrates with the 5′-leader sequence (Fig. 1C), demonstrating that the cross-linked RNA is able to achieve a catalytically active conformation and that the relative position of the cross-linked elements is likely to reflect that of the native reaction. Alkaline hydrolysis of 5′-32P-labeled pre-tRNA cross-linked to unlabeled 4sU69 P RNA produces a ladder that extends 4 nt longer than the 5′-leader sequence, indicating that cross-linking occurs to the subsequent position N(5) in the pre-tRNA acceptor stem since fragments containing the cross-link will be of larger molecular weight and shifted away from the observed base ladder (Fig. 1D). The XL69 cross-link provides direct evidence of the proximity of helix P4 to the pre-tRNA substrate. However, the data above also indicate that the universally conserved bulge and adjacent functional groups involved in metal ion coordination (Frank and Pace 1997; Christian et al. 2002; Crary et al. 2002; Kaye et al. 2002) are several nucleotides distant from the reactive phosphate in the ground state E-S complex.
FIGURE 1.

Intermolecular photocross-linking analysis of P4 position relative to pre-tRNA. (A) Secondary structures of wild-type and mutant forms of helix P4 used in this study. A gray U indicates the position of the bulged uridine. (B) PAGE analysis of the products resulting from 366-nm irradiation of 0.8 μM pre-tRNA (left), 0.4 μM 4sU69 P RNA (middle), and a mixture of 0.8 μM pre-tRNA and 0.4 μM nM 4sU69 P RNA (right) in 1 M NH4Cl, 50 mM MES (pH 5.5), 25 mM CaCl2, 0.02% NP40. The locations of pre-tRNA, P RNA, and the XLU69 intermolecular cross-link are indicated. The position of an intramolecular cross-link is indicated by an asterisk. (C) Analysis of the catalytic activity of the XLU69 cross-link. The gel shows resolution of the gel purified XLU69 RNA (left), the products resulting from incubation of XLU69 in Mg2+ (middle), and a 5′-leader sequence marker (right). (D) Alkaline hydrolysis mapping of 5′-end-labeled pre-tRNA in the XLU69 cross-linked RNA. A 5′-leader RNA is shown as a marker. The positions of nucleotides corresponding to N(+1) and N(+4) relative to the RNase P cleavage site are indicated. (E) Analysis of the products resulting from irradiation of a mixture of 0.8 mM pre-tRNA and 0.4 mM 4sU68 P RNA under the conditions noted in B (middle), and analysis of the catalytic activity of purified XLU68 cross-link as described in C (right). A 5′-leader RNA is shown as a marker (left). (F) Alkaline hydrolysis mapping of 5′-end-labeled pre-tRNA in the XLU68 cross-linked RNA. A 5′-leader RNA is shown as a marker. The position of nucleotide corresponding to N(−2) is indicated.
These results suggest that the metal interactions within P4 are not directly involved in chemical catalysis. To test this hypothesis we examined the effect of helix P4 structure on metal ion binding at the pre-tRNA cleavage site. Previous studies showed that phosphorothioate substitution at the pro-RP oxygen of the scissile phosphate results in catalytic defects that can be overcome or “rescued” by the addition of cadmium (Cd2+) (Warnecke et al. 1996). We, therefore, determined the Cd2+ concentration dependence of rescue of RNase P activity within a constant background of Mg2+ (10 mM) to ensure folding, to measure the apparent affinity of the rescuing metal ion (K 1/2 Cd), and to estimate the minimal number of metal ions involved in rescue (n Hill,) as described in previous studies (Fig. 2; Shan et al. 2001).
FIGURE 2.

Quantitative phosphorothioate rescue analysis of the effects of mutations in the P RNA catalytic core. (A) Cd2+ concentration dependence of k rel (k sulfur/k oxygen) for cleavage of pre-tRNAASP with a cleavage site RP phosporothioate modification by native P RNA (circle), and mutants A248U (square), G332U (triangle), and U69A (diamond) plotted as a function of a nonlinear form of the Hill equation as described previously (Shan et al. 2001). The effect of Cd2+ on rate is expressed as the ratio of the observed reaction rate for the phosphorothioate substituted substrate to the corresponding unmodified control (k rel), which controls for nonspecific effects of Cd2+ binding on the reaction. (B) Cd2+ concentration dependence of k rel for cleavage of the Rp phosporothioate-modified substrate by the Δ69 (circle), Δ69U68 (diamond), and Δ69U70 (triangle) mutant RNase P ribozymes. Solid black line reflects a plot of the data from Δ69U68 RNase P RNA plotted with n Hill fixed at 2. Note that all reactions were carried out in a constant background of 10 mM MgCl2 to ensure RNA folding and that there was essentially no inhibition of the observed reaction rate due to the presence of Cd2+ even at the highest concentrations tested (40 mM).
