

1. Introduction
The culture of neurons from prenatal and neonatal brains of rodents has become semi-routine, but these brains are small and lissencephalic. Neuron cultures have also been developed from a number of other species. Sheep have a gyrencephalic human-like brain, large enough to provide a substantial number of cells, are easy to husband and have low maintenance requirements. More specific reasons to study sheep neurons include neurological diseases in sheep of interest to humans, notably Scrapie and the inherited lysosomal storage disease, ovine CLN6 ceroid lipofuscinosis, studied as a model of the fatal neurodegenerative human neuronal ceroid lipofuscinoses (Batten disease) (Jolly et al., 1989; Palmer et al., 1992; Jolly and Palmer, 1995; Broom et al., 1998).
Previous attempts to culture neurons from sheep foetuses older than 7 weeks were unsuccessful (Richard et al., 1998; Thore et al., 1998). Neural precursor cells cultured from young (26 day) sheep foetuses were differentiated into mainly bipolar neurons with some tripolar cells (Duittoz and Hevor, 2001) but not the more complex cells typical of the developing mammalian brain. Here we describe stable cultures of neurons that do show this complexity, from older (9–15 week old) sheep foetuses.
2. Methods
2.1. Animals
Foetuses were generated in sheep maintained under New Zealand pastoral conditions by artificial insemination (Kay et al., 1999) and pregnancies confirmed by ultrasound scanning (White et al., 1984). Altogether 28 foetuses were harvested; 2 with a foetal age of 7 weeks, 5 aged 8 weeks, 12 aged 9 weeks, 1 aged 11 weeks, 1 aged 12 weeks, 2 aged 13 weeks, 2 aged 14 weeks and 3 aged 15 weeks. Animal procedures accorded to NIH guidelines and the New Zealand Animal Welfare Act, 1999.
Dams were killed with a captive bolt, the foetuses exsanguinated, the brains removed and placed in Dulbecco’s phosphate buffered saline (DPBS) containing 50 U/ml of penicillin and 50 μg/ml of streptomycin, at 4°C.
2.2. Brain cell preparation
The cerebral hemispheres and cerebellum were weighed, the meninges and choroid plexuses removed, the brain chopped finely and incubated overnight, 4°C, in 0.025% trypsin (Sigma, St Louis, MO, USA), in Ca2+-free Krebs Ringer solution (Sigma), then at 37°C, 30 min, with gentle shaking. Digestion was stopped with 0.05% soybean trypsin inhibitor (Sigma) in Krebs Ringer solution, 1:1. After 10 min at room temperature (RT) with 0.003% DNase (Sigma), tissue fragments were gently triturated using a plastic serological pipette (Nunc, Roskilde, Denmark), large fragments allowed to settle, suspended cells harvested by aspiration and gentle centrifugation, then re-suspended in culture medium. Cells that excluded trypan blue were counted.
Immediate dissociation at 37°C for 30 min was compared in four separate experiments as was dissociation at 37°C for 60 min with papain (Sigma), 30 U/g of brain dissolved in 3.7 mM cysteine, 90 mM Na2SO4, 30 mM K2SO4, 15.8 mM MgCl2, 1 mM HEPES, 1 mM kynurenic acid (Sigma) and 10 μg/ml phenol red, pH 7.4.
2.3. Cell culture
Culture plasticware (Nunc) and German glass coverslips (Bellco, Vineland, NJ, USA) were treated overnight in 15 μg/ml poly-L-lysine (Sigma) in water, followed by overnight incubtion in culture medium containing 10% foetal calf serum (FCS, MultiSer, Biosciences, Sydney, Australia), then equilibrated in culture medium. Plating densities ranged from 500 to 1,000 live cells/mm2. Neurons were cultured at 37°C, saturating humidity, 5 % CO2, in complete DMEM and F12 medium, 1:1, (Gibco, Gaithersburg, MD, USA, and Sigma) modified to contain 20 mM KCl (Mattson and Kater, 1988), 2 mM glutamine and 2% B27 supplement (Gibco). Antibiotics, 50 U/ml of penicillin and 50 μg/ml of streptomycin, and a fungicide, 0.125 μg/ml of amphotericin (Gibco), were also added. Half medium changes were made 1–2 times a week. Non-neuronal glial cell growth was controlled with arabinosofuranosyl cytosine (ara-C, Sigma), as required, by the addition of 10 μM to cultures for 48 h followed by a complete medium change.
Cerebellar neurons were cultured using the same conditions with tri-iodothyronine (T3), 1mM in dimethyl sulphoxide (DMSO) added to give the desired final concentrations, 10–25 nM.
2.4. Neuron identification and characterisation
Neurons were identified in vitro by a phase bright halo around the cell body seen with differential interference contrast optics (Modulation Optics, NY, USA), or neuron specific immunostaining used for all quantitative analysis of fixed cells.
(i) Fixation
Cultures were rinsed with phosphate buffered saline (PBS), pH 7.4, 37°C, and fixed for 40 min at RT with 4% paraformaldehyde (PFA, Sigma) in 0.1 M phosphate buffer, pH 7.4. A 5 h fixation with 5% glutaraldehyde and 2% PFA was used for the immunological detections of γ -aminobutyric acid (GABA) and glutamate. Cultures were rinsed 2x with PBS and stained immediately, or stored at 4°C in PBS containing 0.05% sodium azide.
(ii) Antibodies
Neurons were stained with antibodies against β-III-tubulin (1:5,000, monoclonal, Promega, Sydney, Australia), MAP-2 (1: 5,000, monoclonal, Roche, Auckland, New Zealand), GABA (1:2,000, monoclonal 3A12, Dr. P. Streit, Institute of Brain Research, University of Zurich, Switzerland; Matute and Streit, 1986), glutamate (1:2,000, rabbit, Sigma), calretinin (1:2,000, rabbit, Swant, Bellinzona, Switzerland) and calbindin-D28K (1:500, rabbit, Sigma). Binding was detected with alkaline phosphatase or horseradish peroxidase (1:500, Sigma), or Alexa fluorophore (1:1,000, Molecular Probes Eugene, OK, USA) conjugated second antibodies diluted in 1% goat serum in PBS, pH 7.4, containing 0.2% Triton X-100 (BDH, Poole, England) and 0.04% Thimerosal (Sigma).
