

Abstract
Cochlear and vestibular sensory cells undergo apoptosis when exposed to aminoglycoside antibiotics in organ culture, but mechanisms of chronic drug-induced hair cell loss in vivo are unclear. We investigated cell death pathways in a mouse model of progressive kanamycin-induced hair cell loss. Hair cell nuclei showed both apoptotic- and necrotic-like appearances but markers for classic apoptotic pathways (cytochrome c, caspase-9, caspase-3, JNK, TUNEL) were absent. In contrast, drug treatment caused EndoG translocation, activation of μ-calpain, and both the synthesis and activation of cathepsin D. Poly (ADP-ribose) polymerase 1 (PARP1) was decreased, but a caspase-derived 89 kDa PARP1 fragment was not present. The mRNA level of PARP1 remained unchanged. Thus, chronic administration of aminoglycosides causes multiple forms of cell death, without a major contribution by classic apoptosis. These results provide a better understanding of the toxic effects of aminoglycosides and are relevant to design protection from aminoglycoside-induced hearing loss.
Keywords: aminoglycosides, apoptosis, necrosis, calpains, cathepsin D
Abbreviations: AIF, apoptosis-inducing factor; EndoG, endo-nuclease; PARP1, poly (ADP-ribose) polymerase
Introduction
Ototoxic (toxic to the inner ear) aminoglycoside antibiotics can cause the death of the sensory hair cells in both the cochlear and vestibular end organs producing loss of hearing and balance, respectively. The initial trigger of the toxic actions appears to be the formation of reactive oxygen species1 subsequently invoking apoptotic or necrotic pathways. In cell or organ culture and with acute application of the drugs, apoptosis may be a predominant form of cell death. In a clinical situation, however, aminoglycoside ototoxicity develops slowly and sometimes is not manifest until weeks after the cessation of treatment. Information on molecular pathways of chronic cell death is sparse probably because a major animal model has been the guinea pig. Only recently an adult mouse model has been developed2 for which molecular and genetic tools are more readily available.
c-Jun N-terminal kinases (JNKs) and caspase cascades have been suggested as downstream pathways in aminoglycoside cytotoxicity in cochlear and vestibular organ explants.3–6 Caspase-3 plays a central role in the execution phase of classic apoptosis by cleaving poly (ADP-ribose) polymerase (PARP1), a 113 kDa nuclear repair enzyme, into fragments of 89 and 24 kDa.7,8 Caspase-3 can be activated by cytochrome c released from mitochondria upon loss of mitochondrial membrane potential,9 an event that has been implicated in aminoglycoside-mediated hair cell death in organ cultures.10 However, mitochondrial permeability changes may also release apoptosis-inducing factor (AIF) and endonuclease G (EndoG)- triggering cell death. AIF and EndoG represent caspase-independent pathways that can lead to apoptosis-like features of cells following transfer into nuclei.11,12 Recent evidence suggests that AIF may have dual functions: proapoptotic and antiapoptotic, depending on cell type and stimuli.13
Lysosomal pathways of cell death should be of special interest in attempts to explain the toxicity of aminoglycosides since these drugs accumulate in these organelles both in the kidney and the inner ear in vivo.14,15 Lysosomes may trigger different forms of cell death depending on the cell types and the stimuli.16,17 Pathways of lysosomal origin may lead to apoptosis associated with caspases while the release of lysosomal proteases, such as the cysteine cathepsins B and L and the aspartyl cathepsin D, may cause necrosis, apoptosis-or necrosis-like cell death.18,19
Calpains, cytosolic calcium-activated neutral cysteine proteases, are, at least in part, also associated with lysosomes.19,20 Under appropriate stimuli, their active form dissociates from its inhibitor, calpastatin, and translocates to the cytoplasmic membrane where cytoskeletal attachments would be cleaved.21 Active calpains participate in the regulation of both apoptosis and necrosis.9
In this study, we investigate signal transduction pathways of cell death in the inner ear following chronic kanamycin-treatment in the adult mouse.2 In particular, we are probing pathways that have been implicated in aminoglycoside-induced cell death in vitro and those that are associated with a lysosomal origin. Understanding the pathways which are activated by stress in the cochlea will increase our understanding of the complexity of aminoglycoside-induced ototoxicity and may offer additional possibilities to protect against hearing loss induced by these antibiotics.
Results
Chronic treatment with kanamycin induces slow functional damage in mice
Hearing loss was assessed by measuring evoked auditory brain stem responses (ABR). Auditory thresholds were comparable for all animals at the beginning of the study. Saline-injected control animals maintained stable thresholds for the duration of the experiments. After 7 days of kanamycin treatment there were no significant changes in ABR thresholds at 24 kHz. Continued injections of the same dose of kanamycin for 14 days resulted in significant threshold shifts of about 45–50 dB, which stabilized by 5 weeks (Figure 1).
Figure 1.
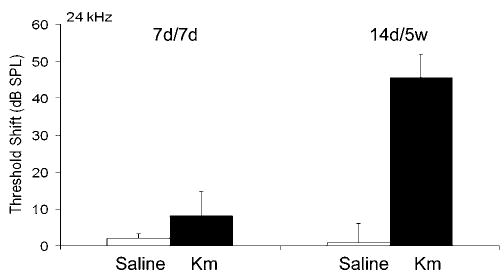
Threshold shifts in mice treated with kanamycin. After 7 days of kanamycin treatment there was no significant change in ABR thresholds at 24 kHz, measured on day 7 (7 d/7 d). Continued injections of kanamycin for 14 days resulted in an increased threshold shift which stabilized around 50 dB at 5 W (14 d/5 W; n = 5 each, P<0.05)
Chronic treatment with kanamycin induces hair cell loss in the cochlea
Cochlear pathology was consistent with the functional deficits determined by the ABR measurements. In saline-treated CBA mice, rhodamine phalloidin staining for actin showed the well-defined outline of outer hair cells (Figure 2a). After treatment with kanamycin for 7 days there was no apparent loss of outer hair cells (Figure 2b) but continued injections resulted in the beginning of death of outer hair cells at 11 days (Figure 2c). After 14 days about 30% of outer hair cells were lost in the basal turn (Figure 2d) and 1 week after the end of the drug administration almost all outer hair cells in the basal turn had disappeared (Figure 2e).
Figure 2.