To ask whether P4 structure specifically affects active site metal binding, we examined whether mutations within helix P4 altered the observed affinity of rescuing Cd2+ at the scissile phosphate (Shan et al. 2001). For the native P RNA the concentration dependence of Cd2+ rescue is cooperative, with apparent affinity for rescuing Cd2+ (K 1/2 Cd) of 6.7 ± 2.0 mM, and an n Hill of 1.8 ± 0.3, consistent with previous studies (Beebe et al. 1996; Warnecke et al. 1996; Fig. 2A, solid line). Mutation of the universally conserved bulged uridine to A (U69A), which has been shown to cause only modest effects on binding and catalysis (Kaye et al. 2002), has no effect on Cd2+ binding affinity or cooperativity (K 1/2 Cd = 4.4 ± 1.2 mM, n Hill of 1.6 ± 0.3). Similarly, individual mutations within two other universally conserved regions in RNase P RNA that have been proposed to be involved in active site contacts (J5/15 and J18/2) (Kurz and Fierke 2000; Harris and Christian 2003) also had little effect. Specifically, mutation of A248 to U in J5/15, which disrupts a conserved base-pairing interaction with the N(−1) base adjacent to the cleavage site, has only a modest effect on rescuing Cd2+ affinity (K1/2Cd = 18 ± 3 mM, n Hill = 2.0 ± 0.4), while mutation in J18/2 at a position identified by short-range cross-linking from the cleavage site (G332C) is essentially the same as wild-type E. coli RNase P RNA (K 1/2 Cd = 5.6 ± 1.2 mM, n Hill = 1.6 ± 0.3, Fig. 2A). These data indicate that the identity of the universally conserved bulge base and conserved elements in J5/15 and J18/2 proposed to be involved in active site contacts are not directly linked to metal binding at the pre-tRNA cleavage site.
In contrast to the findings above, mutations that remove or change the position of the bulged uridine in helix P4 (Δ69, Δ69U68, and Δ69U70, Fig. 1A), cause significant changes in both affinity and number of rescuing Cd2+ ions (Fig. 2B). These mutations have previously been shown to cause large catalytic defects and reduce apparent Mg2+ binding (Kaye et al. 2002). Here, these mutations show no sign of saturation of thiophilic metal ion binding at the scissile phosphate, reducing the affinity of rescuing Cd2+ by at least sixfold. In addition, there is a significant decrease in the apparent cooperativity of rescuing metal binding at the scissile phosphate, (n Hill = 1) (Fig. 2B). Importantly, these mutations have previously been shown to only alter terbium (Tb+3) cleavage of the P RNA backbone at or immediately adjacent to the site of mutation and are thus unlikely to reflect indirect effects from changes in RNA folding (Kaye et al. 2002). These data indicate that helix P4 plays a role in positioning catalytic metal ions despite its distance from the reactive phosphate.
The close association of helix P4 and the pre-tRNA acceptor stem and the sensitivity of catalytic metal ion affinity to the loss or change in the position of the P4 bulge suggests that helix P4 may act to position the scissile phosphate relative to other active site elements. To test this hypothesis we examined cross-linking in the Δ69U70 and Δ69U68 mutant ribozymes. No cross-linking to the pre-tRNA substrate was observed when a 4sU was placed at a bulge repositioned 1 nt in the 3′ direction in Δ69U70 P RNA (data not shown). However, an intermolecular cross-link (XLU68) was observed when the bulge was moved 1 nt in the 5′ direction in the Δ69U68 P RNA (Fig. 1E). Reaction of the XLU68 cross-link in Mg2+ leads to a decrease in full-length XLU68 and the formation of a new band migrating well above that expected from the release of the 5′-leader sequence observed for XLU69. These data indicate that XLU68 retains catalytic activity and that the site of P4 cross-linking is shifted into the 5′ leader at least 5 nt from the native position within the acceptor stem. Alkaline hydrolysis mapping of the XLU68 cross-link site confirms this interpretation and demonstrates cross-linking at N(−2), upstream of the pre-tRNA cleavage site (Fig. 1F). Thus, P4 mutations that weaken catalytic metal ion binding without large-scale effects on P RNA structure (Kaye et al. 2002) can also change the position of the pre-tRNA cleavage site within the ground state enzyme–substrate complex.