(iii) Chromogenic staining
Primary antibodies were incubated in a humid chamber, 4°C, 16 h. All other steps were at RT on a rocking platform, each followed by 3× 10 min washes with 0.2% Triton X-100 in PBS. Secondary antibody incubations were for 2 h. Avidin-biotin amplification systems were sometimes used. Anti-rabbit IgG Vectastain Elite ABC kit reagents (Vector, Burlingame, CA, USA) were diluted 4x more than recommended. Biotinylated anti-mouse IgG and Extravidin–horse radish peroxidase (Extravidin-HRP, Sigma) conjugated reagents were diluted to 1:1,000. Substrate incubations with BCIP/NBT (Sigma) for alkaline phosphatase, or diaminobenzidine (DAB, Sigma) for HRP lasted 10 min. Stained glass coverslips were mounted with PBS:glycerol (1:1) on microscope slides and sealed with Glyceel (BDH). Cultures in plastic dishes were covered with glycerin jelly (7.6% gelatin, 54% glycerol and 0.2% phenol stored at 4°C).
(iv) Fluorescent staining
Primary antibodies were bound to cultures on glass coverslips as above. Secondary antibodies were goat anti-mouse IgG or goat anti-rabbit IgG conjugated to Alexa 488 (green) or Alexa 594 (red) fluorophores (Molecular Probes, 1:1,000). Primary antibodies were applied together for double immunofluorescent labelling, as were the secondary antibodies conjugated to the different fluorophores. Coverslips were mounted on glass microscope slides with Fluoromount (DAKO, Carpinteria, Ca, USA). A Spot RT colour digital camera (Diagnostic Instruments, Sterling Heights, MI, USA) was used or a Leica DMLD photomicrography system on an inverted DMRB microscope (Leica, Wetzler, Germany).
Nuclei were stained with Hoechst 33342 (Molecular Probes), 1 μg/ml in the penultimate rinse, 15 min, and counted to determine cell numbers.
(v) GABAergic and glutamatergic neurons
Immunoreactivities to GABA and glutamate were determined by fluorescent double immunolabelling. 630x images of 10 representative fields of duplicate cultures on 12 mm round coverslips were taken for each filter set. Exposure and gain settings were kept uniform and merged images generated with Spot RT digicam software. β-III-tubulin positive neurons were counted in 10 fields of duplicate coverslips.
(vi) Calcium binding protein distribution in neurons
Calcium binding proteins were detected with alkaline phosphatase conjugated secondary antibody (Sigma) and BCIP/NBT (blue), followed by incubation with the second primary antibody detected with HRP and DAB (yellow, Sigma). Calcium binding protein positive and β-III-tubulin positive neurons were counted in 10 fields in duplicate wells. Fluorescent stainings were performed as above.
2.5. Cell freezing and thawing
Aliquots, 0.5 or 1ml, of 5–10 ×106 live cells/ml suspended in 8% DMSO in FCS were equilibrated at RT, 10 min, then buried in powdered dry ice until deep frozen and stored at −130°C. Cells were thawed at 37°C, diluted 10x in complete DMEM/F12, pelleted and resuspended in culture medium. More sophisticated freezing protocols tested involved suspending cells in 10% glycerol or DMSO in culture medium and cooling them to −35°C at −1°C/min in a Planer R204 cell freezer (UK) and storing them in liquid nitrogen.
2.6. Neuron counts
Neuron numbers were calculated from cell counts along diagonals across β-III-tubulin immunostained dishes. At least 30 areas were counted, about 1% of the culture surface, and enough to ensure that the standard error of the mean (SEM) between field counts was less than 1% of the total cells counted.
2.7. Time-lapse photography
The undersides of Nunc culture dishes were scored with a fine needle to create reference points, the dishes treated with poly-L-lysine and 9-week-old foetal cryopreserved neurons plated out at 300–700 cells/mm2, and cultured in complete DMEM/F12 medium. After 3 days moderately populated fields were photographed at 30 min intervals over 8 h using differential interference contrast optics, 400x magnification. Dishes were returned to the incubator when not being photographed. Cultures were then immunostained for β-III-tubulin. A grid was aligned with the reference points on sequential photographs to locate the same fields and trace neuron movements.
2.8. Estimation of cell cycle length
An average length of the cell cycle of 480 hours was estimated from the DNA doubling time of the 9-week-old foetal sheep brain (McIntosh et al., 1979).
2.9. Mean neurite length
A grid/intersect method was used to determine the average neurite length (McIntyre and Piris, 1981). Fields of β-III-tubulin stained neurons were photographed at 100x and intersections between neurites and 5×5 mm grid lines counted on them along with the number of cells. A calibration curve was generated from grid intersections of randomly distributed rods of known length. The correlation between total test line length and total intersections was R2 = 0.997. Average neurite lengths were estimated from 20 to 293 neurons.
3. Results
3.1. Foetal harvest and cell yields
The results presented here are a summary over 100 different sets of culture experiments of cells from 28 foetuses, frozen cells from some foetuses being used on several different occasions.
Foetuses were harvested at different times of gestation, between 7 and 15 weeks, and a standard protocol for cell dissociation developed, overnight incubation with trypsin at 4°C followed by a 30 min incubation at 37°C, and trituration. Dissociated cells were harvested by gentle centrifugation. At this stage neurons were indistinguishable from non-neuronal cells as tissue dissociation resulted in a complete loss of neurites. About 95% of the cells were viable, as indicated by trypan blue exclusion.
This method was the one adopted routinely, because it fitted in with the timing of foetal harvesting but the overnight incubation at 4°C was not necessary, nor was trypsin the only effective protease. Immediate 30 min trypsin dissociations at 37°C were tested in four experiments as were and papain dissociations. All three incubation protocols resulted in good single cell suspensions, which attached and started neurite formation 24–48 hours after plating. Papain treatment of the older foetal brains yielded preparations with less debris than trypsin digestion. This debris tended to obscure viewing in the first days of culture but it was removed in media changes during the first week and caused no ongoing problems.
The period of foetal development surveyed is one of rapid brain growth, resulting in a steady increase in the total number of cells (neurons and glia) recovered per brain with age; from 100 million cells from cerebral hemispheres of 7-week-old foetuses to 500 million cells from 15-week-old foetuses (Fig. 1). This rapid growth of the brain disguised a drop in the number of cells recovered per gram, from approximately 230 million per gram at 7 weeks to only 18 million per gram at 15 weeks.
Figure 1.
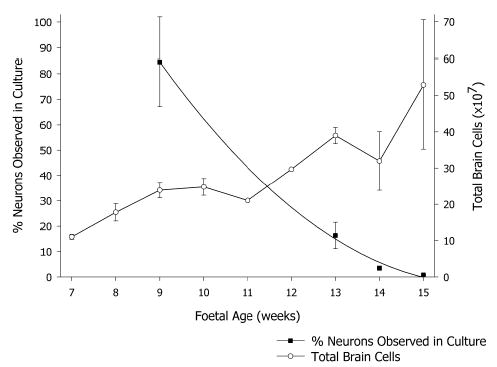
Cell recovery from foetal brains of different ages. Total viable cells (○) recovered from foetuses from 7 to 15 weeks of age (mean ± sem, n ranges from 1 to 9) and the percentage of cells identified as neurons (▪) after 3 weeks in culture (mean ± sem, n ranges from 5 to 13).