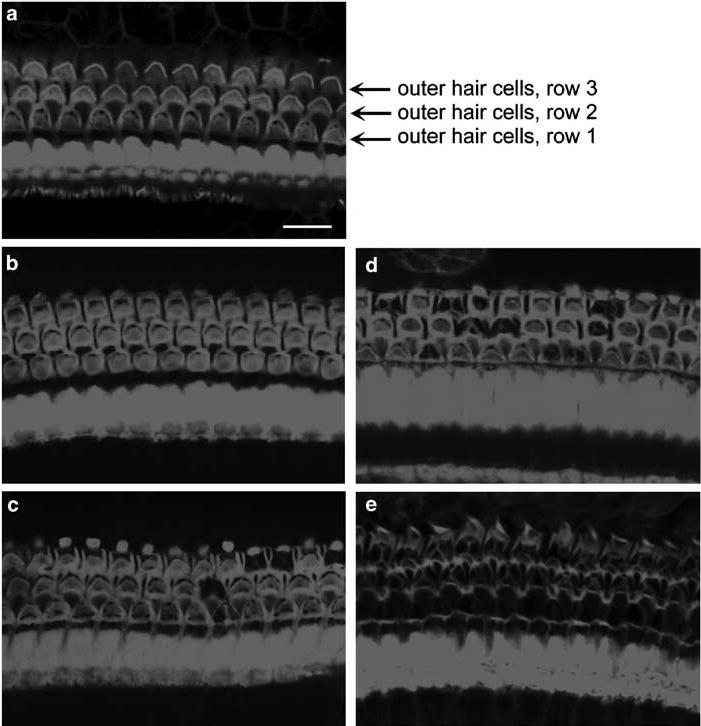
Hair cell loss in the cochlea. a: In the saline-treated CBA mice, rhodamine phalloidin staining for actin showed the well-defined outline of outer hair cells. b: After treatment with kanamycin for 7 days there was no apparent loss of outer hair cells. c: After treatment with kanamycin for 11 days some outer hair cells had disappeared in the basal turn. d: After 14 days of treatment with kanamycin, about 30% of outer hair cells were lost in the basal turn. e: At 1 week after the end of a 14-day treatment, almost all outer hair cells in the basal turn had vanished. The figure is representative of three individual animals at each time point. Scale bar: 10 μm
Outer hair cell death by multiple pathways in the organ of Corti
Transmission electron microscopy showed both apoptotic and necrotic features in the same cochlea beginning at 11 days of kanamycin-treatment. Incomplete and lumpy chromatin condensation (Figure 3b) indicated apoptotic or apoptotic-like cell death, while cellular and nuclear swelling with scattered chromatin condensation suggested necrosis or necrotic-like cell death (Figure 3c). Surveying a series of sections taken through entire cochleae suggested the presence of more (about four times as many) apoptotic- than necrotic-like nuclei. Saline control animals showed normal nuclei (Figure 3a).
Figure 3.
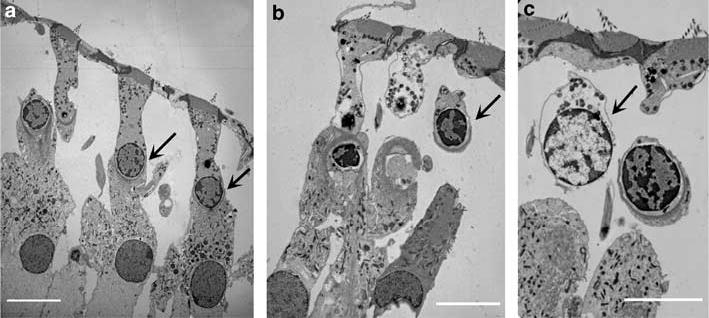
Electron microscopic characterization of outer hair cell death. Ultrathin sections of mice cochlea at 11 days of treatment with saline (a) and kanamycin (b and c) were observed under electron microscopy. Nuclear morphology of outer hair cells in the basal turn of the saline group was normal (a, arrow); kanamycin treatment caused apoptotic-like cell death (b, arrow) indicated by incomplete and lumpy chromatin condensation as well as necrosis or necrotic-like cell death (c, arrow) indicated by cellular and nuclear swelling, and scattered chromatin condensation. This figure is representative of 2 individual animals for each condition. Scale bar: 10 μm
In agreement with the morphological assessment, nuclear staining in the organ of Corti did not indicate any dying cells in saline controls or after 7 days of kanamycin-treatment (Figure 4a and b). After 11 days of drug treatment nuclei of outer hair cells began to exhibit mixed morphological features including apoptosis, apoptotic-like cell death, and necrosis or necrotic-like cell death (Figure 4c). The number of missing or abnormal nuclei was increased at 14 days of kanamycin treatment. However, only a few cells over the entire length of the cochlea stained with TUNEL during the treatment (Figure 4d). Six cochleae each were examined after 3, 7, 11 and 14 days; some reactive cells were found after 11 days of treatment. In contrast, intense noise (106 dB SPL, 2–20 kHz for 2 h) produced a consistent TUNEL signal in the outer hair cell area at 30 min, 2, 3 and 5 h after exposure (Figure 4e).
Figure 4.
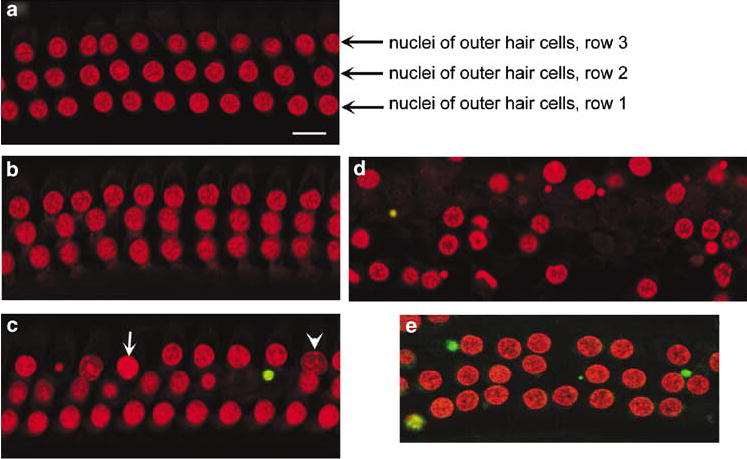
Nuclear morphology and TUNEL staining. Surface preparations of the organ of Corti in the basal turn were stained with TUNEL (green) and for nuclei with propidium iodide (red). a: Normal morphology and no TUNEL-positive staining was found in the outer hair cells in saline-treated control animals. b: After 7 days of kanamycin treatment the tissue appears normal. c: After 11 days of kanamycin treatment PI stained a number of cells with nuclear condensation (arrow) and swelling (arrow head), but only occasional cells were stained with TUNEL (green). d: At 14 days of kanamycin treatment loss of outer hair cells had increased. The sections shown are representative of six individual animals at each time point. In contrast, 3 h after noise exposure cochlear preparations consistently showed positive TUNEL stain (e: green). Scale bar: 10 μm
Kanamycin treatment does not cause the release of cytochrome c, activation of caspase-9 and -3 or JNK in the organ of Corti
Since the morphology of dying cochlear cells had shown apoptotic-like features, we determined whether constituents of classic apoptotic pathways were activated by kanamycin. We analyzed cochleae after drug treatment for 3, 7, 11 and 14 days; in addition, we analyzed caspase-3 on days 3, 7 and 14 days after the end of the 14-day drug regimen. There was no evidence for the release of cytochrome c by Western blot and immunohistochemistry (data not shown) nor for the activation of caspase-3, which was probed by immunohistochemistry with antibodies from two different companies (Figure 5-1a). Active caspase-9 (by Western blot) was also absent at all times except in one of 5 cochleae at the 14-day treatment point. Again in contrast, noise exposure (106 dB SPL, 2–20 kHz for 2 h) led to positive staining for caspase-3 in outer hair cells 3 h after the exposure (Figure 5-1b).
Figure 5.
