

Summary
Mutations at CPC1 disrupt assembly of a central pair microtubule-associated complex and alter flagellar beat frequency in Chlamydomonas. Sequences of wild-type genomic clones that complement cpc1, and of corresponding cDNAs, reveal the gene product to be a 205 kDa protein with two predicted functional domains, a single EF hand motif near the C-terminus and an unusual centrally located adenylate kinase domain. Homologs are expressed in mammals (testis and tracheal cilia) as well as ciliated lower eukaryotes. Western blots confirm that Cpc1 is one of six subunits in a 16S central pair-associated complex. Motility defects associated with cpc1 alleles in vivo are partially rescued in vitro by reactivation of axonemes or cell models in saturating concentrations of ATP; thus the Cpc1 complex is essential for maintaining normal ATP concentrations in the flagellum.
Keywords: Cilia, Flagella, Central pair, Motility, Chlamydomonas, Adenylate kinase
Introduction
The central pair complex of 9+2 eukaryotic cilia and flagella is an essential regulator of axonemal dynein activity. Absence of the entire central pair complex causes complete lack of motility as seen in both humans (Baccetti et al., 1979) and Chlamydomonas (Warr et al., 1966; Witman et al., 1978). Studies of the pattern and rate of outer doublet sliding in protease-treated axonemes support models in which proteins projecting from central pair microtubules interact with radial spokes; these interactions in turn transmit signals that regulate dynein activity on outer doublet microtubules (Smith and Sale, 1992; Yoshimura and Shingyoji, 1999; Wargo and Smith, 2003). Ultimately regulation of dynein activity involves spoke-dependent modulation of protein kinases and phosphatases associated with outer doublets (Habermacher and Sale, 1997; Porter and Sale, 2000; Smith, 2002a; Smith, 2002b). However, the mechanism of signal transduction from the central pair to radial spokes is presently unknown. Few central pair-associated proteins have been identified and the transient interaction between central pair and radial spoke proteins remains uncharacterized. Here we focus on Cpc1, a protein essential for assembly of a large central pair projection complex that may interact with radial spokes.
The central pair consists of two microtubules and at least 25 associated proteins (Adams et al., 1981; Dutcher et al., 1984). Central pair-associated proteins link the two microtubules together and form projections that support a cylindrical cage of filaments surrounding the microtubules (for reviews, see Smith and Lefebvre, 1997; Smith and Yang, 2004). This cage is discontinuous in specific radial positions around the circumference of the central pair cylinder, and each discontinuity is hypothesized to be the location of a specific central pair-radial spoke interaction site (Mitchell, 2003a). Mutations at the CPC1 locus disrupt assembly of a large portion of the central pair complex that includes structures attached to both the C1 and C2 microtubules (Mitchell and Sale, 1999). These structures repeat every 16 nm along the central pair and could potentially transmit signals to radial spokes and dyneins distributed along the entire flagellum (Mitchell, 2003a). The cpc1 mutation results in flagella that beat with near normal waveform but reduced beat frequency (38 Hz compared with the normal 60 Hz during forward swimming in this organism).
Biochemical comparisons of cpc1 mutant and wild-type flagella by SDS-PAGE revealed the absence of at least four central pair proteins in the mutant. When these proteins were extracted from wild-type flagella, they co-sedimented on sucrose density gradients as a large (16S) complex containing six polypeptides, none of which have been characterized at the molecular level (Mitchell and Sale, 1999). In the hope that further characterization of the Cpc1 protein might increase our understanding of mechanisms that regulate flagellar dynein activity, we have cloned and sequenced the CPC1 gene, raised antibodies against the gene product that identify it as a subunit of the 16S complex and further characterized motility defects associated with the cpc1 mutation. One conserved domain within Cpc1 is homologous to adenylate kinases, which function in many ciliary and flagellar systems to regenerate ATP from ADP. We present evidence that the phenotype of cpc1 cells results in part from an inability to maintain normal ATP concentrations within the flagellar compartment.
Materials and Methods
Chlamydomonas strains and growth
Chlamydomonas reinhardtii strains 137c (CC 124), arg2 (CC 1930), arg7 (CC 1931), pf16 (CC 624) and pf18 (CC 1036) were obtained from Elizabeth Harris at the Chlamydomonas genetics center, Duke University. Both mating types of uni1-1 were obtained from David Luck. Mutant strains cpc1-1 (CC 3707) and cpc1-2 (CC 3708), described previously (Mitchell and Sale, 1999), were backcrossed at least four times into the 137c background before further crosses with arg2, arg7, or uni1-1 to create double-mutant strains. All in vitro reactivation and biochemical studies used the cpc1-2 allele. Both cpc1-1 and cpc1-2 alleles were used for transformation rescue experiments. All genetic manipulations followed standard procedures (Harris, 1989). Glass bead-mediated transformation and selection of transformants used ARG plasmid pARG7.8, as previously detailed (Mitchell and Sale, 1999).
Molecular biology
Hybridization probes were labeled with digoxygenin and detected by chemiluminescence (Roche, Indianapolis, IN) on Biomax ML film (Kodak, Rochester, NY). Wild-type genomic clones for the CPC1 gene were identified by hybridization of a bacterial artificial chromosome (BAC) library filter (Genome Systems Inc, St Louis) with a unique 710 bp MscI-PstI fragment of the insertion site clone from mutant allele cpc1-2 (Mitchell and Sale, 1999). To isolate cDNA clones, gel-purified restriction fragments of genomic subclone pES10 were digoxygenin-labeled and used to probe filter lifts of a lambda gt10 cDNA library (Wilkerson et al., 1994). All selected cDNAs were excised from lambda gt10 with EcoRI and subcloned into the EcoRI site of pBluescript KS(+) (Stratagene, LaJolla, CA). Genomic and cDNA clones were sequenced by the University of Iowa DNA core facility. Sequence assembly and analysis of predicted translation products, including determination of similarity and identity, used Vector NTI Advance, version 9.0 (Invitrogen, Carlsbad, CA); database comparisons used BLAST (Altschul et al., 1997) at NCBI with default parameters. Expressed sequence tags (ESTs) that aligned with CPC1 were identified through analysis of tracks on the Chlamydomonas Genome v1.0 (http://genome.jgi-psf.org/chlre1/chlre1.home.html) and confirmed by BLAST searches of Chlamydomonas ESTs. Sequence data available on the genome browser were produced by the US Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) and are provided for use in this publication only. Secondary structure predictions were based on the PROF program as accessed through the PredictProtein server (Rost and Sander, 1993; Rost and Liu, 2003). Coiled coil structures were predicted using the programs Paircoil and Multicoil (Berger et al., 1995; Wolf et al., 1997).
Generation of antibodies
Polyclonal antibodies to Cpc1 were raised against two fusion proteins that encoded regions near the N-terminus (GSTCpc1A) or C terminus (GSTCpc1D), created as follows. A 951-bp region of cDNA clone pCPC1A, encoding amino acids (a.a.) 63–379 of Cpc1, was released with PstI and cloned into the PstI site of pBS KS(+) to make pBCPC1A. The pBCPC1A insert was then released with BamHI and XhoI and cloned between BamHI and XhoI sites of GST expression vector pGEX-4T-2 (Amersham Pharmacia Biotech, Piscataway, NJ) to make pCPC1ExA. The entire insert (930 bp) and flanking vector sequences of cDNA clone pCPC1D, encoding a.a. 1110–1419 of Cpc1, was released with SmaI and EcoRV and cloned into SmaI-digested pGEX-2T to generate pCPC1ExD. Fusion proteins expressed in Escherichia coli BL21 (DE3) pLysS cells (Stratagene) were gel-purified and used to raise polyclonal antibodies in rabbits (Covance, Princeton, NJ). Specific antibodies were affinity purified using blot strips of fusion protein as an affinity matrix. An approved animal use protocol is on file with the SUNY Upstate institutional Committee for the Humane Use of Animals.