Most of the viable cells from younger foetuses became adherent neurons, immediately recognised by phase bright halos seen with differential interference contrast optics (Fig 2), but the proportion declined with foetal age, from close to 100% of cells from 7-week-old foetuses. This decline was more than the overall increase in total cell numbers. Only 9 × 106 neurons were recovered in cultures from 15-week-old foetuses, 2% of the viable cells plated (Fig 1).
Figure 2.
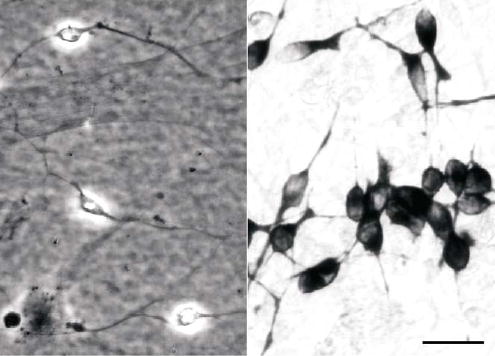
Identification of neurons in culture. Neurons recognised by phase-bright halos seen with differential contrast optics at 2 days in culture (left), and immunostaining against β-III-tubulin after 12 days in culture (right). Scale bar, 20 μm.
Weak perinuclear immunostaining for β-III-tubulin was observed after 2–5 days in culture. Some neurons stained less intensely than others although fine neurites were clearly visible. Antibodies to MAP-2 stained the same cells. Staining for both markers became intense and uniform by 12 days in culture (Fig 2). β-III-tubulin immunostaining for was used for subsequent identification and quantification of neurons.
3.2. Cryopreservation
Each brain yielded far more cells than could be used in immediate experiments. Cryopreservation protocols were examined for saving these for later use. Most effective was simple snap freezing suspensions in 8% DMSO in FCS, by covering the tubes with powdered dry ice. A substantial proportion of cells retained viability through freezing, −130°C storage, and thawing. Viable cell recoveries increased from 23±0.7% (n=2) for cells from 9-week-old foetuses to 48±3.9% (n=6) for cells from 15-week-old foetuses. Freezing and thawing did not affect the proportion of β-III-tubulin positive cells in subsequent culture and the same marked drop in the proportion of neurons in cultures from older foetuses was observed as in fresh cultures, down to under 2% of cells from 15 week old foetuses. This method was adopted routinely. The more sophisticated time-dependent temperature reductions in different concentrations of DMSO or glycerol were not as effective. Four separate trials resulted in unworkably low yields of cells viable in culture, and attempts at these methods were discontinued.
3.3. Neuron cultures
Neurons adhered to and grew well on poly-L-lysine treated plastic and German glass, but neurons from foetuses 9 weeks and older were prone to lift off either surface after 3 to 4 weeks in culture, apparently because an underlying glial cell layer reached confluence, rapidly depleted the medium and lowered the pH. Supplement B27 (Gibco) used routinely in media in place of serum was effective in suppressing this in cultures of cells from 9-week-old foetal brains, but cultures of cells from older foetuses often showed excessive glial cell replication in spite of it. If detected early this proliferation could be controlled with a pulse of arabinosofuranosyl cytosine, (ara-C), but not with a continuous low concentration of ara-C, 0.4 μM, which caused the loss of all neurons within 16 days.
An apparent increase in the number of neurons in some cultures led to an examination of mitotic activity. Simultaneous staining with anti-β-III-tubulin and Hoechst 33342 to detect chromatin revealed telophase nuclei in β-III-tubulin positive cells (Fig. 3). Active division was found in nuclei of 0.06% (42 out of 69936) β-III-tubulin positive cells from a 9-week-old foetus after 3 days in culture.
Figure 3.
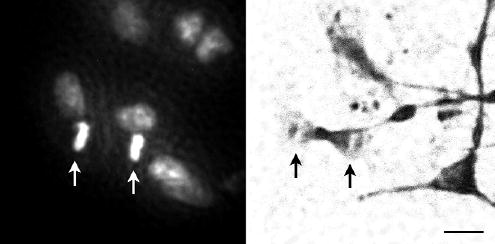
Telophase nucleus (arrows) in a neuron from a 9 week old foetus observed by fluorescence of Hoechst 33342 staining (left) and immunostaining for β-III-tubulin (right). Both pictures are of the same group of cells. Note the presence of nuclei in non-tubulin positive cells. Scale bar, 10 μm.
These divisions were followed by time-lapse photography (Fig. 4). Dividing cells spent about an hour in telophase. Given an average cell cycle length of 480 hours (see Methods) about 30% of cells from 9-week-old foetuses were mitotically active at some stage in culture. Time-lapse photography also revealed random cell mobility on the culture surface (Fig. 5), resulting in a net movement of between 0.55 – 4.9 μm in each 30 min interval. No relationship was obvious between cell mobility and cell morphology.
Figure 4.
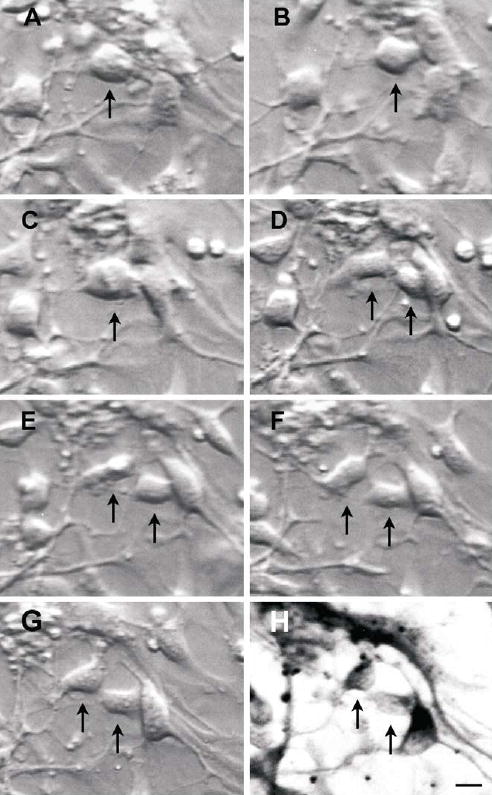
Time-lapse photography of a dividing cell from a 9 week old foetus (↑ ), using differential contrast microscopy at 1h (B), 2.5 h (C) then at hourly intervals (D–G), over 6.5 hours, and the resultant daughter cells immunostained for β-III-tubulin (H). Note the relatively light β-III-tubulin staining, indicative of “new” neurons. Scale bar, 10 μm.