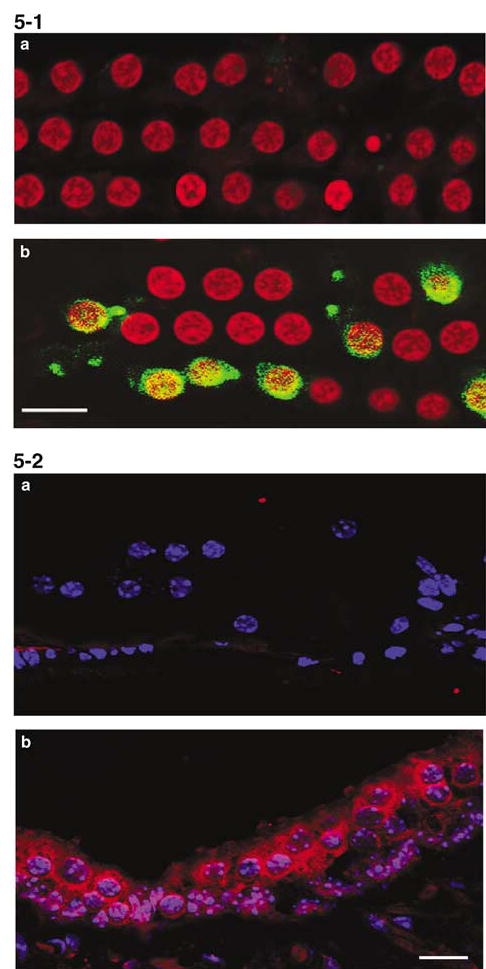
Activation of caspase-3 and JNK is not evident in the organ of Corti after kanamycin treatment. 5-1: Surface preparations of the organ of Corti were stained with active caspase-3 antibody (green) and propidium iodide (PI, red). a: Example of kanamycin treatment for 11 days. Caspase-3 immunoreactivity was not found at any of time points tested (during kanamycin treatment for 3, 7, 11 and 14 days as well as 3 days, 1 week, 2 weeks after the end of a 14-day treatment). Six animals were investigated at each time point, all without staining for caspase-3. b: In contrast, noise exposure produced a very strong signal 3 h after the end of exposure. Scale bar: 10 ím. 5-2: Sections from kanamycin-treated mice stained with phospho-JNK1/2 (red) and Heochest 33342 (blue). a: Organ of Corti after kanamycin treatment for 11 days. Phospho-JNK1/2 immunoreactivity showed negative results in the organ of Corti at all time points tested and in all six animals that were tested at each individual time point. b: Phospho-JNK1/2 immunoactivity was expressed in the saccule. Scale bar: 10 μm
Next, we used a phospho-JNK1/2 antibody which recognizes both JNK1 and JNK2 phosphorylation to detect activation of JNKs after the kanamycin insult. There was no positive staining of phospho-JNK1/2 at any time in the organ of Corti (Figure 5-2a). The phospho-JNKs, however, were strongly expressed in the sacculus of the vestibular organ where the staining was located in the cytosol of hair cells (Figure 5-2b).
Mitochondrial dysfunction in outer hair cells following kanamycin treatment
To investigate whether some of the ototoxicity of kanamycin was due to caspase-independent cell death pathways, we examined the localization of mitochondrial proteins, EndoG and AIF. In control animals, immunofluorescent staining located EndoG to the mitochondria of outer hair cells (Figure 6a). After 7 days of kanamycin treatment, EndoG was still present in mitochondria of most outer hair cells in the basal turn, but a few cells already showed diffusion of EndoG (Figure 6b). Continued treatment with kanamycin for 11 days clearly caused EndoG translocation into the nuclei of outer hair cells (Figure 6c). The level of AIF in the cochlea was decreased on Western blots and immunostaining, but there was no translocation into nuclei of outer hair cells following kanamycin treatment (data not shown).
Figure 6.
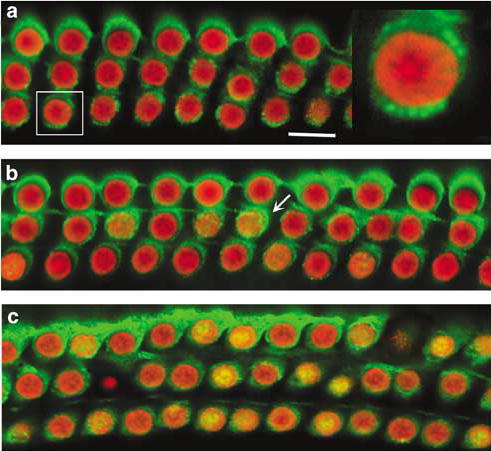
Immunoreaction for EndoG in the cochlea. Surface preparations of the organ of Corti were stained for EndoG (green) and nuclei (PI, red). a: EndoG was located in the cytosol of outer hair cells in saline control animals, and EndoG-like immunoreactivity seemed to be limited to mitochondria (a right, magnified outer hair cell). b: At the 7th day of kanamycin treatment EndoG was still associated with mitochondria of most outer hair cells, but already appeared diffuse in a few cells (b, arrow). c: After kanamycin for 11 days, EndoG was translocated into the nuclei (c, yellow). This figure is representative of three individual animals at each time point. Scale bar: 10 μm
Kanamycin triggers activation of μ-calpain
Activation of calpains can be triggered by stress to the endoplasmic reticulum. Western blot analysis indicated two bands (80 and 78 kDa) of μ-calpain in cochlear extracts but the total level of μ-calpain did not change with kanamycin treatment (data not shown). However, μ-calpain re-distributed in the organ of Corti, especially in outer hair cells following kanamycin injections. μ-calpain immunostaining was broadly localized in the cells of saline control animals (Figure 7a), but took on a punctuate appearance outlining the cell membranes following kanamycin administration. This translocation to the plasma membrane of outer hair cells began after 7 days of kanamycin treatment (Figure 7b) and became more intense after 11 days (Figure 7c) and 14 days of kanamycin treatment (Figure 7d). m-Calpain was mostly expressed in supporting cells and did not change with treatment (data not shown).
Figure 7.