Flagellar isolation and fractionation
Cells grown in acetate-enriched medium (Sager and Granick, 1953) were deflagellated with 5 mM dibucaine (Witman, 1986). Membranes were removed from flagella by resuspension in HMDEK [30 mM HEPES, 5 mM MgSO4, 1 mM DTT, 0.5 mM EGTA, 25 mM potassium acetate, 1 mM phenylmethylsulfonyl fluoride (PMSF), pH 7.4] and mixing with an equal volume of HMDEK containing 0.4% NP-40. To prepare pf16 axonemes that displayed instability of the C1 microtubule, HMDEK was replaced by HMDEN (10 mM HEPES, 5 mM MgSO4, 1 mM DTT, 0.5 mM EDTA, 30 mM NaCl, 1 mM PMSF, pH 7.4). To remove C2, axonemes were extracted with HMDEK containing 0.6 M NaCl for 30 minutes on ice, centrifuged for 20 minutes at 4°C and resuspended in HMDEK before addition of SDS-PAGE sample buffer. The C1 microtubule and its associated structures were solubilized by treating NaCl-extracted axonemes with 0.2 M KI in HMDEK (Mitchell and Sale, 1999). Proteins in the KI extract were further separated by dialysis into HMDEK and centrifugation on a 5-20% sucrose gradient (prepared with HMDEK) at 155,000 g (average of 218,000 g at bottom of tube) for 16 hours in an SW40 rotor. Gradients were collected from the bottom of the tube in 0.5 ml fractions. Protein composition was determined by SDS-PAGE on 6% acrylamide gels stained with Coomassie Blue or silver (Wray et al., 1981), as indicated. Dynein heavy chains (average Mr 500,000) and Benchmark Protein Ladder (Life Technologies) were used as protein size-standards. For immunoblotting, proteins were transferred to Immobilon-P membranes (Millipore, Bedford, MA) and probed with affinity-purified polyclonal anti-Cpc1A at 1:100 dilution. Antibodies were detected with peroxidase-labeled goat-anti-rabbit (BioRad, Hercules, CA) developed with ECL reagents (Amersham). For electron microscopy, axonemes were fixed and processed as previously described (Mitchell and Sale, 1999). Negatives taken on a JEOL 100CX II were digitized on a UMAX (Taiwan) PowerLook II flat-bed scanner. Images were adjusted for uniform density and contrast in Photoshop v6.0 (Adobe).
Motility
Motility of transformant colonies was determined by transferring arg+ colonies with sterile toothpicks into microtiter wells containing 200 μl minimal medium. After 24 hours, velocity was visually compared with the wild type and cpc1 controls under dark-field illumination on an Olympus SZ60 stereo microscope. Flagellar and axonemal beat frequencies were measured by tuning the frequency of stroboscopic illumination under dark-field illumination on a Zeiss Axioskop microscope, as previously described (Mitchell and Kang, 1991). A 630 nm red cut-off filter was used to reduce phototactic responses during live cell beat frequency measurements. Cell model reactivation (Horst and Witman, 1998) utilized reactivation buffer that contained 1 mM ATP or ADP and 10–8 M Ca2+. Some model reactivation experiments were performed on uniflagellated cells produced in the uni1-1 mutant background. Reactivation of axonemes was performed by resuspension of detergent-extracted axonemes in 10–8 M Ca2+ reactivation buffer lacking nucleotide, followed by dilution of axonemes 1:10 into reactivation buffer containing 1 mM ADP or ATP. Frequency measurements were restricted to axonemes beating freely in solution parallel to the cover slip or slide surface. The microscope stage was maintained at 22°C with an HCC-100A temperature controller attached to a stage-mounted HE-102 heat exchanger (Dagan Corp, Minneapolis, MN).
Adenylate kinase assays
Adenylate kinase activity was assayed essentially as described (Gibbons, 1966). The assay was initiated by adding 9 volumes assay buffer to 1 volume of axonemes in HMDEK to produce final concentrations of 30 mM HEPES, pH 7.4, 3 mM MgSO4, 10 mM glucose, 2 mM ADP, 1 mM NADP and 5 U/ml of a 2:1 mixture of hexokinase and glucose-6-phosphate dehydrogenase. Assay buffer was pre-incubated at room temperature for 10 minutes before the start of the reaction to eliminate any contaminating ATP in the ADP preparation. Activity was monitored by the linear rate of increase in absorbance at 340 nm. All assay components were from Sigma. Axonemal protein concentration was estimated using a dye-binding assay (BioRad).
Results
Cloning the CPC1 locus
To identify the gene product of CPC1, we began with analysis of sequences flanking the insertion site in one mutant allele, cpc1-2, which was previously cloned by plasmid rescue of a 4.2 kb PstI fragment from mutant DNA (Mitchell and Sale, 1999). The 1.4 kb insert of this plasmid consisted of two Chlamydomonas genomic fragments separated by 200 bp of pUC vector sequences (Fig. 1A). Hybridization of part of this insert (60A8L) to genomic Southern blots generated restriction fragment length polymorphisms between wild-type strain 137c and both of our insertional alleles, cpc1-1 and cpc1-2 (Fig. 1B), indicating that insertions in both alleles occurred close to the position of this probe. The 60A8L fragment was therefore chosen as a marker for the CPC1 gene.
Fig. 1.
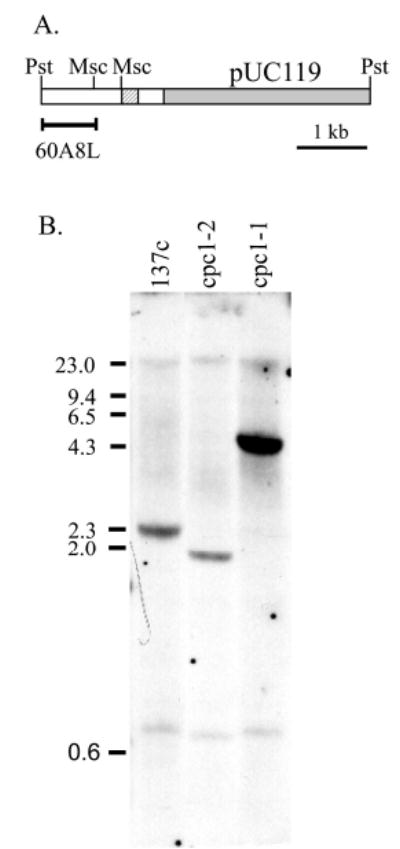
(A) Restriction map of the plasmid rescued from insertional allele cpc1-2, which contains a complete pUC119 vector (shaded) and another small fragment of pUC119 (hatched) embedded within Chlamydomonas genomic sequences (unshaded). (B) Southern blot of genomic DNA from Chlamydomonas wild-type strain 137c and from two insertional alleles of cpc1, digested with PvuII and probed with PstI-MscI fragment labeled 60A8L in (A). Hybridization of this probe to bands that differ from the wild type in both mutants indicates that 60A8L is close to the insertion site in both alleles.