Figure 5.
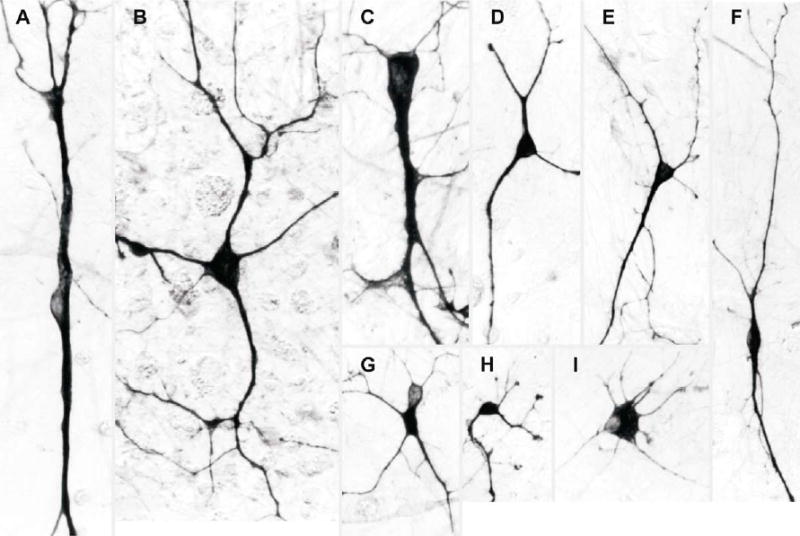
Examples of different neuron morphologies observed. (A). A giant bipolar cell with an oval body 10–12 μm in diameter and coarse neurites of equal thickness. (B). A giant multipolar regular star-shaped neuron with a body 20 μm in diameter with five neurites of variable thickness radiating from it. (C). An asymmetrical triangular giant cell with an elongated body, 30 × 14 μm, a thick neurite on each apex and fine neurites from the base. (D). A regular sized tripolar neuron with a symmetrical rounded body with a longest axis of 8 μm and neurites of equal thickness. (E). A cell with an elongated triangular body with a longest axis of 14 μm, a thick neurite on each apex and fine neurites from the base. (F). A bipolar fusiform cell with a spindle shaped cell body 6–8 μm in diameter and neurites of equal thickness. (G). A multipolar neuron with a darkly stained cell body with a lightly stained blister and two fine “cat whisker-like” neurites. (H). A multipolar neuron with knobbed neurite tips and an oval cell body, 8 μm in diameter. (I). A multipolar neuron with a flat cell body, 20 μm in diameter, and short radiating neurites.
3.4. Morphological characteristics of neurons in culture
Cells from younger foetuses attached overnight and neurite formation began within two days. This sometimes took 3–4 days for cells from foetuses older than 13 weeks. Most neurons were bipolar, tripolar or multipolar (Figs 2, 3, 4, 7 and 8). Various more complex morphologies, (Fig 5), were evident as early as 2 days after plating, but relatively simple bi- and tripolar neurons were still present after several weeks.
Figure 7.
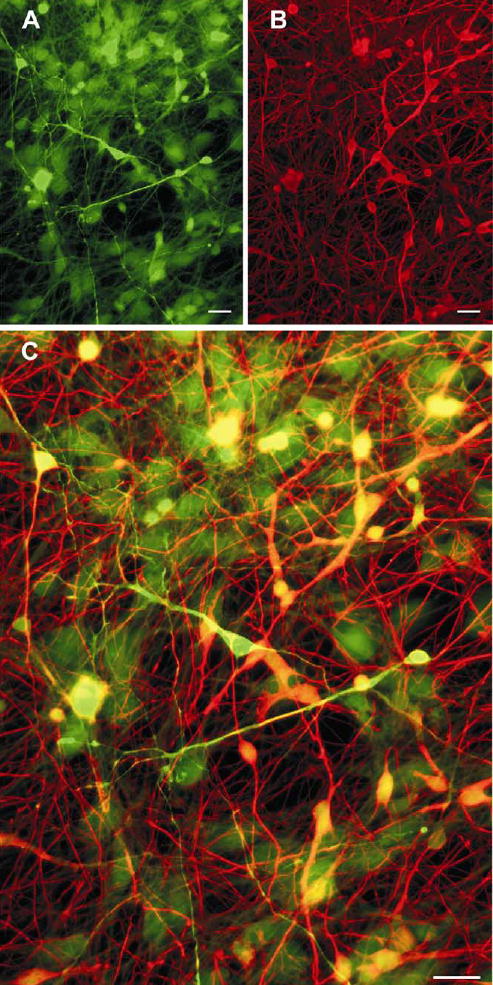
Cultured neurons showing distinct neurotransmitter phenotypes. Double immunolabelling of GABA (green, A) and glutamate (red, B) of neurons from a 10-week-old fetus cultured for 30 days. These images are merged in C. Neurons immunoreactive for GABA only remained red, for glutamate only remained green, and those immunoreactive for both became yellow. Scale bars, 20 μm.
Figure 8.
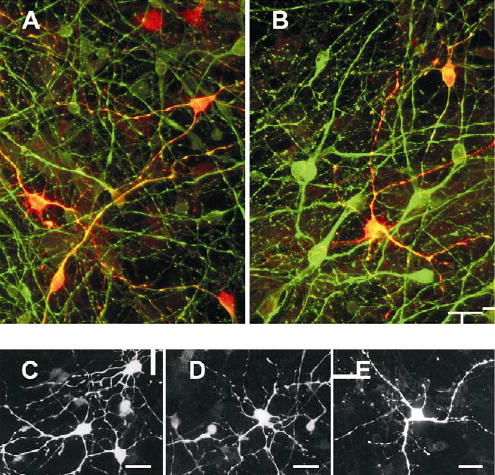
Calcium binding proteins. These are expressed in subsets of neurons from a 10-week-old foetus (A, B), and a 9-week-old foetus (C – E) cultured for 30 days. A and B are merged images double immunolabelled for either calretinin or calbindin-D28k respectively (red), together with β-III-tubulin (green). More complex individual neurons stained for calretinin (C,D) or calbindin (E). Scale bars, 20 μm.