The data above thus provide insight into three fundamental elements of RNase P function. First, our data indicate that the metal binding pocket in helix P4 is sufficiently distant from the cleavage site in the ground state to make its direct participation in catalytic metal binding unlikely. These data provide a key distance constraint between the ribozyme and the middle of the pre-tRNA acceptor stem, as well as functional data that is consistent with enzyme–substrate interactions predicted from recent crystallographic data (Kazantsev et al. 2005; Torres-Larios et al. 2005; Fig. 3). Second, the phosphorothioate-rescue studies above show that changes in P4 structure can nevertheless have dramatic effects on metal ion binding at the reactive phosphate. The indirect effect of helix P4 on catalysis provides a concrete example of metal binding at one position altering metal binding and structure at a different position in an RNA complex. Specifically, the data suggest that structural metal binding in helix P4 leads to specific but indirect stabilization of catalytic metal ions at the scissile phosphate, illustrating potentially profound effects of coupled metal ion interactions on RNA structure and function. Third, we show that the perturbation of the position of the bulged U in P4, which inhibits catalytic metal ion biding (Kaye et al. 2002), leads to a change in the position of pre-tRNA within the active ribozyme–substrate complex. This finding suggests a helix P4-metal interaction with the acceptor stem that positions the reactive phosphate in the active site, providing for optimal catalytic metal ion coordination. Taken together, these new data help to resolve conflicting observations about the roles of helix P4, metal ions, and substrate binding in RNase P (Smith and Pace 1993; Frank and Pace 1997; Christian et al. 2002; Crary et al. 2002; Kazantsev et al. 2005; Torres-Larios et al. 2005).
FIGURE 3.

Three-dimensional representation of Type B RNase P and tRNA adapted from Kazantsev et al. (2005, © National Academy of Sciences, USA). Colored dots in blue, green, orange, and purple represent interactions previously identified between ribozyme and substrate. Yellow dots represent positions involved in the XLU69 cross-link obtained here using E. coli (Type A) RNase P. A black arrow indicates the site of pre-tRNA cleavage.
MATERIALS AND METHODS
RNA synthesis and ribozyme reactions
The synthesis of wild-type and site-specifically modified RNAs have been described previously (Christian et al. 2000). Briefly, wild-type or mutant RNase P ribozyme (E. coli) and wild-type substrate (Bacillus subtilis pre-tRNAASP) were transcribed using T7 polymerase and DNA plasmid under standard conditions. RNAs containing site-specific phosphorothioate (PS) or 4sU modifications were made by ligating two RNA fragments using the method of Moore and Sharp (1992). PS or 4sU modifications were incorporated into short (11-nt) oligonucleotides by chemical synthesis (Dharmacon) and subsequently ligated to transcribed RNA fragments containing the remaining sequence of ribozyme or substrate. Individual stereoisomers of PS modified oligonucleotides were separated by reverse phase HPLC as described by Slim and Gait (1991) prior to ligation.
UV cross-linking
Cross-linking was done under conditions of excess substrate ([S]/[E] > 5) in the presence of CaCl2 at low pH (5.5) to allow RNA folding and substrate binding while suppressing catalysis during the cross-linking reaction. Ribozymes containing 4sU at positions 68, 69, or 70 (see text) and 5′-32P-labeled substrate pre-tRNAs were resuspended separately in 1 M NH4Cl, 50 mM MES (pH 5.5), 0.02% NP40 to a concentration of 200 nM and 1 μM, respectively, and renatured in a PTC-100 programmable thermal controller (MJ Research; 85°C for 3 min, cooled to 65°C at 0.2°C/sec, maintained at 65°C for 5 min, cooled to 50°C at 0.25°C/sec, maintained at 50°C for 15 min, cooled to 37°C at 0.2°C/sec, maintained at 37°C for 15 min). CaCl2 was then added to a final concentration of 25 mM and the RNAs incubated for an additional 15 min at 37°C. Equal volumes (200 μL) of ribozyme and substrate RNAs were combined and allowed to incubate at 37°C for 2 min and then set on ice. Aliquots (12 μL) were placed on a parafilm-covered aluminum block on ice and the samples were irradiated at 366 nm (UVP model UVGL-58) for 15 min through a ⅛-in. glass plate at a distance of 3 cm (Harris et al. 1997). Individual aliquots were then pooled, phenol extracted, and the RNA recovered by ethanol precipitation. Cross-linked RNAs were separated from uncross-linked material on a 6% (19:1) polyacrylamide 8 M urea gel and recovered as described above but with the addition of glycogen (10 μg/mL final concentration) to facilitate precipitation. The locations of individual RNA–RNA cross-links were mapped by alkaline hydrolysis of purified cross-linked RNAs in the presence of 0.1 M NaOH at 37°C for 3 min, followed by the analysis of the cleavage products on a 22.5% (19:1) polyacrylamide 8 M urea gel. Alkaline hydrolysis produces a discrete cleavage ladder from the 32P-labeled 5′ end of the pre-tRNA substrate to the nucleotide immediately adjacent to the site of cross-linking and is separated from slow migrating pre-tRNA fragments cross-linked to RNase P RNA. The site of 4sU cross-linking to the pre-tRNA substrate is thus one nucleotide position further from the 5′-32P-labeled end of pre-tRNA than that indicated by the largest band in the alkaline ladder. The ability of cross-linked RNA to undergo self cleavage was tested by renaturing purified fractions of XL68 and XL69 as described above with the exception that MgCl2 (25 mM final concentration) was added in place of CaCl2 and the RNA allowed to incubate for an additional 30 min at 37°C. Reactions were terminated and analyzed by PAGE as described above and quantified with a Molecular Dynamics Phosphorimager.