Hybridization screening of a large-insert bacterial artificial chromosome (BAC) library of wild-type genomic DNA with the 60A8L fragment selected six BAC clones, which were restriction mapped and tested for their ability to rescue the cpc1-2 allele to a wild-type swimming speed. These BACs formed a 125-kb contig. As shown in Fig. 2A, complementation experiments identified BAC 28H16 as the smallest BAC that complemented the mutation, and showed that a 15-kb region of overlap between BACs 28H16 and 6H3 contained a functional copy of the CPC1 gene. Although the co-transformational rescue rate was below 4% for most of these experiments (last column in Fig. 2A), we never observed rescue to wild-type motility in the absence of BAC DNA or in the presence of DNA from unrelated BAC clones (negative control transformations were included in every experiment). Mapping of the relationships between these six BAC clones was aided by a database of BAC end sequences (http://bahama.jgi-psf.org/prod/bin/chlamy/plate2seq.cgi), which also provided the orientation of each BAC clone (designated as x and y ends in the database and in Fig. 2A). The CPC1 gene has been mapped to linkage group III between PTX2 and AC208 (Kathir et al., 2003) and its chromosomal orientation was determined by maps (http://www.biology.duke.edu/chlamy_genome/BAC/index.html) that link the orientation of the BACs shown in Fig. 2 to LG III markers near CPC1 (arrows in Fig. 2A).
Fig. 2.
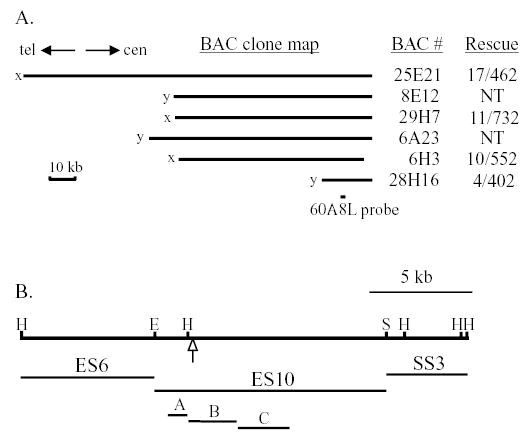
(A) Map of overlapping bacterial artificial chromosome (BAC) clones selected with cpc1 insertion site probe 60A8L. Indicated to the right are the number of fast-swimming (rescued) transformants and the total number of arg+ colonies screened. NT, not tested. All four tested BAC clones rescued the cpc1 mutant phenotype, indicating that the CPC1 gene lies in the region of overlap between BACs 6H3 and 28H16. The orientation of this BAC contig on chromosome III is indicated by arrows pointing toward the telomere (tel) and centromere (cen). (B) Restriction map of BAC 28H16 (top line) shows the location of the insertion site in mutation cpc1-2 (arrow). Subclones ES6, ES10 and SS3, and three smaller fragments of ES10 (A, B, C) shown below the restriction map were tested for their ability to rescue the mutation. Rescue was only seen with ES10. H, HindIII; E, EcoRI; S, SalI.
Transformation with BAC 28H16 DNA that had been digested with restriction enzymes showed that the gene was disrupted by HindIII but not by EcoRI or SalI (Table 1). When three plasmids containing the three fragments generated by EcoRI and SalI digestion of BAC 28H16 (Fig. 2B) were tested for their ability to complement the cpc1-2 mutation, only the largest of these, ES10, rescued mutant cells to wild-type motility (15 wild type out of 672 colonies screened). Flagellar beat frequency of rescued strains was indistinguishable from that in the wild type (57±4 Hz, n=15). As a further test of the ability of this plasmid to rescue the cpc1 defect, axonemes from transformants were examined by thin section electron microscopy. As shown in Fig. 3, C1-associated projection 1b, which is missing in mutant axonemes, is restored in ES10 transformants of cpc1-2. Identical results were obtained by transformation of a cpc1-1 strain (unpublished data). All necessary coding regions for a functional CPC1 gene product are therefore present on the ES10 fragment.
Table 1.
Restriction digestion to delimit the CPC1 gene on BAC28H16
| Transforming DNA | Number rescued/number testeda |
|---|---|
| – | 0/372 |
| Uncut | 4/336 |
| HindIII | 0/276 |
| SalI | 5/256 |
| EcoRI | 27/960 |
The number of arg+ colonies that swam at wild-type velocity when no DNA (–) or 10 μg of BAC 28H16 DNA, treated as noted in the first column, was mixed with 2 μg of ARG plasmid and co-transformed into cpc1-2arg2 cells.
Fig. 3.

Thin-section electron micrographs of wild-type Chlamydomonas axonemes (WT), cpc1-2 axonemes and axonemes from cpc1 cells transformed with plasmid ES10 [cpc1-2(ES10)]. Diagram to the right summarizes central pair structures as seen in cross sections (modified from Mitchell and Sale, 1999). Central pair structures missing in cpc1 are fully restored in transformants (arrows). Bar, 100 nm.
The ES10 insert was fully sequenced, and three smaller fragments indicated as A, B and C in Fig. 2B were used to screen a size-selected lambda gt10 cDNA library. Five cDNA clones spanning a total of 4.5 kb were sequenced and found to overlap, except for a short gap (9 nucleotides) between the ends of cDNA clones a and c. The sequence of this gap was determined directly from sequence analysis of ES10. These cDNAs did not contain the 3′ end of a coding region, but sequences from clones c and d overlapped with expressed sequence tag (EST) AV626984. We obtained the corresponding cDNA clone from the Kazusa DNA Research Institute (Asamizu et al., 1999) and used its sequence to identify ESTs that extend our cDNA sequences in the 3′ direction by an additional 1.7 kb for a predicted total mRNA size of 6.2 kb (Fig. 4A, right-facing arrow). Genomic clone pES10 extends an additional 420 bp upstream of cDNA a and 1.8 kb downstream of the furthest 3′ EST and overlaps part of the 3′ UTR of an adjacent gene (left-facing arrow in Fig. 4A), as determined by sequence identity with two ESTs (BI726615 and BQ824695). The 3′ ends of these two transcription units are separated by 77 bp.
Fig. 4.
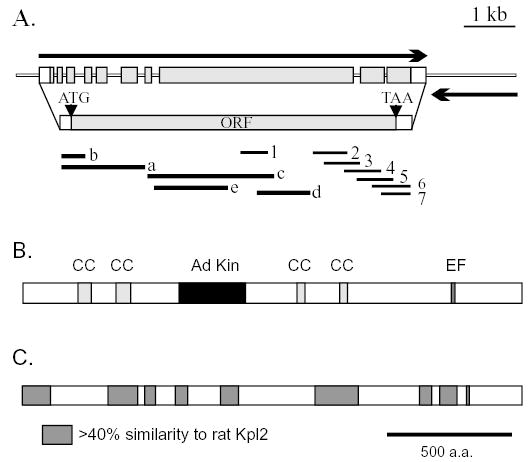
Diagram of transcription units in ES10 and the domain structure of the predicted translation product, Cpc1. (A) Directional arrows show one complete and one partial transcription unit in ES10. The left-pointing partial transcription unit was identified from BLAST searches of EST databases and with the ES10 sequence and includes only the 3′ untranslated region of a gene as reported in the Chlamydomonas genome database v1.0 (predicted product genie 3.63). The right-pointing complete transcription unit and corresponding ORF were identified from sequence analysis of ES10, cDNA clones a-e (see text) and EST sequences 1-7 (corresponding to AV626984, BU650028, BE337526, BG857322, BG859077 and BE337525, respectively). (B) Distribution of sequences predicted to form α-helical coiled coils (CC) and sequences with similarity to adenylate kinases (Ad Kin) and to calcium binding domains (EF). (C) Distribution of regions with highest similarity to a mammalian homolog, rat Kpl2 (AAD56310). Sequences of genomic clone ES10 and the CPC1 cDNA have been deposited in GenBank (Accession no. AY601881 and AY601882).