Twelve morphological types were consistently recognisable, classified according to the form of the cell body. The frequencies of these cells were estimated by counting them as a proportion of 168 cells. Bipolar cells with oval bodies 8–10 μm in diameter and neurites of equal thickness, and bipolar asymmetrical cells with oval bodies 8–10 μm in diameter and one very fine neurite were common, 12% and 18% respectively (Figs 2, 7 and 8). Giant cells included bipolar cells with oval bodies 10–12 μm in diameter and coarse neurites of equal thickness, 1.2% (5A); multipolar regular star-shaped neurons with cell bodies 20 μm in diameter and five or more neurites of variable thickness radiating from them, 0.6% (5B); and asymmetrical triangular giant cells with elongated bodies, 30 × 14 μm, a thick neurite on each apex and fine neurites from the base, 0.6% (5C). Regular sized tripolar morphologies included neurons with symmetrical rounded triangular bodies with a long axis of 8 μm and neurites of equal thickness, 7% (5D) and cells with elongated triangular bodies with a longest axis of 14 μm, a thick neurite on each apex and fine neurites from the base, 19% (5E). Bipolar fusiform cells were observed, with spindle shaped bodies 6–8 μm in diameter and neurites of equal thickness, 8% (5F).
Complex morphologies included multipolar neurons with star-shaped cell bodies 10 μm in diameter, and five or more neurites of variable thickness radiating from them, 8% (Fig 8C–E); multipolar neurons with darkly stained cell bodies, 18–27 μm in length, each with a lightly stained blister-like nuclear compartment, and with two fine “cat whisker”-like neurites extending from it, 5% (Fig 5G); multipolar neurons with knobbed neurite tips and oval cell bodies, 8 μm in diameter, with some short neurites terminating in flattened feet, 1.2% (5H); and irregular multipolar neurons with flat cell bodies, 20 μm in diameter, and short radiating neurites, 3.6% (5I).
Variable miscellaneous neuron morphologies were also observed, 16%. Cultures of cells from younger foetuses lacked the giant neurons. Cryopreserved neuron cultures contained the same range of morphologies as fresh ones.
3.5. Neurite extension
Neurites grew faster from neurons from foetuses 9 weeks old than from neurons from 14 –15 week old foetuses, reaching a range of lengths of about 125–170 μm after 12 days in culture, which declined to 80–100 μm after 30 days, as a consequence of a noticeable fasciculation. Neurites of cells from foetuses older than 13 weeks ranged between 50 and 100 μm after 30 days in culture. Initial neurite growth was slightly slower from cryopreserved neurons but growth rates were similar from 12 days.
3.6. Beading
β-III-tubulin positive beading of neurites was common, was seen as early as 5 days in culture, and was not associated with any particular morphology. Both beaded and non-beaded neurons occurred in the same culture (Fig. 6). Beads were regularly spaced, the majority at 5–8μm intervals and the extent and pattern of beading was independent of foetal age. Some beading was at branch points and some not. The beading patterns of freshly plated and cryopreserved neurons were the same.
Figure 6.
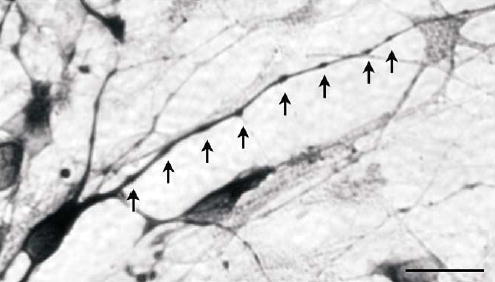
Neurite beading. Beads (↑) on a neuron from a 9-week-old foetus after 18 days in culture. Note the regularity of the beading. Scale bar, 20 μm.
3.7. Neurotransmitters
Individual cells in cultures from 9 and 10-week-old foetuses responded differently to simultaneous GABA and glutamate immunostaining (Fig. 7). GABAergic neurons, defined as those with mainly GABA positive perikarya and neurites, and a compact nuclear morphology were mostly tripolar or multipolar. Relatively few were bipolar. The proportion of GABAergic neurons changed with time in culture. Only 3% of neurons were GABAergic after one week. This rose to 18% after 3 weeks, then declined to 8 ±1.2% (n=4) after 4 weeks.
A second group of mainly bipolar and tripolar cells were immuno-reactive for both GABA and glutamate. Glutamate staining dominated a third population of neurons that included many simple bipolar cells. An extensive network of neurites, and complex pyramidal and multipolar neurons were glutamate positive after 3 weeks in culture (Fig. 7). Astrocytes, recognised by large and flattened nuclei, were weakly immunoreactive to glutamate and GABA.
3.8. Calcium binding proteins
Calcium binding proteins were evident in neurons from 9-week-old foetuses (Fig. 8), 24% being calretinin positive after 5 days in culture and 2.4 % being calbindin positive. These proportions were maintained for 17 days when 25 ± 2.3% ( n=2) were calretinin positive and 2.5 ±0.8% (n=2) calbindin positive. Both proteins were mainly in tripolar and multipolar neurons and their processes, but were not linked to any particular morphology (Fig 8C–E) and many tri- and multipolar neurons remained unstained.
A lower proportion, 4 ±0.8% (n=2), of neurons cultured from 15-week-old foetuses stained for calretinin after 6 days in culture. The number of positive cells increased with time, while the overall number of neurons declined, so that 40 ±6.2% (n=2) of neurons where immunoreactive after 30 days in culture. Occasional calretinin positive neurons with giant multipolar cell bodies and neurites extending several mm were observed. Comparable proportions of calretinin positive neurons, increasing with time, were observed in cultured frozen cells.
3.9. Cerebellar cell cultures
Both fresh and frozen cerebellar granule cells adhered within 24 h and developed neurites. They stained for β-III-tubulin and aggregated in the presence of 10 to 25 nM T3 for 30 days (Fig 9).
Figure 9.
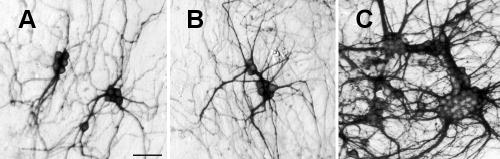
Cerebellar granule neurons from a 14 week foetus showing dose dependent clustering in response to tri-iodothyronine (T3) after 30 days in culture. (A) No added T3, (B) 10 nM T3, (C) 25 nM T3. Scale bar, 25 μm.