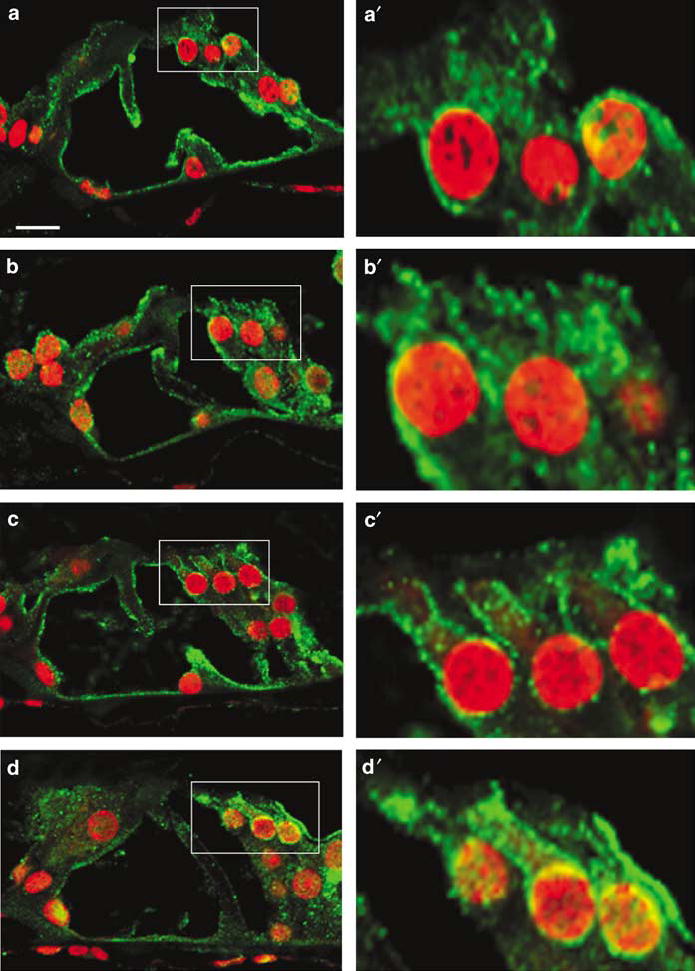
Activation of calpain in the cochlea. The sections were stained with μ-calpain antibody (green) and PI (red). a: In saline control animals, μ-calpain was localized in both the cytoplasm and cell membrane. b, c, d: Following kanamycin administration for 7 days (b), 11 days (c) and 14 days (d), μ-calpain was gradually translocated to the plasma membrane of outer hair cells. Right panels are high magnifications of outer hair cells (boxed) from each corresponding image. All sections were taken from the basal turn. This figure is representative of three individual animals at each time point. Scale bar: 10 μm
Kanamycin increases cathepsin D in the organ of Corti
Western blots showed two bands immunoreactive for cathepsin D of 52 and 42 kDa (Figure 8-1), molecular sizes consistent with procathepsin and its processed form, respectively.22 Both bands increased as early as 7 days after kanamycin treatment. To characterize the localization of cathepsin D, we labeled the cochlear sections with anti-cathepsin D and a nuclear stain, Hoechest 33342. Cathepsin D immunoreactivity (Figure 8-2) was located in all cell types of the organ of Corti, including outer hair cells (box), inner hair cells (arrowhead), and supporting cells (arrow). In tissues from control animals the immunoreactivity had a diffuse cytoplasmic localization and the staining in outer hair cells was weaker than in inner hair cells and supporting cells (Figure 8-2a). Staining intensity increased with kanamycin treatment in all of the cells (Figure 8-2b).
Figure 8.
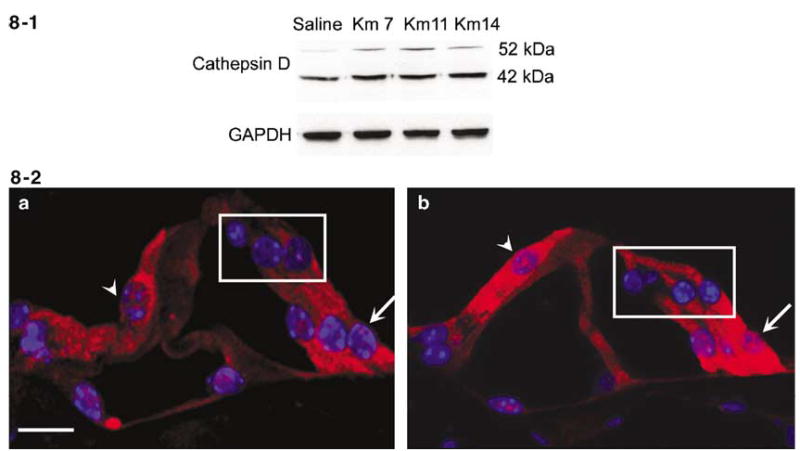
Kanamycin promotes expression and activation of cathepsin D in the organ of Corti. 8-1: Western blot analysis of cochlear extracts indicated that the precursor (52 kDa) and processed cathepsin D (42 kDa) were increased following kanamycin treatment for 7 days as well as for 11 and 14 days. Saline indicates saline-treated control animals; Km 7, Km 11 and Km 14 designate kanamycin treatment for 7, 11 and 14 days. GAPDH is a control for the loading amount of protein. This figure is representative of three individual experiments. 8-2: Immunocytochemical localization of cathepsin D. Cathepsin D is stained red, nuclei are blue with Hoechst-33342. a: Cathepsin D staining had a diffuse cytoplasmic localization in all cell types of the organ of Corti, including outer hair cells (box), inner hair cell (arrowhead) and supporting cells (arrow). b: The intensity of staining was increased by kanamycin treatment for 7 days; cathepsin D immunostaining was similar at 11 and 14 days of treatment. All images were taken from the basal turn in the organ of Corti. This figure is representative of five individual mice at each time point. Scale bar: 10 μm
Kanamycin reduces PARP1 protein in outer hair cells without changing the level of mRNA
PARP1, a 113 kDa nuclear enzyme, participates in the regulation of both DNA repair and transcription. PARP1 is cleaved into 89 and 24 kDa fragments by caspase-3 during classic apoptosis, and yields a 50 kDa fragment during necrosis. Western blot (Figure 9-1) analysis of cochlear extracts showed the 113 kDa band decreased following kanamycin treatment for 11 and 14 days, but no 89 kDa fragment of PARP1 appeared. Immunostaining (Figure 9-2) with a polyclonal antibody recognizing both the 113 kDa active enzyme and the 89 kDa cleavage fragment localized PARP1 in the nuclei of outer hair cells (box), inner hair cell (arrowhead) and supporting cells (arrow). Kanamycin treatment specifically decreased immunoreactivity in the outer hair cells, while inner hair cells and supporting cells remained unaffected, even after 14 days of drug treatment.
Figure 9.
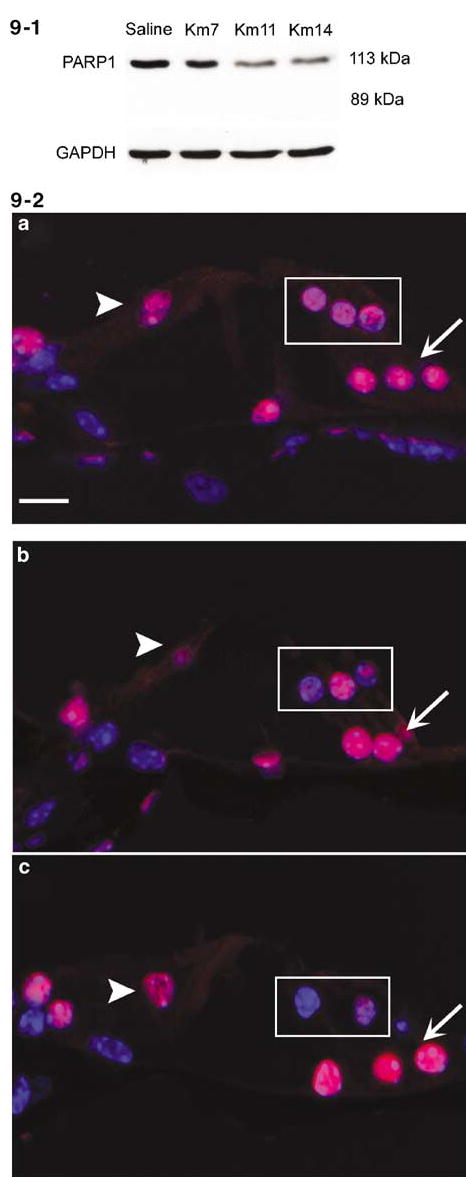
Kanamycin leads to PARP1 degradation in the organ of Corti. 9-1: Western blot analysis of cochlear extracts showed that the 113 kDa of PARP1 was decreased after 7 days of kanamycin treatment, and more strongly decreased after treatment for 11 and 14 days. No 89 kDa fragment of PARP1 was present. Saline indicates saline control animals; Km 7, Km 11 and Km 14 indicates kanamycin treatment for 7, 11 and 14 days. GAPDH is a control of the loading amount of protein. This figure is representative of four individual experiments 9-2: Sections of organ of Corti were stained with anti-PARP1 antibody (red) and Hoechest-33342 (blue). a: In saline control animals immunostaining showed PARP1 localized in the nuclei of outer hair cells (a, box), inner hair cell (a, arrowhead) and supporting cells (a, arrow). b: After kanamycin treatment for 7 days PARP1 immunoreactivity was decreased in the nuclei of outer hair cells of the basal turn (b, box). c: PARP1 immunoreactivity further decreased after 11 days (c, box). This figure is representative of five individual mice at each time point. Scale bar: 10 μm
In order to analyze possible causes for the decreased level of PARP1 protein, we quantified mRNA of PARP1 in the cochleae of animals treated with saline or kanamycin for 11 days. The mRNA ΔCT values were essentially identical at 6.56 and 6.63, respectively.