Based on the presence of a single complete transcription unit in pES10, on the ability of this clone to complement cpc1 and on the site of the insertion that generated allele cpc1-2, marked by an arrowhead in Fig. 2B, this transcription unit is the CPC1 gene. It contains a single large ORF (Fig. 4A) whose predicted translational start site fits well to the consensus based on other Chlamydomonas genes. The gene spans ten exons and has a 5′ UTR of at least 191 nucleotides and a 3′ UTR of 284 nucleotides. A single polyadenylation signal sequence of TGTAA appears 14 bp upstream of the polyadenylation site in the CPC1 EST sequences.
Characterization of the CPC1 gene product
The CPC1 transcript encodes a predicted protein of 205 kDa. Structure prediction and homology search programs reveal several distinct domains within a predominantly (66%) α-helical protein. Four blocks of sequence are predicted to form α-helical coiled-coils at more than 50% probability with one or more prediction algorithms (Fig. 4B) which suggests that much of Cpc1 functions as an extended structural protein. However, between the second and third coiled-coil domains is a sequence with several blocks of homology to adenylate kinase family members, and near the carboxyl terminus is a sequence that matches the consensus for a single calcium binding (EF hand) domain. The EF hand domain (Fig. 5A) retains all of the residues common to this motif, including those involved in coordination of Ca2+ (underlined in Fig. 5A), and occurs in the context of the helix-loop-helix pattern typical of Ca2+-binding EF hands.
Fig. 5.
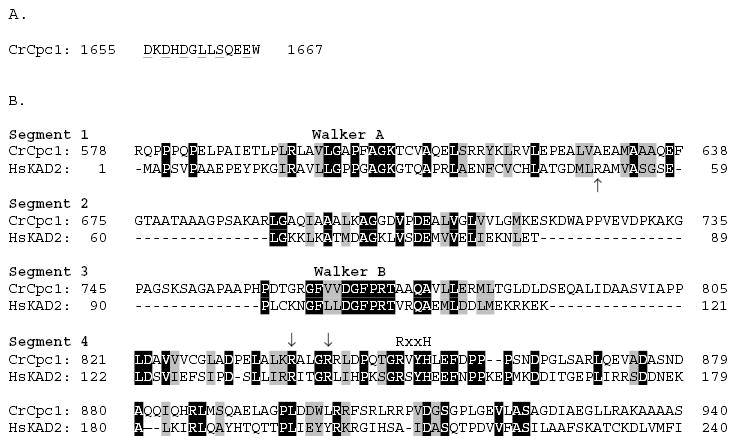
(A) Cpc1 sequences predicted to fold into an EF hand, with residues important for coordination of Ca2+ underlined. (B) Alignment of portions of the Cpc1 adenylate kinase domain with the complete sequence of human adenylate kinase isoform 2 (AAC52061). Identical residues are shaded black, conservative replacements in grey. Cpc1 sequences homologous to adenylate kinase are separated into four segments, with segment 1 spanning the phosphate binding loop (P-loop, Walker A motif) and segment 3 encompassing the highly conserved Walker B motif. Arrows (segments 1 and 4) indicate arginine residues that may be important for substrate interactions. The RxxH sequence in segment 4, part of the LID domain, is common to most eukaryotic adenylate kinases.
BLAST searches with Cpc1 sequences outside the region of adenylate kinase similarity identified homologs in several mammalian species, most represented by ESTs from tissues with motile cilia or flagella (trachea, lung, brain, testis). The best match, Kpl2, represents the predicted product of a previously characterized full length cDNA sequence that was selected in a screen for mRNAs upregulated during ciliogenesis in rat tracheal explants (Ostrowski et al., 1999), whereas the remainder are all partial sequences of hypothetical proteins based on genomic predictions or cDNA sequences. The human Kpl2 homolog was also identified as a ciliary protein using proteomics (Ostrowski et al., 2002). Fig. 4C indicates that regions of Chlamydomonas Cpc1 with greater than 40% similarity to rat Kpl2 are found over most of the length of these large proteins. These regions include the 100 N-terminal amino acids, two of the predicted coiled coil domains, conserved regions of the adenylate kinase domain, and the region of homology to EF hand proteins. Queries with the N-terminus (a.a. 1–110) of Cpc1 produced single significant BLAST hits in genomes of a fish, Fugu rubripes (E value 2E-09), a sea urchin, Stronggylocentrotus purpuratus (4E-14), a ciliate, Tetrahymena thermophila (8E-04), and an apicomplexan flagellate, Toxoplasma gondii (8E-06), but not in a fungus (Saccharomyces cerevisiae), an insect (Drosophila melanogaster) and a round worm (Caenorhabditis elegans). Homologs of Cpc1 are therefore likely to be present in most organisms with motile cilia or flagella.
Alignment of the Cpc1 adenylate kinase domain with the complete sequence of a more typical adenylate kinase (human KAD2) shows that this domain is separated in Cpc1 into four segments by linker sequences of low complexity (Fig. 5B), but it still retains all the sequence features that place it closer to the adenylate kinase family of P-loop kinases than to another NMP kinase family or any of the families of the GTPase clade. The Cpc1 Walker A motif, which retains the hallmarks of P-loops common to both kinases and nucleotidases (Walker et al., 1982), ends in a threonine (GKT) and thus more closely resembles the P loops of other NMP kinases such as UMP kinases, whereas glycine (GKG) is found in the same position in most AMP kinases (Leipe et al., 2003). Significance of this variant residue has not been characterized but GKT P-loops appear in the sequences of adenylate kinase homologs of some prokaryotes (e.g. Campylobacter jejuni, GenBank Accession no. Q9PHM8) and lower eukaryotes (Leishmania donovani, GenBank Accession no. AAD39831). The Walker B motif of Cpc1 includes a perfect copy of the DGFPRT sequence, which is only found among AMP kinases and has been used as an identifying signature of this P-loop kinase family. The two arginine residues marked with arrows in segment 4 interact with the adenine base moiety and gamma phosphate respectively, of ATP in several families of P-loop kinases (Leipe et al., 2003). Conservation of these residues in Cpc1 supports functional interaction with ATP for this protein. The region immediately following these arginines includes an RxxH sequence, which is a conserved feature in the base-interacting LID domain of eukaryotic and some bacterial AMP kinases, but is not present in Archaeal AMP kinases or in UMP or CMP kinases (Leipe et al., 2003). Its presence further supports the identification of Cpc1 as a member of the eukaryotic AMP kinase family. One feature missing from Cpc1 is an arginine, present in most adenylate kinases including human KAD2, located carboxyterminal to the Walker A motif (arrow in Fig. 5B, segment 1), which has been shown to bind the alpha phosphate of AMP in the crystal structure of the E. coli enzyme (Muller and Schulz, 1992). Other basic residues in the primary sequence of Cpc1 may have taken over this function, as arginines located carboxyterminal to the Walker B motif fulfil this function in other NMP kinases (Leipe et al., 2003).
The most striking divergence of Cpc1 from the typical adenylate kinases is the location of this enzymatic domain within a much larger protein and the presence of three extended linker sequences between the conserved motifs. Extended amino-terminal α-helical coiled-coil domains and linker domains similar to those in Cpc1 appear in all of the close homologs of Cpc1, such as rat Kpl2, as well as in the vertebrate KAD7 family members (e.g., human KAD7, GenBank Accession no. Q96M32). These linkers may have little effect on the structure of catalytic regions in Cpc1. The lengths of sequences that connect catalytically important structural domains among the many families of P-loop kinases and nucleotidases are quite variable, even though the basic P-loop fold shows little divergence in three-dimensional architecture (Muller and Schulz, 1992; Cheek et al., 2002).