4. Discussion
Ovine cerebral cortical neurons from 9 to 15-week-old foetuses and cerebellar neurons from 12 to 15-week-old foetuses were routinely maintained in culture for 3 to 4 weeks, and can be maintained for longer. Cultures from older foetuses are less reliable and culture characteristics depend on the age of the foetus. Nearly all the cells from the younger foetuses adhered as neurons (Fig. 1), but there was a rapid foetal age related drop in the proportion of cells that attached as neurons, and an increasing risk of cultures being overtaken by non neuronal cell proliferation. However the cultures from the older foetuses contained more complex cell types (Fig. 5), perhaps indicative of the increasing complexity of the developing foetal brain, or sensitivity to the growth factor milieu created by accompanying glial cells. β-III-tubulin reactivity marked the same cells as MAP-2 staining and is generally simultaneous with the earliest phase of differentiation to neurons in mammals (Moody et al., 1996), but was delayed while the sheep neurons established themselves in culture. This is a property of sheep neurons, not of technique. Mouse neurons we have cultured using the same techniques were β-III-tubulin positive on adhesion. In this respect the sheep cells behave like amphibian neurons that do not stain for β-III-tubulin until they develop neurites (Moody et al., 1996).
We have clarified the foetal brain age window useful for ovine neuron culture. Brains from foetuses less than 8 weeks old yield relatively few neurons because of the small size, foetuses around 9 weeks are ideal because of the high numbers of neurons obtained that are viable in culture. Older foetuses yield progressively fewer neurons that are increasingly difficult to maintain, but have the advantage that they contain more complex cells and cerebellar neurons can be cultured from them. We did not explore specific culture conditions that may make it easier to maintain the neurons from older foetuses.
Previous studies reported difficulty in culturing cells from sheep foetuses older than 7 weeks (Richard et al., 1998; Thore et al., 1998) and it is not clear why the cultures from older foetuses reported here were much more stable. They open up the study of more complex neurons in culture.
There are several advantages in using sheep foetuses as a source of neurons, instead of the usually used rodents. The number of cells obtained from a single foetus compensates for the extra effort required for its generation, once a satisfactory system of foetal production is in place. Sheep are largely self sufficient in pastoral conditions. Many more simultaneous experiments can be performed with 150 million genetically identical cells from a single 9 week old sheep foetus, compared with 20–60 million genetically heterogeneous cells from a litter of neonatal mice.
Gyri developing in foetal sheep brains provide valuable signposts to identify particular regions. They do not develop in rat brains, which, unlike gyrencephalic sheep and human brains, remain lissencephalic throughout life. Developing sheep brains are big enough for substantial numbers of cells to be obtained from specific regions isolated by dissection.
From about 11 weeks, the cerebellum is large enough to be easily separated from the brain stem and provide large numbers of neurons. Cerebellar neurons isolated from 15 week old foetuses aggregated in the presence of T3 (Fig 9), as do rat cerebellar neurons (Sarafian and Verity, 1986), and remained similarly sedentary under hypothyroid conditions. This is probably reflective of normal brain development, which is dependent on thyroid hormone (Dussault and Ruel, 1987).
The utility of the large cell yield was extended by snap-freezing cells. Viability of the thawed cells, 23–48%, was within 12–60% range reported for freezing rat (Mattson and Kater, 1988, Swett et al., 1994) and human cells (Silani et al., 1988) by various methods, and much better than obtained using more sophisticated programmed time-dependent temperature reductions in cryopreservatives. Neuron cultures from frozen stocks and freshly cultured cells contained similar proportions of the various neuron types. Lifetimes in culture, tubulin staining, neurite growth and beading, calcium binding protein distributions and morphologies were all similar, in keeping with reports of cultures from frozen foetal rat brains (Petite and Calvet, 1995; 1997; Mattson and Kater, 1988).
There is a large variation in the stage of development of mammalian brains at birth, sheep brains being particularly mature. Most neuron culture studies use brains from neonatal rodents, about day 17. These brains are at a similar stage of development as the 9-week-old foetal sheep brains, towards the end of the neurogenic phase in which neuroblasts asymmetrically divide to neurons and the beginning of the glial cell proliferative phase (Temple, 2001; Barlow, 1969). This peaks in sheep around 90 days and is over well before birth (McIntosh et al., 1979). Gyri develop during this period.
The ages of the foetuses used in this study spanned this growth spurt and the cells cultured from them largely reflected the in vivo developmental changes. Cells from the 9-week-old foetuses were mainly neurons or final stage neuroblasts, easy to culture, and cultures contained few non-neuronal cells. A much larger number of cells were obtained from the older foetuses, but a smaller number of neurons, and many more non-neuronal cells, in line with rapid brain growth driven by glial cell production. Cultures of cells from foetuses 13 weeks old and older usually required mitotic inhibitor treatment to prevent overgrowth by non-neuronal cells, serum deprivation alone being insufficient.
Morphologies of the cultured ovine neurons fall into three major classes, bipolar, tripolar and multipolar, based on the shapes of the cell bodies and the number of neurites emanating from them. These are similar to the morphologies of neurons in vivo. Older foetuses also yield more complex neuron types not present in cultures from younger foetuses, notably giant cells, (Fig 5A–C). Stratification of the sheep cerebral cortex takes place between 8–14 weeks (Åström, 1967). These giant neurons resemble the Cajal-Retzuis neurons of layer I having large cell bodies and thick irregular dendrites (Peters and Jones, 1984; Meyer et al., 1998).
Changes in neurite length during culture also mimicked the in vivo situation. The length of both axons and dendrites depends on the neuron type and in which layer it is found. After reaching a peak average length at 12 days in culture, the average neurite length declined, probably due to increasing fasciculation, as has been observed before (Fayaz and Tator, 2000).
Beading is another in vivo phenomenon mimicked in culture, where both beaded and non-beaded neurites are observed (Fig. 6), on cells from all foetal ages. Similar beading occurs on axons and dendrites of a wide range of neurons in vivo (Peters and Jones, 1984).
The ovine neuron cultures also showed the complexity in neurotransmitter class and calcium binding protein content observed in vivo. Neurons fall into two categories, inhibitory (e.g. GABAergic) and excitory (e.g. glutamatergic), and immunostaining for these neurotransmitters has been used to differentiate between the two phenotypes in situ (Marc et al., 1995). Some of the cultured sheep neurons were glutamatergic, some GABAergic, and some were immunoreactive for both (Fig. 7). The initial proportion of neurons that were GABAergic was low, but increased with time in culture to 18%, similar to the proportion found among mammalian cortical neurons in vivo, in the 15–25% range (Ren et al., 1992; Glezer et al., 1993; Amaral, 2000).