Discussion
The salient result of this study is the evidence for multiple pathways of cell death in outer hair cells in response to chronic administration of aminoglycosides in vivo. Both apoptotic- and necrotic-like morphology was evident during the period of ongoing cell death between the first indications of hair cell loss and the stabilization of a population of surviving cells. However, molecular features of classical apoptosis, for example the release of cytochrome c from the mitochondria, activation of caspase-9 and -3 or c-jun N-terminal kinase, could not be detected during this period. In contrast, significant activation of calpains and cathepsin D was apparent. Interestingly, active caspase-3 and JNK phosphorylation were identified in vestibular tissues, underscoring differences in cell death pathways between the two inner ear structures that are targeted by aminoglycoside antibiotics.
It may seem surprising that caspase-3 and c-jun N-terminal kinases (JNKs) are not involved as a major mechanism of toxic damage to the cochlea. A considerable body of literature reports caspase- and JNK-dependent apoptotic pathways in hair cell death. However, most of those studies were carried out in cell or organ cultures derived from neonatal animals. Around the time of cochlear maturation, a ‘susceptible period’ to aminoglycoside antibiotics exists, clearly differentiating the response in a neonatal organ culture from that in mature animals.23 Furthermore, in acute models of toxicity the application of aminoglycosides causes cell death within hours, a situation significantly different from chronic drug treatment in vivo where days or weeks may pass without overt signs of ototoxicity. Support for the concept of distinctly different responses to aminoglycosides in vivo and in vitro also exists for their nephrotoxic side effects. Tubular necrosis is a confirmed route of cell death in vivo24 while renal cell lines show apoptosis in response to aminoglycosides.25
Multiple organelles are involved in the ototoxic cascade following chronic administration of aminoglycosides. Mitochondria sequester several factors that have been associated with caspase-independent apoptotic-like cell death such as AIF and EndoG,11,26,27 and EndoG indeed seems to be involved in the cochlea in vivo, since it appeared in the nuclei of outer hair cells during aminoglycoside treatment. In contrast, AIF was not translocated to the nucleus suggesting that it does not serve as a proapoptotic factor. However, AIF also has antioxidant properties, and the decline in its protein levels may indirectly contribute to cell death by affording less protection against kanamycin-induced ROS formation.
The data presented here also implicate calpains in aminoglycoside-induced cell death. Calpains, which participate in multiple cellular functions including apoptosis and necrosis, can be activated through stress to the endoplasmic reticulum.28,29 The best characterized calpains are two ubiquitously expressed isozymes, μ- and m-calpain, hetero-dimeric proteins composed of a specific catalytic subunit (80 kDa) and a common regulatory subunit (30 kDa). Localization of activated calpains to membranes may induce membrane rupture,30 especially in lysosomes with the resultant leakage of cathepsins which in turn can cleave the DNA repair enzyme PARP1, triggering necrotic- or apoptotic-like cell death.31 In particular, μ-calpain maybe an important mediator for cell fate in drug treatment, since relocalization of activated μ-calpain was evident in the cytosolic membranes of outer hair cells. Calpain inhibitors also protect hair cells from gentamicin ototoxicity in cochlear culture.32
In agreement with the predominant cellular localization of aminoglycosides in lysosomes, we find the synthesis and activation of the lysosomal protease cathepsin D increased. Lysosomes also sequester iron,33,34 and the presence of both aminoglycosides and iron could lead to the formation of ROS which in turn cause destabilization of lysosomal membrane.35 Cathepsin D can be involved in the induction of various forms of cell death including apoptosis, necrosis, necrosis- or apoptosis-like cell death. Specifically, the calpain–cathepsin cascade is a major pathway regulating necrosis or necrotic-like cell death.9,36
One of the targets of a calpain–cathepsin cascade and of caspase-3 is PARP1. Hydrolysis by these two proteases yields different cleavage fragments and results in different forms of cell death.31,37 Consistent with a calpain–cathepsin action, the 113 kDa enzyme decreased in the nuclei of outer hair cells with kanamycin treatment without generating the fragmentation products associated with caspase actions (89 and 24 kDa). The unchanged level of mRNA for PARP1 further corroborates that kanamycin reduces PARP1 in a protein cleavage process and not by downregulation of the PARP1 gene. Both the activation of calpain–cathepsin and the cleavage of PARP1 are consistent with necrosis or necrotic-like death in outer hair cells.
Classic caspase-mediated apoptosis is certainly involved in other auditory pathologies, and even its contribution to chronic drug-induced ototoxicity should not be completely ruled out. The demonstration of TUNEL staining and caspase activation after acute noise overexposure clearly attest to differential responses to stressors and also eliminates the question whether methodological problems may have caused the failure to find clear apoptotic markers in chronic aminoglycoside ototoxicity. We should consider, however, that timing is crucial and cells may die quickly and express apoptotic markers for only a short time. Although we have studied an extensive time sequence from 3 to 14 days of treatment, a complete measure of apoptotic reactions may not have been possible.
In any case, our data show that cell death in chronic ototoxicity is complex. The coexistence of multiple cell death mechanisms in the same tissue is not uncommon and may even occur in the same cell population.38 A most painstaking morphological day-by-day evaluation of kanamycin ototoxicity39 suggested the occurrence of both apoptotic and necrotic features. Following chronic aminoglycoside treatment Nakagawa et al.40 did not see apoptotic labels at the primary site of aminoglycoside toxicity, namely the basal turn of the cochlea. They speculated that ‘outer hair cells of the basal turn may degenerate predominantly by necrosis’. Ylikoski et al.41 observed hair cells in vivo whose ‘mode of death … remains to be elucidated’. The results presented here clarify these issues and delineate pathways that contribute to cell death in drug-induced hearing loss.