Biochemistry of the Cpc1 complex
Location of the Cpc1 protein within flagellar fractions was determined with western blots using antibodies directed against GST fusion protein Cpc1A. On western blots of demembranated axonemes from wild-type flagellar samples, this antibody detected a single protein that migrates at approximately 265 kDa (Fig. 6). The 265 kDa band is absent from axonemal proteins of cpc1 or central pair assembly mutant pf18, and is reduced in abundance in axonemes from pf16, a mutation that renders the C1 central pair microtubule unstable during axoneme isolation (Dutcher et al., 1984). We then used this antibody to test the solubility of Cpc1 following successive extractions of wild-type axonemes with 0.5 M NaCl, which removes the C2 microtubule and 0.2 M KI, which solubilizes the C1 microtubule. As anticipated, Cpc1 remains in the pellet after extraction with sodium chloride, but fails to pellet after extraction with potassium iodide (Fig. 7A,B). Our antibody identifies Cpc1 co-migrating with the smallest of the four HMW central pair microtubule-associated bands (CP1-CP4) extracted by potassium iodide. We previously showed that CP4 co-purifies with several other proteins in a 16S complex, and that additional material of the same apparent size as CP4 sedimented in a second peak at about 9S (Mitchell and Sale, 1999). To see if Cpc1 is identical to CP4, the KI soluble material (Fig. 7A, lane KS) was sedimented through a sucrose gradient and resulting fractions were analyzed by SDS-PAGE and immunoblotting (Fig. 8). Western blots show that most Cpc1 sediments near the bottom of the gradient (fractions 5 and 6) but a second minor peak sediments above the middle of the gradient (fraction 10), corresponding to the16S and 9S peaks of CP4 (Mitchell and Sale, 1999). Of the five proteins that co-sediment with Cpc1 at 16S (apparent sizes of 350 kDa, 265 kDa, 135 kDa, 79 kDa and 56 kDa; arrows in Fig. 8B), only the 265 kDa (Cpc1) and 56 kDa bands sediment in the 9S region. None of these bands are seen in KI extracts from cpc1-2 axonemes (Fig. 8A).
Fig. 6.
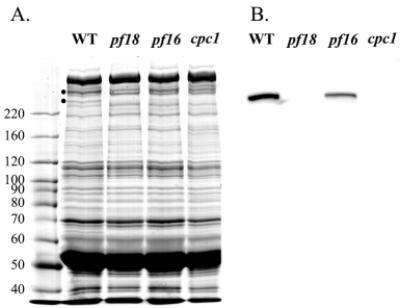
Western blotting of flagellar axonemes from wild-type cells and three central pair defective Chlamydomonas strains. (A) 6% SDS polyacrylamide gel of wild-type and pf18, pf16 and cpc1 mutant axonemes stained with Coomassie blue. Dots next to the wild-type sample mark two high molecular weight proteins, CP3 and CP4, missing from central pair assembly-defective strain pf18. The sizes of molecular weight standards (in kDa) are shown along the left edge. (B) Western blot of the gel in (A) probed with anti-Cpc1A. A single band is seen in the wild type and at reduced amounts in pf16 axonemes. The immunoreactive band aligns with the location of CP4 in (A).
Fig. 7.
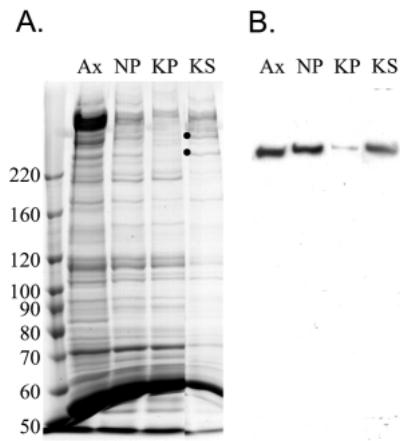
Purification of Cpc1 by Chlamydomonas flagellar fractionation. (A,B) 6% SDS polyacrylamide gel (A) and corresponding western blot (B) of wild-type axonemes (Ax), the insoluble material following successive extractions with 0.5 M NaCl (NP) and 0.2 M KI (KP) and the material solubilized by KI (KS). Western blotting with anti-Cpc1A shows that Cpc1 is extracted by KI. Stoichiometric amounts were loaded in each lane. Dots in (A) mark the location of high molecular weight central pair proteins CP3 and CP4.
Fig. 8.
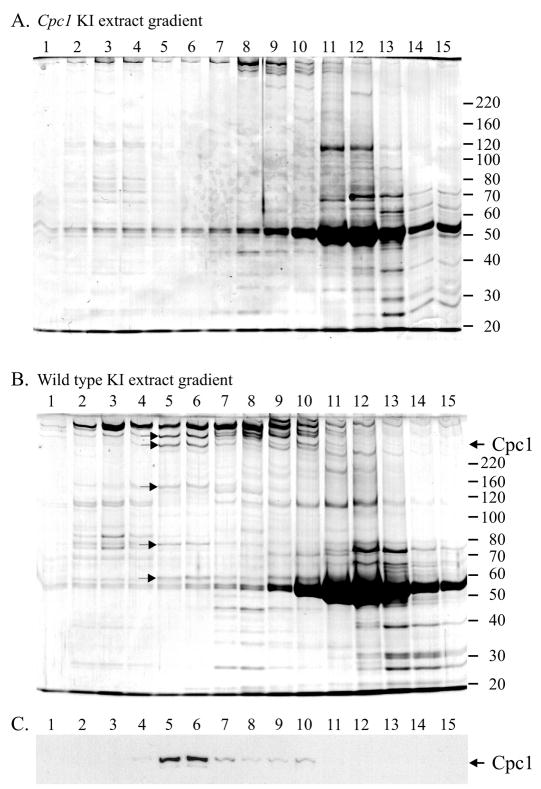
Sucrose density gradient analysis of KI extracts from Chlamydomonas axonemes. (A,B) cpc1 axonemes (A) and wild-type axonemes (B) were sedimented on 5-20% sucrose gradients and resulting fractions were analyzed by SDS-PAGE (bottom of gradient to the left). Gels were stained with Coomassie blue. The sizes of molecular weight standards (in kDa) are shown along the right edge of each gel. (C) Western blotting of gel in (B) with anti-Cpc1A shows that Cpc1 sediments in two peaks with approximate sedimentation coefficients of 16S and 9S. Five proteins [marked in (B) with arrows], including CP3, CP4 (Cpc1) and bands of 135 kDa, 79 kDa, and 56 kDa co-sediment with Cpc1 in the faster sedimenting (16S) peak. None of these bands are seen in KI extracts from cpc1 axonemes (A).