The calcium binding proteins, calbindin and calretinin, marking different types of GABAergic interneurons, are found mainly in non-pyramidal neurons (Celio, 1990; Kawaguchi et al., 1995; DeFelipe, 1997). The tripolar and multipolar morphologies of the cultured ovine calretinin and calbindin positive neurons (Fig. 8) resemble interneurons and are similar to those observed in rat neuron cultures (Isaacs et al., 2000). Calbindin and calretinin are also expressed in migrating neuroblasts during development (Meyer et al., 1998; Ulfig, 2002; Ang et al., 2003). Many immature neurons in the rat cerebral cortex transiently express calretinin towards the end of the neuronal migration phase (Schierle et al., 1997). Cajal-Rezius neurons and subpial pyriform cells from the marginal zone of the developing cerebral cortex are also immunoreactive for calretinin (Meyer et al., 1998). These cell types are likely to be represented in ovine neuron cultures from older foetuses.
Two caveats need to be raised to avoid over-interpreting in vivo - in vitro connectedness. Firstly all the cells are rounded at dissociation and it is not clear to what extent this is a de-differentiating event. For many of the older cells it is fatal, shown by much lower adhesion numbers. Proportionalities of cell types and ages will reflect this.
Secondly cells are dividing and dying at all ages. The number of neurons in some cultures increased to exceed the number of cells originally plated, as a result of single cell divisions, observed by time-lapse photography (Fig. 4) and the presence of telophase nuclei (Fig. 3). These are probably the final divisions of neuronal precursors. About 30% of the cultured 9-week-old foetal sheep cells divide to yield tubulin positive neurons in this way, compared to the division of 23% of neurons from the rat ganglion (DiCicco-Bloom et al., 1990). The time related increase in the proportion of calretinin positive cells may be related to an increase in the proportion of younger cells in the cultures. The eventual drop in neuron numbers in culture probably occurs because most final divisions have taken place, exposing a steady loss of neurons as the determinant of neuron number.
These cultures were originally developed to study neurons cultured from sheep affected with CLN6 ovine ceroid lipofuscinosis (Batten disease). Initial experiments suggested pathology in these neurons, in the form of lysosome-derived storage bodies characteristic of the disease (Hughes et al.,1999). Subsequent experiments using cells cultured by the methods here have revealed that this process is more complex, and that there is a glial cell involvement. This mimics the development of pathology in vivo (Oswald et al., 2005). Studies on the interactions of glia and neurons are on-going. Studies on affected cerebellar cells revealed a suppression of the tri-iodothyronine-induced aggregation of cells (Palmer et al., 2002). The cultures will be used to study the ovine CLN6 gene product in neurons. Sheep neuron cultures could be useful for study of other central nervous system diseases such as Scrapie, and for general studies where a large number of cells are advantageous.
Acknowledgments
This work was supported by grants from the New Zealand Neurological Foundation (GWK, DNP), the United States National Institutes of Health, grant NS 40297 (GWK, MJO, DNP) the Canterbury Medical Research Foundation, New Zealand (DNP) and a Leslie Averill Research Fellowship from the Canterbury Medical Research Foundation (MJO). The authors thank John Wynyard, Nadia Mitchell and Nigel Jay for their expert technical assistance.
References
- Amaral DG. The anatomical organisation of the central nervous system. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. 4th ed. New York: McGraw-Hill. 2000: 317–336.
- Ang ESBC, Jr, Haydar TF, Gluncic V, Rakic P. Four-dimensional migratory coordinates of GABAergic interneurons in the developing mouse cortex. J Neurosci. 2003;23:5805–5815. doi: 10.1523/JNEUROSCI.23-13-05805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åström K. On the early development of the isocortex in fetal sheep. Prog Brain Res. 1967;26:1–59. [PubMed] [Google Scholar]
- Barlow RM. The foetal sheep: Morphogenesis of the nervous system and histochemical aspects of myelination. J Comp Neurol. 1969;135:249–262. doi: 10.1002/cne.901350302. [DOI] [PubMed] [Google Scholar]
- Broom MF, Zhou C, Broom JE, Barwell KJ, Jolly RD, Hill DF. Ovine neuronal ceroid lipofuscinosis: a large animal model syntenic with the human neuronal ceroid lipofuscinosis variant CLN6. J Med Genet. 1998;35:717–721. doi: 10.1136/jmg.35.9.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio MR. Calbindin D28-K and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. Types of neurons, synaptic connections and chemical characteristics of cells immunoreactive for calbindin-D28K, parvalbumin and calretinin in the neocortex. J Chem Neuroanat. 1997;14:1–19. doi: 10.1016/s0891-0618(97)10013-8. [DOI] [PubMed] [Google Scholar]
- DiCicco-Bloom E, Townes-Anderson E, Black IB. Neuroblast mitosis in dissociated culture: Regulation and relationship to differentiation. J Cell Biol. 1990;110:2073–2086. doi: 10.1083/jcb.110.6.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duittoz AH, Hevor T. Primary culture of neural precursors from the ovine central nervous system (CNS) J Neurosci Methods. 2001;107:131–140. doi: 10.1016/s0165-0270(01)00363-6. [DOI] [PubMed] [Google Scholar]
- Dussault JH, Ruel J. Thyroid hormones and brain development. Annu Rev Physiol. 1987;49:321–334. doi: 10.1146/annurev.ph.49.030187.001541. [DOI] [PubMed] [Google Scholar]
- Fayaz I, Tator CH. Modelling axonal injury in vitro: injury and regeneration following acute neuritic trauma. J Neurosci Methods. 2000;102:69–79. doi: 10.1016/s0165-0270(00)00282-x. [DOI] [PubMed] [Google Scholar]
- Glezer II, Hof PR, Leranth C, Morgane PJ. Calcium-binding protein-containing neuronal populations in mammalian visual cortex: a comparative study in whales, insectivores, bats, rodents, and primates. Cereb Cortex. 1993;3:249–272. doi: 10.1093/cercor/3.3.249. [DOI] [PubMed] [Google Scholar]