Aside from establishing basic mechanisms, our findings also have implications for attempts to attenuate or prevent drug-induced hearing loss through manipulation of apoptotic pathways. Inhibition of a single pathway that may dominate in cell or organ culture may not be sufficient to block cell death. As a case in point, CEP-1347, an inhibitor of the c-Jun N-terminal kinase apoptotic pathway, provided almost complete protection from aminoglycoside-induced hair cell loss in culture4 but only achieved a modest 30% reduction in hair cell loss in vivo.41
In summary, this study suggests that in a chronic model of aminoglycoside ototoxicity – which we prefer for its relevance to the clinical situation – cell death is complex. Multiple cellular organelles may trigger several pathways that may act independently or in concert. Given this complexity of cell death pathways, an intervention in the early stages of ototoxicity (such as the generation of reactive oxygen species) may be a more promising approach to the prevention of drug-induced hearing loss in a clinical setting.
Materials and Methods
Materials
Kanamycin A sulfate was purchased from USB Corporation (Cleveland, OH, USA; Cat #17924; Lot #110755), ketamine (Ketaset®) from Fort Dodge Animal Health (Fort Dodge, IA, USA), xylazine (TranquiVed®) from Vedco Inc. (St. Joseph, MO, USA), ECL™ for Western blotting detection reagents from Amersham Pharmacia Biotech (Piscataway, NJ, USA). BenchMark™ Protein ladders from Invitrogen™ Life Technologies (Carlsbad, CA, USA). Polyclonal primary antibodies for the measurement of AIF, PARP1, anticathepsin D came from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA), anticalpain I (μ-calpain) mouse monoclonal antibody, anticalpain II (m-calpain) and anti-Endo G rabbit polyclonal antibodies from Chemicon International, Inc. (Temecula, CA, USA), antiphospho-JNK1/2 rabbit polyclonal antibody from Cell Signaling Technology Inc. (Beverly, MA, USA), anti-active caspase-3 rabbit polyclonal antibody from Promega Corporation (Madison, WI, USA) and Cell Signaling Technology Inc. (Beverly, MA, USA), monoclonal anticytochrome c (for Western) and rabbit polyclonal anticaspase-9 from BD Biosciences Pharmingen (San Diego, CA, USA), monoclonal anticytochrome c (for immunohistochemistry) from Promega Corporation (Madison, WI, USA). Secondary antibodies for Western blotting were purchased from Jackson Immunoresearch Laboratories (West Grove, PA, USA), secondary fluorescence antibodies (Alexa 488 and Alexa 546), rhodamine phalloidin, Hoechest 33342 and propidium iodide (PI) from Molecular Probes Inc. (Eugene, OR, USA). Complete™ mini EDTA-free protease inhibitor cocktail tablets from Roche Diagnostic GmbH (Mannheim, Germany). Total RNA isolation was carried out with the RNeasy®Micro Mini Kit purchased from Qiagen Inc. (Valencia, CA, USA), SuperScript™ First-Strand Synthesis Kit was purchased from Invitrogen Corporation (Carlsbad CA, USA), PARP1 and S16 TagMan probes from AB applied Biosystems (Foster city, CA, USA), and Master Mixture from Qbiogene (Carlsbad, CA, USA). All other reagents came from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA).
Animals and drug administration
Male CBA/J mice at an age of 4 weeks were received from Harlan Sprague Dawley Co. (Indianapolis, IN, USA) and divided into saline control and kanamycin treatment groups. The animals had free access to water and a regular mouse diet (Purina 5025, St. Louis, MO, USA) and were acclimated for 1 week before experimental procedures were performed. All experimental protocols were approved by the University of Michigan Committee on Use and Care of Animals. Animal care was under the supervision of the University of Michigan’s Unit for Laboratory Animal Medicine (ULAM).
Experimental mice received 700 mg of kanamycin/kg body weight by subcutaneous injection twice daily. Saline control mice received equivalent volumes of saline. The animals were killed and the cochleae were removed for immunocytochemistry or the extraction of total cochlear protein. Cochleae were collected 3 h after the last injection on the 3rd, 5th, 7th, 11th and 14th day as well as the 3rd, 7th and 14th day after the end of treatment.
Evaluation of auditory function
Auditory thresholds were measured by evoked auditory brainstem responses (ABR). Thresholds were taken for each animal prior to the beginning of the study, at 7 days and 2 weeks after the start of drug treatment, and then after 3 and 5 weeks. ABR measurements were performed as described previously. In brief, the mice were anesthetized with an intramuscular injection of 100 mg of ketamine and 5 mg of xylazine/kg body weight and kept warm with a heating pad. The positive needle electrode was subdermally inserted at the vertex, the midline of the scalp between the external auditory canals. The negative electrode was placed below the pinna of the left ear and the ground electrode was inserted contralaterally. Tone bursts of 12 and 24 kHz (10 ms duration, 1 ms rise and fall time) were generated using a SigGen® software package (Tucker-Davis Technologies, Gainsville, FL, USA) and delivered to the left external auditory meatus in a closed acoustic system through an ear bar connected to a Beyer DT-48 transducer (Beyer Dynamic, Farmingdale, NY, USA). The output was fed to an amplifier, viewed with an oscilloscope and recorded. The average responses from 1020 stimuli were obtained for each frequency by reducing the sound intensity in 5 dB steps until threshold. Thresholds were verified at least twice and defined as the lowest intensity to yield a reproducible deflection in the evoked response trace. Threshold shifts were calculated for individual animals by comparison to their prestudy thresholds. The ABR score of each animal was given and interpreted by an observer without knowledge of the treatment.
Histopathology
Three mice were randomly chosen from saline and kanamycin treatment group, after 7,11 and 14 days treatment and 1 week after the end of 14 days of treatment, the cochleae were immediately fixed with 4% paraformaldehyde in 0.1 M. PBS (pH 7.4) overnight at 4°C and then decalcified with 4% EDTA for 3 days. Following decalcification, the otic capsule was removed and the samples were stained with rhodamine phalloidin (a stock solution of 200 U/ml methanol diluted 1 : 100 in PBS) for 50 min. Thereafter, the organ of Corti was separated from the lateral wall and modiolus, microdissected into individual turns, and mounted on glass sides in Gel-mounting solution (Biomeda Corp., Foster City, CA, USA) and photographed with a Zeiss laser confocal microscope.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
DNA fragmentation was detected by the TUNEL technique, which detects DNA fragmentation during apoptosis. Cochleae were immediately fixed with 4% paraformaldehyde overnight at 4°C. The surface preparations of the organ of Corti were incubated in 0.5% Triton X-100 in sodium citrate for 15 min at room temperature, then washed twice with PBS. Subsequently, 100 μl of TUNEL reaction mixture (Roche Diagnostics Corporation, IN, USA) containing dUTP-FITC and TdT was added on each surface preparation for 60 min incubation at 37°C in the dark. The surface preparation were then washed and incubated again with PI-staining solution (2 μg/ml PI in PBS) for 30 min at room temperature. Finally, the surface preparation was dissected into individual turn and mounted and photographed with the laser confocal microscope (Zeiss).