Motility
Because the Cpc1 protein is predicted to encode an adenylate kinase, which catalyzes the reversible reaction 2ADP ↔ ATP + AMP, one function of Cpc1 may be to increase ATP concentrations locally. The long, narrow flagellar compartment has been hypothesized to restrict direct supply of ATP by diffusion, requiring other methods of locally boosting the ATP concentration enough for normal dynein activity. In some organisms, ATP can be generated directly within the ciliary or flagellar compartment by transfer of phosphates from high-energy storage compounds (creatine phosphate or arginine phosphate) to ADP (Noguchi et al., 2001) and perhaps even by glycolysis (Gitlits et al., 2000). In addition, ATP concentration can be maintained locally by adenylate kinases (Schoff et al., 1989; Nakamura et al., 1999), which, if their forward reactions are favored by removal of AMP, effectively allow each ATP to be used twice. Chlamydomonas flagella can be reactivated in the presence of ADP alone (Hyams and Borisy, 1978) and are known to possess adenylate kinase activity, but to lack creatine and arginine kinase activities (Watanabe and Flavin, 1976). Therefore, the reduced beat frequency of cpc1 cells could result in part from inability to maintain a high enough ATP concentration within the flagellar compartment to support wild-type motility. To test this possibility, we reactivated permeabilized cell models at a saturating ATP concentration (1 mM) and measured the resulting flagellar beat frequency. As summarized in Table 2, cpc1 flagellar beat frequency is only 64% of that of the wild-type frequency in live cells, but increases to almost 90% of the wild-type value in reactivated cell models. Thus an inability to maintain a normal ATP concentration is likely to contribute to the reduced beat frequency of cpc1 flagella in live cells. This result suggests that the adenylate kinase domain in Cpc1 is catalytically active and important for flagellar motility. The remaining 10% difference in beat frequency seen at saturating ATP concentrations suggests that the Cpc1 complex contributes directly to flagellar motility regulation, but may result from an overall change in the relative orientation of C1 and C2 microtubules noted previously in this mutant (Mitchell and Sale, 1999).
Table 2.
Beat frequency of cpc1 flagella in vivo and in vitro
| Beat frequency ± s.d. (n), (Hz)
|
||||
|---|---|---|---|---|
| Sample | Nucleotide added | Wild type | cpc1 | % cpc1/wild type |
| Live cells | – | 58±5 (45) | 37±5 (45)a | 64% |
| Cell model | 1 mM ATP | 43±7 (40) | 38±5 (40)a | 88% |
| Cell model | 1 mM ADP | 20±2 (20) | 19±3 (20) | (100%)b |
| Axonemes (Exp. 1) | 1 mM ATP | 58±3 (40) | 50±5 (40)a | 86% |
| Axonemes (Exp. 2) | 1 mM ATP | 52±5 (96) | 49±4 (96)a | 94% |
| Axonemes | 1 mM ADP | 22±3 (40) | 23±3 (36) | (100%)b |
Significantly different from the wild-type beat frequency (P<0.01, two-tailed t test).
No significant difference between mutant and wild-type beat frequency.
If Cpc1 were the only adenylate kinase available in flagella, then cpc1 cell models should not be reactivated in the presence of ADP alone. In contrast, cpc1 cell models did swim when reactivated in the presence of 1 mM ADP alone (Table 2). Beat frequency in ADP was about 45% of the wild-type frequency in ATP, and did not differ significantly between wild-type and cpc1 cell models. This lack of difference between wild-type and cpc1 beat frequencies could result from release of an adenylate kinase activity into solution, either from flagella or from cell bodies, during detergent treatment. Such solubilized adenylate kinase could act upon the bulk ADP in solution and might produce enough ATP to mask the lack of microtubule-bound Cpc1 adenylate kinase activity in the mutant flagella. We therefore isolated flagella away from cell bodies, removed membranes from the flagella with detergent, washed the resulting axonemes to remove any residual soluble or membrane-associated adenylate kinases and reactivated the purified axonemes in either ATP or ADP. In ATP alone, cpc1 axonemes beat at 90% of the wild-type value (average of the two experiments shown in Table 2), similar to the result with cell models. This confirms that one major reason for decreased beat frequency in cpc1 cells is lack of adequate ATP concentration. However, both wild-type and cpc1 axonemes continued to beat in ADP alone, with no significant difference between strains (Table 2). Therefore cpc1 axonemes must retain other adenylate kinases capable of generating ATP from ADP. A direct assay for adenylate kinase activity in wild-type and cpc1 axonemes did not detect a significant difference in enzymatic activity (data not shown). Lack of Cpc1 protein, although associated with a highly significant frequency reduction in live cells, does not have a significant effect on the synthesis of ATP from ADP in isolated axonemes under the reactivation conditions tested.
To determine the approximate concentration of ATP in vivo in cpc1 flagella, beat frequencies of wild-type axonemes were measured at varying ATP concentrations. A double-reciprocal plot of the data (Fig. 9) was used to estimate that a normal ATP concentration in wild-type flagella is approximately 1.2 mM. In contrast, the ATP concentration in cpc1 flagella that would produce the observed beat frequency of 37 Hz is 0.2 mM.
Fig. 9.
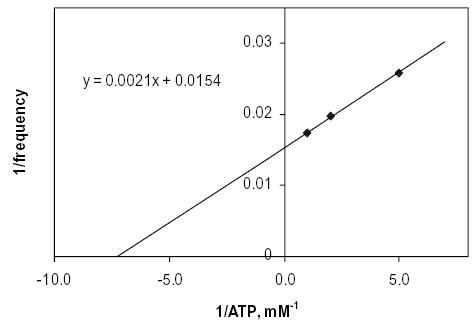
Double-reciprocal plot of the variation of in vitro Chlamydomonas flagellar beat frequency with ATP concentration. Intercepts provide a Frequencymax of 64.9 Hz and a Km(freq) of 0.14 mM ATP.
Discussion
The clearly essential function of central pair-associated proteins in flagellar motility has been difficult to understand, both because the function of most individual central pair proteins has not been explored and because most mutations that disrupt central pair structure generate the null phenotype (paralyzed flagella). Here we characterize the CPC1 locus and show that it encodes a large scaffold protein with an embedded adenylate kinase domain. We also show that Cpc1 is essential for maintaining normal beat frequency, mainly because mutant flagella lack normal concentrations of ATP.
The protein encoded by the CPC1 locus has an estimated mass of 205 kDa (apparent gel size of 265 kDa) and corresponds to the CP4 band previously characterized as a central pair protein (Dutcher et al., 1984). Western blots confirm our previous conjecture that Cpc1/CP4, when extracted with 0.2 M KI, sediments in two peaks, one at 9S, which may contain Cpc1 alone, and one at 16S, in which Cpc1 co-sediments with five additional proteins. Other proteins in this complex, including CP3 (350 kDa), fail to assemble into cpc1 axonemes (Mitchell and Sale, 1999), suggesting that Cpc1 acts as a link between the proteins of this complex and the central pair microtubules. Alternatively, proteins in this complex may depend on formation of the complex for their individual stability or their transport into the flagellar compartment (Fowkes and Mitchell, 1998; Qin et al., 2004). In either case, it is reasonable to surmise that smaller subunits of the 16S complex use one of the two larger subunits as a scaffold. Sedimentation of approximately half of Cpc1 at 9S, separate from the other subunits (except perhaps the 56 kDa band) suggests that the smaller proteins may form a tighter association with 350 kDa CP3 than with 265 kDa Cpc1.
Similarity searches show that Cpc1 is retained in many organisms that have motile cilia or flagella, including vertebrates (humans, rats, puffer fish), echinoderms (sea urchins), and ciliates (Tetrahymena), but not in organisms that lack such motile organelles (C. elegans, S. cerevisiae). Distribution of sequence similarities over the entire length of the rat and algal homologs suggests that the need for Cpc1 function arose early in eukaryotic evolution and has changed little since. Our inability to find Cpc1 homologs in the databases for D. melanogaster, an organism that has motile flagellated 9+2 gametes, may indicate that the need for Cpc1 is not universal to flagellar motility, but could also reflect gaps in available sequences for this organism.