Analysis of catalytic metal ion affinity
Experiments to measure apparent metal binding affinity at the pre-tRNA cleavage site took advantage of the observation that Cd2+ can rescue inhibition of reaction rate due to PS substitution of the cleavage site pro-RP oxygen (Warnecke et al. 1999). Quantitative thiophilic metal ion rescue analysis was used to analyze the apparent affinity of the rescuing Cd2+ ions (Shan and Herschlag 1999). In the current work the rate of cleavage (k obs) for pre-tRNA substrate with and without PS substitution was examined in the context of wild-type and mutant (see text) E. coli RNase P RNA. All reactions were single-turnover, with ribozyme (1.5 μM) in excess over 5′-32P-labeled pre-tRNAASP substrate (2 nM). Reactions were carried out at 37°C in 50 mM buffer (MES at pH 6, HEPES at pH 7, 7.5), 0.02% NP40 with a constant background of 10 mM MgCl2 to insure folding of ribozyme and substrate (Pan et al. 1999), and varing concentrations (0–40 mM) of CdCl2. Ribozyme and substrate were annealed separately as described above with the exception that MgCl2 and CdCl2 were added in place of CaCl2. Reactions were initiated by combining annealed ribozyme and substrate RNA. Ten 2-μL aliquots were removed from the reaction mixture (40 μL) at specific times to insure representative analysis of at least three reaction half-lives (≥3 t 1/2). Reaction of the aliquot was quenched by mixing with 6 μL of stop solution (90% formamide, 125 mM EDTA, 0.005% xylene cyanole, 0.01% bromphenyl blue, and 1 mM Tris at pH 7.5). Reaction products were separated on a 22.5% (19:1) polyacrylamide 8 M urea gel and the conversion to product at each time point quantified with a Molecular Dynamics PhosphorImager.
Analysis of reaction kinetics
The pseudo-first-order rate constant (k obs) was determined by fitting plots (KaleidaGraph, Synergy Software) of the fraction of RNA cleaved as a function of time to an equation for a single exponential:
 |
where [P] is the concentration of product formed at time t, [S]total is the initial substrate concentration added to the reaction, A represents the maximal extent of the reaction, and B is the amplitude of the exponential.
To control for nonspecific effects on ribozyme structure due to changes in divalent ion concentration, the relative reaction rate (k rel) was calculated from the ratio of the observed reaction rates (k obs) of molecules that differ by the single sulfur substitution (e.g., k rel = k obs s/k obs o). Plots of k rel versus added Mn2+ concentration ([Cd2+]) were fit with a nonlinear form of the Hill equation for cooperative binding:
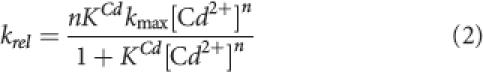 |
where n is the Hill coefficient that gauges the cooperativity of Cd2+ binding (n = 1 represents no cooperativity) and K Cd represents the Cd2+ concentration required to attain half the maximal rate (k max) of self-cleavage at saturating Cd2+ levels. It is important to note that, for the reactions performed here in the presence of a constant background of 10 mM Mg2+, there was essentially no inhibition of the observed reaction rate due to the presence of Cd2+ even at the highest concentrations tested (40 mM).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.158606.