- Hughes SM, Kay GW, Jordan TW, Rickards GK, Palmer DN. Disease-specific pathology in neurons cultured from sheep affected with ceroid lipofuscinosis. Mol Genet Metab. 1999;66:381–386. doi: 10.1006/mgme.1999.2816. [DOI] [PubMed] [Google Scholar]
- Isaacs KR, Wolpoe ME, Jacobowitz DM. Vulnerability to calcium-induced neurotoxicity in cultured neurons expressing calretinin. Exp Neurol. 2000;163:311–323. doi: 10.1006/exnr.2000.7347. [DOI] [PubMed] [Google Scholar]
- Jolly RD, Shimada A, Dopfmer I, Slack PM, Birtles MJ, Palmer DN. Ceroid-lipofuscinosis (Batten's disease): Pathogenesis and sequential neuropathological changes in the ovine model. Neuropathol Appl Neurobiol. 1989;15:371–383. doi: 10.1111/j.1365-2990.1989.tb01236.x. [DOI] [PubMed] [Google Scholar]
- Jolly RD, Palmer DN. The neuronal ceroid-lipofuscinoses (Batten disease): Comparative aspects. Neuropathol Appl Neurobiol. 1995;21:50–60. doi: 10.1111/j.1365-2990.1995.tb01028.x. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson C, Augood S, Emson P. Striatal interneurons: chemical physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kay GW, Hughes SM, Palmer DN. In vitro culture of neurons from sheep with Batten disease. Mol Genet Metab. 1999;67:83–88. doi: 10.1006/mgme.1999.2849. [DOI] [PubMed] [Google Scholar]
- Marc RE, Murry RF, Basinger SF. Pattern recognition of amino acid signatures in retinal neurons. J Neurosci. 1995;15:5106–5129. doi: 10.1523/JNEUROSCI.15-07-05106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Kater SB. Isolated hippocampal neurons in cryopreserved long-term cultures: Development of neuroarchitecture and sensitivity to NMDA. Int J Dev Neurosci. 1988;6:439–452. doi: 10.1016/0736-5748(88)90050-0. [DOI] [PubMed] [Google Scholar]
- Matute C, Streit P. Monoclonal antibodies demonstrating GABA-like immunoreactivity. Histochemistry. 1986;86:147–157. doi: 10.1007/BF00493380. [DOI] [PubMed] [Google Scholar]
- McIntosh GH, Baghurst KI, Potter BJ, Hetzel BS. Foetal brain development in the sheep. Neuropathol Appl Neurobiol. 1979;5:103–114. doi: 10.1111/j.1365-2990.1979.tb00664.x. [DOI] [PubMed] [Google Scholar]
- McIntyre RLE, Piris J. Length measurements by means of a grid. J Clin Path. 1981;34:519–523. doi: 10.1136/jcp.34.5.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Soria JM, Martinez-Galan JR, Martin-Clemente B, Fairén A. Different origins and developmental histories of transient neurons in the marginal zone of the fetal and neonatal rat cortex. J Comp Neurol. 1998;397:493–518. [PubMed] [Google Scholar]
- Moody SA, Miller V, Spanos A, Frankfurter A. Developmental expression of a neuron–specific β-tubulin in frog (Xenopus laevis): A marker for growing axons during the embryonic period. J Comp Neurol. 1996;364:219–230. doi: 10.1002/(SICI)1096-9861(19960108)364:2<219::AID-CNE3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Oswald MJ, Palmer DN, Kay GW, Shemilt MJA, Rezaie P, Cooper JD. Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6) Neurobiol Dis. 2005;20:49–63. doi: 10.1016/j.nbd.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, Jolly RD. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten Disease) Am J Med Genet. 1992;42:561–567. doi: 10.1002/ajmg.1320420428. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Oswald MJ, Westlake VJ, Kay GW. The origin of fluorescence in the neuronal ceroid lipofuscinoses (Batten disease) and neuron cultures from affected sheep for studies of neurodegeneration. Arch Gerontol Geriatr. 2002;34:343–357. doi: 10.1016/s0167-4943(02)00011-0. [DOI] [PubMed] [Google Scholar]
- Peters A, Jones EG. Cerebral cortex Vol 1: Cellular components of the cerebral cortex, Plenum Press, NY. 1984.
- Petite D, Calvet M-C. Cryopreserved neuronal cells in long-term cultures of dissociated rat cerebral cortex: Survival and morphometric characteristics as revealed by immunocytochemistry. Brain Res. 1995;669:263–274. doi: 10.1016/0006-8993(94)01269-n. [DOI] [PubMed] [Google Scholar]
- Petite D, Calvet M-C. Cryopreserved GABAergic neurons in cultures of rat cerebral cortex and mesencephalon: A comparative morphometric study with anti-GABA antibodies. Brain Res. 1997;747:279–289. doi: 10.1016/s0006-8993(96)01287-5. [DOI] [PubMed] [Google Scholar]
- Richard O, Duittoz AH, Hevor TK. Early, middle, and late stages of neural cells from ovine embryo in primary cultures. Neurosci Res. 1998;31:61–68. doi: 10.1016/s0168-0102(98)00024-8. [DOI] [PubMed] [Google Scholar]
- Ren JQ, Aika Y, Heizmann CW, Kosaka T. Quantitative analysis of neurons and glial cells in the rat somatosensory cortex, with special reference to GABAergic neurons and parvalbumin-containing neurons. Exp Brain Res. 1992;92:1–14. doi: 10.1007/BF00230378. [DOI] [PubMed] [Google Scholar]
- Sarafian T, Verity MA. Influence of thyroid hormones on rat cerebellar cell aggregation and survival in culture. Brain Res. 1986;391:261–270. doi: 10.1016/0165-3806(86)90290-7. [DOI] [PubMed] [Google Scholar]
- Schierle GS, Gander JC, D'Orlando C, Ceilo MR, Vogt Weisenhorn DM. Calretinin-immunoreactivity during postnatal development of the rat isocortex: A qualitative and quantitative study. Cereb Cortex. 1997;7:130–142. doi: 10.1093/cercor/7.2.130. [DOI] [PubMed] [Google Scholar]
- Silani V, Pizzuti A, Strada O, Falini A, Buscaglia M, Scarlato G. Human neuronal cell viability demonstrated in culture after cryopreservation. Brain Res. 1988;473:169–174. doi: 10.1016/0006-8993(88)90331-9. [DOI] [PubMed] [Google Scholar]
- Swett JW, Paramore CG, Turner DA. Quantitative estimation of cryopreservation viability in rat fetal hippocampal cells. Exp Neurol. 1994;129:330–334. doi: 10.1006/exnr.1994.1175. [DOI] [PubMed] [Google Scholar]
- Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- Thore CR, Beasley TC, Busija DW. In vitro and in vivo localization of prostaglandin H synthase in fetal sheep neurons. Neurosci Lett. 1998;242:29–32. doi: 10.1016/s0304-3940(98)00040-8. [DOI] [PubMed] [Google Scholar]
- Ulfig N. Calcium-binding proteins in the human developing brain. Adv Anat Embryol Cell Biol. 2002;165:1–92. [PubMed] [Google Scholar]
- White IR, Russel AJF, Fowler DG. Real-time ultrasonic scanning in the diagnosis of pregnancy and the determination of fetal numbers in sheep. Vet Rec. 1984;115:140–143. doi: 10.1136/vr.115.7.140. [DOI] [PubMed] [Google Scholar]