The cpc1 mutation alters flagellar motility by reducing beat frequency, rather than altering waveform, and does not affect the responsiveness of frequency and waveform to changes in calcium ion concentration (Mitchell and Sale, 1999). Other mutations that disrupt the central pair have a qualitatively similar effect in that they all reduce beat frequency, but different mechanisms are probably involved. Mutations such as pf15 and pf20 that completely disrupt central pair assembly result in complete flagellar paralysis, whereas pf6, which only blocks assembly of the C1a projection, allows very slow flagellar bend propagation at up to a few beats per second (Rupp et al., 2001). In the case of cpc1, reduction in beat frequency apparently results from two separate functions of the Cpc1 complex. The adenylate kinase activity of Cpc1 (or the function of another protein in the Cpc1 complex) is required to maintain localized ATP concentrations, as most of the lost beat frequency is restored when mutant axonemes are reactivated in saturating concentrations of ATP. Other in vitro tests of Chlamydomonas flagellar motility mechanisms, based on doublet sliding of protease-treated axonemes, also failed to show a significant difference between cpc1 and the wild type at 1 mM ATP (Smith, 2002b). This phenotype is so far quite specific to cpc1. Axonemes from paralyzed central pair assembly mutations such as pf20 cannot be reactivated with 1 mM ATP, although they can be reactivated to low frequency beating by very low ATP concentrations (e.g. 50 μM) (Omoto et al., 1996). The remaining 10% difference in beat frequency between cpc1 axonemes and wild-type axonemes reactivated in 1 mM ATP must reflect loss of a function unrelated to ATP concentration. This difference could result directly from absence of Cpc1, indirectly from lack of one or more of the other proteins in the 16S Cpc1 complex, or from an overall change in central pair structure.
The in vivo beat frequency reduction that accompanies the cpc1 mutation is similar in a wild-type background (38%) and in a pf28 background (i.e. in the absence of outer row dyneins; 30%) (Mitchell and Sale, 1999). If the effect of cpc1 is primarily to lower in vivo intraflagellar ATP concentrations, then lack of outer row dyneins should reduce ATP consumption and reduce the effect of this mutation. However, as shown by the data in Fig. 9, the estimated ATP concentration in cpc1 flagella is only 0.18 mM, close to the Kmfreq of 0.14 mM taken from the X intercept. Although the rate of ATP consumption may be reduced by the absence of outer row dyneins, this difference is clearly insufficient to allow full recovery of ATP concentrations to wild-type levels.
Location of an adenylate kinase in the central pair complex may indicate a specific requirement for ATP as substrate for a central pair-associated enzyme. The only ATPase known to be associated with the central pair, kinesin-like protein Klp1, is not located near Cpc1 within the central pair structure but rather along the side of the C2 microtubule as determined by immunogold localization (Bernstein et al., 1994) and more recently by thin-section electron microscopy of axonemes from a Klp1 RNAi knockdown strain (D. R. Mitchell and R. Yokoyama, 2003, ‘Structural analysis of central pair function in Chlamydomonas flagella’, presented at the 43rd Annual Meeting of the American Society for Cell Biology in San Francisco, December 2003). Thus far no other enzymes that might require a high ATP concentration, such as ATPases or protein kinases, have been localized to the central pair structure, but our knowledge of the protein composition of the flagellum is still quite incomplete. No significant difference in adenylate kinase activity was seen between wild-type and cpc1 axonemes, confirming that the contribution of the Cpc1 adenylate kinase domain is negligible under these conditions. We have not tested the enzymatic activity of a purified Cpc1 protein, and it remains possible that the adenylate kinase domain is not enzymatically active under any conditions. If that were the case, then other proteins in the Cpc1 complex would have to contribute to the maintenance of wild-type ATP concentrations. A preliminary proteomic analysis of proteins that co-sediment at 16S with Cpc1 has identified two other proteins with domains predicted to have enzymatic activities, including one additional adenylate kinase domain (in CP3) and an enzyme essential for glycolysis (enolase); both of these proteins could contribute to maintenance of ATP concentrations in flagella (unpublished data). The Cpc1 protein also has an EF hand domain that may confer a calcium requirement on Cpc1 enzymatic activity. More detailed analysis of mutations that selectively disrupt parts of the central pair complex, together with ultrastructural analysis of the relationship between central pair orientation and dynein activity (Wargo and Smith, 2003; Mitchell, 2003a; Mitchell, 2003b) will be needed to understand fully the mechanisms through which the central pair complex regulates dyneins.
Acknowledgments
We thank Anthony Koutoulis and Yvonne Vucica for sharing preliminary data on insertional alleles of CPC1, Carolyn Silflow and Pushpa Kathir for mapping the CPC1 gene and Kimberly Brown, Karly Judson, Judy Freshour and Masako Nakatsugawa for technical assistance. This work was supported by grants to D. Mitchell from the National Science Foundation (MCB9982062) and the National Institutes of Health (GM44228).
References
- Adams GMW, Huang B, Piperno G, Luck DJL. Central-pair microtubular complex of Chlamydomonas flagella: polypeptide compostition as revealed by analysis of mutants. J Cell Biol. 1981;91:69–76. doi: 10.1083/jcb.91.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asamizu E, Nakamura Y, Sato S, Fukuzawa H, Tabata S. A large scale structural analysis of cDNAs in a unicellular green alga, Chlamydomonas reinhardtii. I Generation of 3433 non-redundant expressed sequence tags. DNA Res. 1999;6:369–373. doi: 10.1093/dnares/6.6.369. [DOI] [PubMed] [Google Scholar]
- Baccetti B, Burrini AG, Maver A, Pallini V, Renieri T. “9 + 0” immotile spermatozoa in an infertile man”. Andrologia. 1979;11:437–443. [PubMed] [Google Scholar]
- Berger B, Wilson DB, Wolf E, Tonchev T, Milla M, Kim PS. Predicting coiled coils by use sf pairwise residue correlations. Proc Natl Acad Sci USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein M, Beech PL, Katz SG, Rosenbaum JL. A new kinesin-like protein (Klp1) localized to a single microtubule of the Chlamydomonas flagellum. J Cell Biol. 1994;125:1313–1326. doi: 10.1083/jcb.125.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek S, Zhang H, Grishin NV. Sequence and structure classification of kinases. J Mol Biol. 2002;320:855–881. doi: 10.1016/s0022-2836(02)00538-7. [DOI] [PubMed] [Google Scholar]
- Dutcher SK, Huang B, Luck DJL. Genetic dissection of the central pair microtubules of the flagella of Chlamydomonas reinhardtii. J Cell Biol. 1984;98:229–236. doi: 10.1083/jcb.98.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowkes ME, Mitchell DR. The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol Biol Cell. 1998;9:2337–2347. doi: 10.1091/mbc.9.9.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons IR. Studies on the adenosine triphosphatases activity of 14 S and 30 S dynein from cilia of Tetrahymena. 1966;241:5590–5596. [PubMed] [Google Scholar]
- Gitlits VM, Toh BH, Loveland KL, Sentry JW. The glycolytic enzyme enolase is present in sperm tail and displays nucleotide-dependent association with microtubules. Eur J Cell Biol. 2000;79:104–111. doi: 10.1078/S0171-9335(04)70012-6. [DOI] [PubMed] [Google Scholar]
- Habermacher G, Sale WS. Regulation of flagellar dynein by phosphorylation of a 138-kD inner arm dynein intermediate chain. J Cell Biol. 1997;136:167–176. doi: 10.1083/jcb.136.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, E. H. (1989). The Chlamydomonas sourcebook, San Diego, CA: Academic Press.