Electron microscopy
Two mice were randomly chosen from saline- and kanamycin- treated groups after 11 days of treatment. After opening the auditory bullae, the cochleae were immediately fixed with cold 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M PBS (pH 7.4) at 4°C for 4 h. The auditory bullae were decalcified in 4.13% EDTA pH 7.3 for 3 days at 4°C. Following washes in 0.1 M cacodylate buffer, the tissue was postfixed in 1% OsO4 for 1.5 h at room temperature, dehydrated to 70% ethanol then en bloc stained with saturated uranyl acetate in 70% ethanol before further dehydration and embedding in plastic resin. The entire bulla was embedded. Sections parallel to the cochlear modiolus were cut at 6–8 levels through the intact cochlea. At each level a series of 1 μm sections were collected and stained with toluidine blue for light microscope examination. At each of those levels a series of thin sections for electron microscopy were also collected, some of which were mounted on formvar-coated single slot grids to enable uninterrupted views of the section of the whole cochlea. This procedure enables assessment of almost the whole length of the organ of Corti. Thin sections were stained with uranyl acetate and lead citrate and all thin sections were examined in a JEOL 1010 transmission electron microscope. Photographic negatives of the micrographs were digitized on a flatbed scanner and the images imported into Adobe Photoshop. The digital images were processed to obtain contrast reversal and optimal contrast and brightness.
Extraction of protein
Cochleae were rapidly removed and dissected in ice cold 10 mM PBS. Tissue from one mouse cochlea was homogenized in ice cold RIPA buffer by using a micro Tissue Grind pestle for 30 s. After incubation for 20 min on ice, the homogenates were centrifuged at 15 000 × g at 4°C for 10 min, and the supernatant of total protein was collected. For extraction of the cytosolic component, tissues from two mice cochleae were pooled and homogenized in cytoplasmic lysis buffer (10 mM sodium HEPES buffer (pH 7.9) additionally containing 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 5 mM DTT, 10 mM each of the phosphatase inhibitors NaF and sodium β-glycerophosphate (GP), 1 μg/ml (p-amidinophenyl) methanesulfonyl fluoride (PMSF) and 1/10 tablet/ml of Complete™ Mini EDTA-free protease inhibitor cocktail) by using a micro Tissue Grind Pestle (Kontes Glass Company, NJ, USA) for 10 s. The homogenates were kept on ice for 15 min, then centrifuged at 500 × g at 4°C for 10 min to obtain a first supernatant as the cytosolic component with mitochondria. This supernatant was centrifuged at 15 000 × g at 4°C for 10 min, removed from the pellet and the second supernatant was collected as cytosolic protein without mitochondria. The protein concentrations were measured by the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA).
Isolation of cochlear total RNA and quantitative RT-PCR (Q-PCR)
Total RNA was isolated from individual mouse cochleae by using RNeasy® Micro Mini Kit (Qiagen Inc. Valencia, CA, USA) following the protocol provided by the manufacturer. Cochleae were dissected under a dissection microcope in RNAlater® (Ambion, Inc., Austin, TX, USA). Quantification and quality of RNA was determined on a Model 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA).
Total RNA (1 μg) was reverse transcribed to cDNA by using SuperScript III reverse transcriptase (Invitrogen) following the protocol from the manufacturer. The first strand cDNA was diluted (1:10) with DEPC-H2O and stored as aliquots at −20°C. TaqMan primer and probes were obtained from the ‘Assays by Design’ (PARP1) or ‘Assays on Demand’ services (S16) (ABI, Foster City, CA, USA). A 96-well plate was used for the PCR reactions. For each gene, triplicate PCR reactions were run for each RNA sample. A housekeeping gene, S16, was also determined on each plate. The CT value (the number of cycles at which the PCR reaction reaches an arbitrary threshold value) was calculated for each reaction.42
Western blot analysis
Total protein or cytosolic protein with or without mitochondria (50 μg each) was separated by SDS-PAGE. A 10% polyacrylamide gel was used for the separation of AIF, cathepsin D, caspase-9 and phospho-JNK; an 8% gel for calpain I and II, a 7.5% gel for PARP1, and a 6% gel for cytochrome c. After electrophoresis the proteins were transferred onto nitrocellulose membranes (Pierce, Rockford, IL, USA), and blocked with 5% nonfat dry milk in PBS-0.01% Tween 20 (PBS-T). The membranes were then incubated with anti-AIF (1 : 250 dilution), anticathepsin D (1 : 250 dilution), anti-PARP1 (1 : 500 dilution), anti-phospho-JNK1/2 (1 : 100), anti-calpain II (1 : 200), and anticaspase-9 (1 : 1000) rabbit polyclonal antibodies, as well as anticalpain I (1 : 200), anticytochrome c (1 : 500) mouse monoclonal antibodies for 2 h. After washing three times with PBS-T buffer, the membranes were incubated with secondary antibody (goat anti-rabbit IgG or rabbit anti-mouse IgG) at a concentration of 1 : 10 000 for 1 h. Following extensive washing of the membrane, the immunoreactive bands were visualized by enhanced chemiluminescence (ECL) according to the instructions of the manufacturer (Amersham Pharmacia Biotech, Piscat-away, NJ, USA). The membranes were then stripped and restained with anti-GAPDH at a concentration of 1 : 20 000 for testing the loading amount.
Immunocytochemistry
Cochleae were fixed immediately with 4% paraformaldehyde overnight at 4°C. Cryostat sections of 5 μm and surface preparations of the organ of Corti were incubated in 0.5% Triton X-100 for 15 min at room temperature. After being washed three times with PBS, the blocking solution of 10% goat serum was added to the sections for 30 min at room temperature, followed by the primary antibody at 4°C for 72 h. Concentrations of anti-AIF, anti-PARP1, anticathepsin D, anticalpain I (μ-calpain) and anticalpain II (m-calpain) were 1 : 100, antiactive caspase-3 and antiphospho-JNK1/2 were 1 : 50, and anti-EndoG and anticytochrome c were 1 : 500. The sections or surface preparations were washed three times with PBS and the secondary antibody (Alexa 488 or Alexa 546 conjugated) at a concentration of 1 : 500 was applied at 4°C overnight in darkness. Some sections were finally incubated with Hoechst 33342 or PI (2 μg/ml in PBS) at room temperature for 40 min for illumination of the nucleus. In addition, PI staining was also used to distinguish necrosis and apoptosis.43 After washing with PBS, the slides were mounted and photographed with the laser confocal microscope (Zeiss).
Statistical analysis
Data were statistically evaluated by Student’s t test and by analyses of variance with Student–Newman–Keuls test for significance (P<0.05). using Primer of Biostatistics software (McGraw-Hill Software, New York, NY, USA).
Acknowledgments
This research was supported by research Grant R01 DC-03685 and a core center Grant P30 DC-05188 from the National Institute on Deafness and Other Communication Disorders, National Institutes of Health.