REFERENCES
- Beebe J.A., Kurz J.C., Fierke C.A. Magnesium ions are required by Bacillus subtilis ribonuclease P RNA for both binding and cleaving precursor tRNAAsp. Biochemistry. 1996;35:10493–10505. doi: 10.1021/bi960870m. [DOI] [PubMed] [Google Scholar]
- Christian E.L., Kaye N.M., Harris M.E. Helix P4 is a divalent metal ion binding site in the conserved core of the ribonuclease P ribozyme. RNA. 2000;6:511–519. doi: 10.1017/s1355838200000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian E.L., Kaye N.M., Harris M.E. Evidence for a polynuclear metal ion binding site in the catalytic domain of ribonuclease P RNA. EMBO J. 2002;21:2253–2262. doi: 10.1093/emboj/21.9.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary S.M., Kurz J.C., Fierke C.A. Specific phosphorothioate substitutions probe the active site of Bacillus subtilis ribonuclease P. RNA. 2002;8:933–947. doi: 10.1017/s1355838202025025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedor M.J., Williamson J.R. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- Frank D.N., Pace N.R. In vitro selection for altered divalent metal specificity in the RNase P RNA. Proc. Natl. Acad. Sci. 1997;94:14355–14360. doi: 10.1073/pnas.94.26.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.E., Christian E.L. Recent insights into the structure and function of the ribonucleoprotein enzyme ribonuclease P. Curr. Opin. Struct. Biol. 2003;13:325–333. doi: 10.1016/s0959-440x(03)00069-1. [DOI] [PubMed] [Google Scholar]
- Harris M.E., Kazantsev A.V., Chen J.L., Pace N.R. Analysis of the tertiary structure of the ribonuclease P ribozyme-substrate complex by site-specific photoaffinity crosslinking. RNA. 1997;3:561–576. [PMC free article] [PubMed] [Google Scholar]
- Kaye N.M., Zahler N.H., Christian E.L., Harris M.E. Conservation of helical structure contributes to functional metal ion interactions in the catalytic domain of ribonuclease P RNA. J. Mol. Biol. 2002;324:429–442. doi: 10.1016/s0022-2836(02)01094-x. [DOI] [PubMed] [Google Scholar]
- Kazantsev A.V., Krivenko A.A., Harrington D.J., Holbrook S.R., Adams P.D., Pace N.R. Crystal structure of a bacterial ribonuclease P RNA. Proc. Natl. Acad. Sci. 2005;102:13392–13397. doi: 10.1073/pnas.0506662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz J.C., Fierke C.A. Ribonuclease P: A ribonucleoprotein enzyme. Curr. Opin. Chem. Biol. 2000;4:553–558. doi: 10.1016/s1367-5931(00)00131-9. [DOI] [PubMed] [Google Scholar]
- Moore M.J., Sharp P.A. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- Pan T., Fang X., Sosnick T. Pathway modulation, circular permutation, and rapid RNA folding under kinetic control. J. Mol. Biol. 1999;286:721–731. doi: 10.1006/jmbi.1998.2516. [DOI] [PubMed] [Google Scholar]
- Shan S.O., Herschlag D. Probing the role of metal ions in RNA catalysis: Kinetic and thermodynamic characterization of a metal ion interaction with the 2′-moiety of the guanosine nucleophile in the Tetrahymena group I ribozyme. Biochemistry. 1999;38:10958–10975. doi: 10.1021/bi990388e. [DOI] [PubMed] [Google Scholar]
- Shan S., Kravchuk A.V., Piccirilli J.A., Herschlag D. Defining the catalytic metal ion interactions in the Tetrahymena ribozyme reaction. Biochemistry. 2001;40:5161–5171. doi: 10.1021/bi002887h. [DOI] [PubMed] [Google Scholar]
- Slim G., Gait M.J. Configurationally defined phosphorothioate-containing oligoribonucleotides in the study of the mechanism of cleavage of hammerhead ribozymes. Nucleic Acids Res. 1991;19:1183–1188. doi: 10.1093/nar/19.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D., Pace N.R. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- Smith D., Burgin A.B., Haas E.S., Pace N.R. Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J. Biol. Chem. 1992;267:2429–2436. [PubMed] [Google Scholar]
- Torres-Larios A., Swinger K.K., Krasilnikov A.S., Pan T., Mondragon A. Crystal structure of the RNA component of bacterial ribonuclease P. Nature. 2005;437:584–587. doi: 10.1038/nature04074. [DOI] [PubMed] [Google Scholar]
- Warnecke J.M., Furste J.P., Hardt W.D., Erdmann V.A., Hartmann R.K. Ribonuclease P (RNase P) RNA is converted to a Cd(2+)-ribozyme by a single Rp-phosphorothioate modification in the precursor tRNA at the RNase P cleavage site. Proc. Natl. Acad. Sci. 1996;93:8924–8928. doi: 10.1073/pnas.93.17.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke J.M., Held R., Busch S., Hartmann R.K. Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis . J. Mol. Biol. 1999;290:433–445. doi: 10.1006/jmbi.1999.2890. [DOI] [PubMed] [Google Scholar]