- Horst, C. J. and Witman, G. B. (1998). Reactivation of Chlamydomonas cell models. In Cilia and Flagella (ed. W. Dentler and G. Witman), pp. 207–210. San Diego, CA: Academic Press. [DOI] [PubMed]
- Hyams JS, Borisy GG. Isolated flagellar apparatus of Chlamydomonas: characterization of forward swimming and alteration of waveform and reversal of motion by calcium ions in vivo. J Cell Sci. 1978;33:235–253. doi: 10.1242/jcs.33.1.235. [DOI] [PubMed] [Google Scholar]
- Kathir P, LaVoie M, Brazelton WJ, Hass NA, Lefebvre PA, Silflow CD. Molecular map of the Chlamydomonas reinhardtii nuclear genome. Eukaryotic Cell. 2003;2:362–379. doi: 10.1128/EC.2.2.362-379.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipe DD, Koonin EV, Aravind L. Evolution and classification of P-loop kinases and related proteins. J Mol Biol. 2003;333:781–815. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Mitchell DR. Orientation of the central pair complex during flagellar bend formation in Chlamydomonas. Cell Motil Cytoskeleton. 2003a;56:120–129. doi: 10.1002/cm.10142. [DOI] [PubMed] [Google Scholar]
- Mitchell DR. Reconstruction of the projection periodicity and surface architecture of the flagellar central pair complex. Cell Motil Cytoskeleton. 2003b;55:188–199. doi: 10.1002/cm.10121. [DOI] [PubMed] [Google Scholar]
- Mitchell DR, Kang Y. Identification of oda6 as a Chlamydomonas dynein mutant by rescue with the wild-type gene. J Cell Biol. 1991;113:835–842. doi: 10.1083/jcb.113.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DR, Sale WS. Characterization of a Chlamydomonas insertional mutant that disrupts flagellar central pair microtubule-associated structures. J Cell Biol. 1999;144:293–304. doi: 10.1083/jcb.144.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CW, Schulz GE. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 A resolution. A model for a catalytic transition state. J Mol Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Iitsuka K, Fujii T. Adenylate kinase is tightly bound to axonemes of Tetrahymena cilia. Comp Biochem Physiol. 1999;124:195–199. doi: 10.1016/s0305-0491(99)00114-5. [DOI] [PubMed] [Google Scholar]
- Noguchi M, Sawadas T, Akazawa T. ATP-regenerating system in the cilia of Paramecium caudatum. J Exp Biol. 2001;204:1063–1071. doi: 10.1242/jeb.204.6.1063. [DOI] [PubMed] [Google Scholar]
- Omoto CK, Yagi T, Kurimoto E, Kamiya R. Ability of paralyzed flagella mutants of Chlamydomonas to move. Cell Motil Cytoskeleton. 1996;33:88–94. doi: 10.1002/(SICI)1097-0169(1996)33:2<88::AID-CM2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Ostrowski LE, Andrews K, Potdar P, Matsuura H, Jetten A, Netteshein P. Cloning and characterization of KPL2, a novel gene induced during ciliogenesis of tracheal epithelial cells. Am J Respir Cell Mol Biol. 1999;20:675–683. doi: 10.1165/ajrcmb.20.4.3496. [DOI] [PubMed] [Google Scholar]
- Ostrowski LE, Blackburn K, Radde KM, Moyer MB, Schlatzer DM, Moseley A, Boucher RC. A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- Porter ME, Sale WS. The 9+2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J Cell Biol. 2000;151:F37–F42. doi: 10.1083/jcb.151.5.f37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Diener DR, Geimer S, Cole DG, Rosenbaum JL. Intraflagellar transport (IFT) cargo: IFT transports flagellar precursors to the tip and turnover products to the cell body. J Cell Biol. 2004;164:255–266. doi: 10.1083/jcb.200308132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Liu J. The predict protein server. Nucleic Acids Res. 2003;31:3300–3304. doi: 10.1093/nar/gkg508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Sander C. Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- Rupp G, O’Toole E, Porter ME. The Chlamydomonas PF6 locus encodes a large alanine/proline-rich polypeptide that is required for assembly of a central pair projection and regulates flagellar motility. Mol Biol Cell. 2001;12:739–751. doi: 10.1091/mbc.12.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager R, Granick S. Nutritional studies with Chlamydomonas reinhardtii. Ann New York Acad Sci. 1953;466:18–30. doi: 10.1111/j.1749-6632.1953.tb30261.x. [DOI] [PubMed] [Google Scholar]
- Schoff PK, Cheetham J, Lardy HA. Adenylate kinase activity in ejaculated bovine sperm flagella. J Biol Chem. 1989;264:6086–6091. [PubMed] [Google Scholar]
- Smith EF. Regulation of flagellar dynein by calcium and a role for an axonemal calmodulin and calmodulin-dependent kinase. Mol Biol Cell. 2002a;13 :3303–3313. doi: 10.1091/mbc.E02-04-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EF. Regulation of flagellar dynein by the axonemal central apparatus. Cell Motil Cytoskeleton. 2002b;52:33–42. doi: 10.1002/cm.10031. [DOI] [PubMed] [Google Scholar]
- Smith EF, Lefebvre PA. The role of central apparatus components in flagellar motility and microtubule assembly. Cell Motil Cytoskeleton. 1997;38:1–8. doi: 10.1002/(SICI)1097-0169(1997)38:1<1::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Smith EF, Sale WS. Regulation of dynein-driven microtubule sliding by the radial spokes in flagella. Science. 1992;257:1557–1559. doi: 10.1126/science.1387971. [DOI] [PubMed] [Google Scholar]
- Smith EF, Yang P. The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton. 2004;57:8–17. doi: 10.1002/cm.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha and beta subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wargo MJ, Smith EF. Asymmetry of the central apparatus defines the location of active microtubule sliding in Chlamydomonas flagella. Proc Natl Acad Sci USA. 2003;100:137–142. doi: 10.1073/pnas.0135800100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr JR, McVittie A, Randall J, Hopkins JM. Genetic control of flagellar structure in Chlamydomonas reinhardii. Genet Res Camb. 1966;7:335–351. [Google Scholar]
- Watanabe T, Flavin M. Nucleotide-metabolizing enzymes in Chlamydomonas flagella. J Biol Chem. 1976;251:182–192. [PubMed] [Google Scholar]
- Wilkerson CG, King SM, Witman GB. Molecular analysis of the γ heavy chain of Chlamydomonas flagellar outer-arm dynein. J Cell Sci. 1994;107:497–506. doi: 10.1242/jcs.107.3.497. [DOI] [PubMed] [Google Scholar]
- Witman GB. Isolation of Chlamydomonas flagella and flagellar axonemes. Meth Enzym. 1986;134:280–290. doi: 10.1016/0076-6879(86)34096-5. [DOI] [PubMed] [Google Scholar]
- Witman GB, Plummer J, Sander G. Chlamydomonas flagellar mutants lacking radial spokes and central tubules. J Cell Biol. 1978;76:729–747. doi: 10.1083/jcb.76.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray W, Boulikas T, Wray VP, Hancock R. Silver staining of proteins in polyacrylamide gels. Anal Biochem. 1981;118:197–203. doi: 10.1016/0003-2697(81)90179-2. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Shingyoji C. Effects of the central pair apparatus on microtubule sliding velocity in sea urchin sperm flagella. Cell Struct Funct. 1999;24:43–54. doi: 10.1247/csf.24.43. [DOI] [PubMed] [Google Scholar]