References
- 1.Forge A, Schacht J. Aminoglycoside antibiotics. Audiol Neurootol. 2000;5:3–22. doi: 10.1159/000013861. [DOI] [PubMed] [Google Scholar]
- 2.Wu W-J, Sha S-H, McLaren JD, Kawamoto K, Raphael Y, Schacht J. Aminoglycoside ototoxicity in adult CBA, C57BL and BALB mice and the Sprague-Dawley rat. Hear Res. 2001;158:165–178. doi: 10.1016/s0378-5955(01)00303-3. [DOI] [PubMed] [Google Scholar]
- 3.Forge A, Li L. Apoptotic death of hair cells in mammalian vestibular sensory epithelia. Hear Res. 2000;139:97–115. doi: 10.1016/s0378-5955(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 4.Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham LL, Cheng AG, Rubel EW. Caspase activation in hair cells of the mouse utricle exposed to neomycin. J Neurosci. 2002;22:8532–8540. doi: 10.1523/JNEUROSCI.22-19-08532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsui JI, Ogilvie JM, Warchol ME. Inhibition of caspases prevents ototoxic and ongoing hair cell death. J Neurosci. 2002;22:1218–1227. doi: 10.1523/JNEUROSCI.22-04-01218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ricci JE, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol. 2003;160:65–75. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duriez PJ, Shah GM. Cleavage of poly (ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol. 1997;75:337–349. [PubMed] [Google Scholar]
- 9.Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 10.Dehne N, Rauen U, de Groot H, Lautermann J. Involvement of the mitochondrial permeability transition in gentamicin ototoxicity. Hear Res. 2002;169:47–55. doi: 10.1016/s0378-5955(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 11.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 12.Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 13.Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111:147–150. doi: 10.1016/s0092-8674(02)01046-2. [DOI] [PubMed] [Google Scholar]
- 14.Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43:1003–1012. doi: 10.1128/aac.43.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashino E, Shero M, Salvi RJ. Lysosomal targeting and accumulation of aminoglycoside antibiotics in sensory hair cells. Brain Res. 1997;777:75–85. doi: 10.1016/s0006-8993(97)00977-3. [DOI] [PubMed] [Google Scholar]
- 16.Roberg K, Kagedal K, Ollinger K. Microinjection of cathepsin d induces caspase-dependent apoptosis in fibroblasts. Am J Pathol. 2002;161:89–96. doi: 10.1016/S0002-9440(10)64160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ. Cathepsin B contributes to TNF-á-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katunuma N, Murata F, Le QT, Hayashi Y, Ohashi A. New apoptosis cascade mediated by lysosomal enzyme and its protection by epigallo-catechin gallate. Adv Enzyme Regul. 2004;44:1–10. doi: 10.1016/j.advenzreg.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 19.Brunk UT, Dalen H, Roberg K, Hellquist HB. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic Biol Med. 1997;23:616–626. doi: 10.1016/s0891-5849(97)00007-5. [DOI] [PubMed] [Google Scholar]
- 20.Yamashima T, Tonchev AB, Tsukada T, Saido TC, Imajoh-Ohmi S, Momoi T, Kominami E. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus. 2003;13:791–800. doi: 10.1002/hipo.10127. [DOI] [PubMed] [Google Scholar]
- 21.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 22.Ishidoh K, Kominami E. Processing and activation of lysosomal proteinases. Biol Chem. 2002;383:1827–1831. doi: 10.1515/BC.2002.206. [DOI] [PubMed] [Google Scholar]
- 23.Pujol R. Periods of sensitivity to antibiotic treatment. Acta Oto-Laryngologica Suppl. 1986;429:29–33. doi: 10.3109/00016488609122727. [DOI] [PubMed] [Google Scholar]
- 24.Erdem A, Gundogan NU, Usubutun A, Kilinc K, Erdem SR, Kara A, Bozkurt A. The protective effect of taurine against gentamicin-induced acute tubular necrosis in rats. Nephrol Dial Transplant. 2000;15:1175–1182. doi: 10.1093/ndt/15.8.1175. [DOI] [PubMed] [Google Scholar]
- 25.El Mouedden M, Laurent G, Mingeot-Leclercq MP, Tulkens PM. Gentamicin-induced apoptosis in renal cell lines and embryonic rat fibroblasts. Toxicol Sci. 2000;56:229–239. doi: 10.1093/toxsci/56.1.229. [DOI] [PubMed] [Google Scholar]
- 26.van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9:1031–1042. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 27.Penninger JM, Kroemer G. Mitochondria, AIF and caspases-rivaling for cell death execution. Nat Cell Biol. 2003;5:97–99. doi: 10.1038/ncb0203-97. [DOI] [PubMed] [Google Scholar]
- 28.Jäättelä M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat Immunol. 2003;4:416–423. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 29.Ferri KR, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–E263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 30.Ray SK, Neuburger TJ, Deadwyler G, Wilford G, DeVries GH, Banik NL. Calpain and calpastatin expression in primary oligodendrocyte culture: preferntial localization of membrane calpain in cell processes. J Neurosci Res. 2002;70:561–569. doi: 10.1002/jnr.10414. [DOI] [PubMed] [Google Scholar]
- 31.Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- 32.Ding D, Stracher A, Salvi RJ. Leupeptin protects cochlear and vestibular hair cells from gentamicin ototoxicity. Hear Res. 2002;164:115–126. doi: 10.1016/s0378-5955(01)00417-8. [DOI] [PubMed] [Google Scholar]
- 33.Sternlieb I, Goldfischer S. Heavy metals and lysosomes. Front Biol. 1976;45:185–200. [PubMed] [Google Scholar]
- 34.Garner B, Roberg K, Brunk UT. Endogenous ferritin protects cells with iron-laden lysosomes against oxidative stress. Free Radic Res. 1998;29:103–114. doi: 10.1080/10715769800300121. [DOI] [PubMed] [Google Scholar]
- 35.Nilsson E, Ghassemifar R, Brunk UT. Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem J. 1997;29:857–865. doi: 10.1023/a:1026441907803. [DOI] [PubMed] [Google Scholar]
- 36.Tsukada T, Watanabe M, Yamashima T. Implications of CAD and DNase II in ischemic neuronal necrosis specific for the primate hippocampus. J Neurochem. 2001;79:1196–1206. doi: 10.1046/j.1471-4159.2001.00679.x. [DOI] [PubMed] [Google Scholar]
- 37.Gobeil S, Boucher CC, Nadeau D, Poirier GG. Characterization of the necrotic cleavage of poly (ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–594. doi: 10.1038/sj.cdd.4400851. [DOI] [PubMed] [Google Scholar]
- 38.Unal-Cevik I, Kilinc M, Can A, Gursoy-Ozdemir Y, Dalkara T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke. 2004;35:189–194. doi: 10.1161/01.STR.0000136149.81831.c5. [DOI] [PubMed] [Google Scholar]
- 39.Darrouzet J, Guilhaume A. Ototoxicité de la kanamycine au jour le jour. Étude expérimentale en microscopie. électronique Rev Laryng (Bordeaux) 1974;95:601–621. [PubMed] [Google Scholar]
- 40.Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y. Apoptosis of guinea pig cochlear hair cells following aminoglycoside treatment. Eur Arch Otorhinolaryngol. 1998;255:127–131. doi: 10.1007/s004050050027. [DOI] [PubMed] [Google Scholar]
- 41.Ylikoski J, Xing-Qun L, Virkkala J, Pirvola U. Blockade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Hear Res. 2002;166:33–43. doi: 10.1016/s0378-5955(01)00388-4. [DOI] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Cole KK, Perez-Polo JR. Poly (ADP-ribose) polymerase inhibition prevents both apoptotic-like delayed neuronal death and necrosis after H2O2 injury. J Neurochem. 2002;82:19–29. doi: 10.1046/j.1471-4159.2002.00935.x. [DOI] [PubMed] [Google Scholar]